
Abstract
Hereditary hemochromatosis type 4 is an autosomal-dominant inherited disease characterized by a mutation in the SLC40A1 gene encoding ferroportin. This condition can be further subdivided into types 4A (loss-of-function mutations) and 4B (gain-of-function mutations). To date, only a few cases of type 4B cases have been reported, and the treatment has not been clearly mentioned. Here, we report a genotype of hereditary hemochromatosis type 4B involving the heterozygous mutation c.997 T > C (p. Tyr333His) in SLC40A1. The patient was treated with red blood cell apheresis every month for 1 year, followed by oral deferasirox, and the combined therapy was found to be effective.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hereditary hemochromatosis (HH) is a rare disease characterized by dysfunction of the hepcidin–ferroportin axis due to mutations affecting iron metabolism. Autosomal-dominant HH type 4 is characterized by mutations in SLC40A1, which encodes ferroportin (FPN). Adult patients with HH type 4 may present with skin pigmentation, cardiomyopathy, liver cirrhosis, and endocrine dysfunction such as diabetes mellitus; some may not have clinical manifestations and present only with unexplained elevated ferritin levels during annual measurements. The disease can be further divided into type 4A (classical ferroportin disease, FD) and type 4B (FPN-related HH); type 4A is caused by a loss-of-function mutation, while type 4B is caused by a gain-of-function mutation.
Several cases of HH type 4B have been reported worldwide (Supplementary Table 1) [1,2,3,4,5,6,7,8], but treatment plans and efficacy have not been clearly defined. Here, we report a patient with HH type 4B who was received iron chelation leading to a good response.
Case presentation
A 53-year-old woman (proband II:6, Fig. 1) visited our hospital due to unexplained high serum ferritin (SF) and transferrin saturation (TS) found in her annual measurements. She was asymptomatic, but her liver function was impaired with elevated transaminase levels. She had visited several hospitals in the past few years but had failed to obtain a definite diagnosis. The patient had no history of iron supplementations, blood transfusion, or hemolysis.

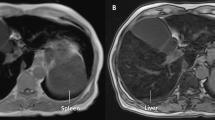
Images of liver MRI (T2WI) before treatment (a) and after treatment (b)
The patient had no family history of skin pigmentation, cardiomyopathy, liver cirrhosis, or endocrine dysfunction; however, her immediate family members, including her 30-year-old daughter, 58-year-old brother, and 62-year-old sister, had similarly elevated SF or TS levels (Table 1). Her father had died of upper gastrointestinal hemorrhage, but it was unknown whether he had a history of liver cirrhosis.
Laboratory examination revealed hemoglobin (Hb) measurements of 13.1 g/dL. The SF and TS levels were up to 5651.33 ng/mL and 99.38%, respectively. Hepcidin was 199.55 ng/mL, alanine transaminase (ALT) was 200 U/L (normal range 0–40 U/L), and aspartate transaminase (AST) was 84 U/L (normal range 0–40 U/L) without evidence of autoimmune hepatitis, viral hepatitis, or a tumor. Tumors and infections were excluded using positron emission tomography and computed tomography (PET/CT). Magnetic resonance imaging (MRI) showed decreased T2 signal intensity in the liver (Fig. 1). The liver iron concentration (LIC) [9, 10], calculated using MRI T2*, was 22.9 mg/g dry weight (normal range < 2 mg/g dry weight), indicating severe iron deposition.
A similar phenotype in the patient’s family members suggested a hereditary iron overload disorder. Next-generation sequencing (NGS) revealed a heterozygous genotype of SLC40A1 (c.997 T > C, p. Tyr333His); no mutations were found in HFE, HJV, HAMP, or TFR2. We confirmed that her daughter and brother carried the same mutation in SLC40A1 using Sanger sequencing (Fig. 2); her sister refused genetic examination. After testing, the woman, her daughter, and her brother were diagnosed with HH type 4B (Fig. 3).

Results of Sanger sequencing

Family tree of the pedigree
The patient underwent red blood cell apheresis once a month for 1 year. Blood tests revealed decreased ALT, AST, SF, and TS levels (Fig. 4). A new MRI was performed showing improved iron deposition in the liver with an LIC of 3.93 mg/g dry weight. She was treated with oral deferasirox (5 mg/kg daily) from August 2019 onward and remained asymptomatic until the last follow-up.

Outcome after treatment. Serum ferritin (a) and aminotransferases (b) decreased during treatment. c TS decreased more than serum iron. d Hb and RET were stable when patient was treated with red blood cell apheresis
Discussion
HH is a rare disease, and patients may visit doctors multiple times due to different initial symptoms. Asymptomatic patients can be easily missed, and diagnosis may be delayed due to an insufficient understanding of the disease. In the case presented here, it took a full year for the patient to be properly diagnosed.
SLC40A1 encodes ferroportin (FPN), a transmembrane protein that exports iron from both the intestinal tract and macrophages to the mammalian bloodstream. FPN is internalized and decomposed by ubiquitination when hepcidin binds to its C-terminal lobe, retaining its outward-open conformation [11, 12]. When the body requires iron, hepcidin levels decrease and FPN is once again expressed. Negative feedback regulation satisfies the need for hematopoiesis while simultaneously avoiding oxidative damage.
HH type 4 is subcategorized into type 4A (classical ferroportin disease, FD) and type 4B (FPN-related HH). Typically, patients with type 4A carry loss-of-function mutation, while patients with type 4B carry gain-of-function mutations that result in iron output deficiency and hepcidin sensitivity [13]. In patients with FD, the accumulation of FPN in lysosomes prevents its further expression on the membrane, leading to iron retention [14, 15]. Patients with FD often develop anemia after phlebotomy because it is difficult for macrophages to provide iron to erythrocytes. Abnormal FPN in HH type 4B patients has difficulty binding to hepcidin; it is internalized because of insufficient ubiquitination at its intracellular end [16, 17]. As hepcidin binds to the FPN C-terminal lobe, patients with HH type 4B show different degrees of hepcidin resistance based on the mutation site. Mutations in the nucleic acids encoding the N-terminal lobe of FPN (such as N144H, G204S, and Y64N) generate partial resistance to hepcidin, whereas mutations in the nucleic acids encoding the C-terminal lobe (including Y333A, C326S, and F324A) lead to strong resistance to hepcidin [11]. Other hepcidin-resistant mutations include V72F and Y501C [18]. Mutations in C326 and H507 affect FPN’s affinity for hepcidin by interacting with cobalt ions, whereas other mutations generally alter binding through changing hydrophobic or backbone atoms [19].
Region F324-S343 of FPN is the suspected binding region of hepcidin [20]. The functional consequences of Y333H mutation were predicted to be deleterious using polymorphism phenotyping v2 (Polyphen-2), sorting intolerant from tolerant (SIFT), and rare exome variant ensemble learner (REVEL). Human FPN Y333 fits into the hydrophobic cavity of hepcidin, forming hydrogen bonds with the hepcidin backbone carbonyl [19]. After a 3-h incubation with hepcidin, wild-type FPN was internalized, but Y333H-mutated FPN remained on the membrane of SLC40A1-expressing 293 T cells. It appears that the Y333H mutation causes FPN hepcidin resistance, resulting in the inability to shut iron channels [5].
Gene mutations involved in HH cause iron overload and excessive iron deposition in the parenchymal organs, resulting in serious oxidative stress, thereby causing patients to develop liver cirrhosis, cardiomyopathy, or endocrine diseases. Although the patient presented in this study was clinically asymptomatic, MRI revealed severe iron deposition in the liver and impaired liver function. The patient’s father had died of upper gastrointestinal hemorrhage, although there was no definite history of liver cirrhosis. Red blood cell apheresis and deferasirox have been used for HH type 2 to reduce iron deposition and prevent disease progression, but this treatment has never been reported for use in HH type 4B [21].
The patient in this case presented with decreased SF and TS levels as well as significantly improved liver T2* during follow-up, indicating an improved iron load both in the circulation and the liver. Her daughter and brother showed a similar phenotype but had relatively low SF. The disease had incomplete penetrance, which may explain the milder iron deposition seen in her daughter and brother [22].
Conclusion
HH should be considered in patients with unexplained high SF and suspected family history, and NGS may be useful for diagnosis. SLC40A1 p.Y333H is a recurrent mutation in HH type 4B patients in China. Red blood cell apheresis and deferasirox treatment can reduce iron deposition in the body and improve organ function.
Data availability
Not applicable.
References
An P, Jiang L, Guan Y, Wang H, Wang J, Tian Y, Yang W, Shi Y, Xue J, Min J, Wang F (2017) Identification of hereditary hemochromatosis pedigrees and a novel SLC40A1 mutation in Chinese population. Blood Cells Mol Dis 63:34–36. https://doi.org/10.1016/j.bcmd.2017.01.002
Wang Y, Du Y, Liu G, Guo S, Hou B, Jiang X, Han B, Chang Y, Nie G (2017) Identification of novel mutations in HFE, HFE2, TfR2, and SLC40A1 genes in Chinese patients affected by hereditary hemochromatosis. Int J Hematol 105(4):521–525. https://doi.org/10.1007/s12185-016-2150-8
Lyu TX, Zhang W, Li XJ, Xu AJ, Zhao XY, Ou XJ, Huang J (2016) Characteristics of gene mutation in Chinese patients with hereditary hemochromatosis. J Clin Hepatol 32(8):1571–1574. https://doi.org/10.3969/j.issn.1001-5256.2016.08.028
Zhang W, Lv T, Huang J, Ou X (2017) Type 4B hereditary hemochromatosis associated with a novel mutation in the SLC40A1 gene: a case report and a review of the literature. Medicine (Baltimore) 96(38):e8064. https://doi.org/10.1097/md.0000000000008064
Zhang W, Xu A, Li Y, Zhao S, Zhou D, Wu L, Zhang B, Zhao X, Wang Y, Wang X, Duan W, Wang Q, Nan Y, You H, Jia J, Ou X, Huang J, China Registry of Genetic/Metabolic Liver Diseases G (2019) A novel SLC40A1 p. Y333H mutation with gain of function of ferroportin: a recurrent cause of haemochromatosis in China. Liver Int 39(6):1120–1127. https://doi.org/10.1111/liv.14013
Zhu CY, Chen Q, Feng ZX (2021) Hereditary hemochromatosis caused by mutation in the SLC40A1 gene: a case report. J Clin Hepatol 37(5):1180–1182. https://doi.org/10.3969/j.issn.1001-5256.2021.05.041
Wu LY, Song ZY, Li QH, Mou LJ, Yu YY, Shen SS, Song XX (2021) Iron chelators reverse organ damage in type 4B hereditary hemochromatosis: case reports. Medicine (Baltimore) 100(13):e25258. https://doi.org/10.1097/md.0000000000025258
Nishina S, Tomiyama Y, Ikuta K, Tatsumi Y, Toki Y, Kato A, Kato K, Yoshioka N, Sasaki K, Hara Y, Hino K (2021) Long-term phlebotomy successfully alleviated hepatic iron accumulation in a ferroportin disease patient with a mutation in SLC40A1: a case report. BMC Gastroenterol 21(1):111. https://doi.org/10.1186/s12876-021-01674-z
Hankins JS, McCarville MB, Loeffler RB, Smeltzer MP, Onciu M, Hoffer FA, Li CS, Wang WC, Ware RE, Hillenbrand CM (2009) R2* magnetic resonance imaging of the liver in patients with iron overload. Blood 113(20):4853–4855. https://doi.org/10.1182/blood-2008-12-191643
Hoffbrand AV, Taher A, Cappellini MD (2012) How I treat transfusional iron overload. Blood 120(18):3657–3669. https://doi.org/10.1182/blood-2012-05-370098
Le Tertre M, Ka C, Guellec J, Gourlaouen I, Ferec C, Callebaut I, Le Gac G (2017) Deciphering the molecular basis of ferroportin resistance to hepcidin: Structure/function analysis of rare SLC40A1 missense mutations found in suspected hemochromatosis type 4 patients. Transfus Clin Biol 24(4):462–467. https://doi.org/10.1016/j.tracli.2017.07.002
Qiao B, Sugianto P, Fung E, Del-Castillo-Rueda A, Moran-Jimenez MJ, Ganz T, Nemeth E (2012) Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab 15(6):918–924. https://doi.org/10.1016/j.cmet.2012.03.018
Majore S, Bonaccorsi di Patti MC, Valiante M, Polticelli F, Cortese A, Di Bartolomeo S, De Bernardo C, De Muro M, Faienza F, Radio FC, Grammatico P, Musci G (2018) Characterization of three novel pathogenic SLC40A1 mutations and genotype/phenotype correlations in 7 Italian families with type 4 hereditary hemochromatosis. Biochim Biophys Acta Mol Basis Dis 1864(2):464–470. https://doi.org/10.1016/j.bbadis.2017.11.006
Sabelli M, Montosi G, Garuti C, Caleffi A, Oliveto S, Biffo S, Pietrangelo A (2017) Human macrophage ferroportin biology and the basis for the ferroportin disease. Hepatology 65(5):1512–1525. https://doi.org/10.1002/hep.29007
Pietrangelo A (2017) Ferroportin disease: pathogenesis, diagnosis and treatment. Haematologica 102(12):1972–1984. https://doi.org/10.3324/haematol.2017.170720
Pietrangelo A (2010) Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology 139(2):393–408. https://doi.org/10.1053/j.gastro.2010.06.013
Altamura S, Kessler R, Grone HJ, Gretz N, Hentze MW, Galy B, Muckenthaler MU (2014) Resistance of ferroportin to hepcidin binding causes exocrine pancreatic failure and fatal iron overload. Cell Metab 20(2):359–367. https://doi.org/10.1016/j.cmet.2014.07.007
Mayr R, Janecke AR, Schranz M, Griffiths WJ, Vogel W, Pietrangelo A, Zoller H (2010) Ferroportin disease: a systematic meta-analysis of clinical and molecular findings. J Hepatol 53(5):941–949. https://doi.org/10.1016/j.jhep.2010.05.016
Billesbolle CB, Azumaya CM, Kretsch RC, Powers AS, Gonen S, Schneider S, Arvedson T, Dror RO, Cheng Y, Manglik A (2020) Structure of hepcidin-bound ferroportin reveals iron homeostatic mechanisms. Nature 586(7831):807–811. https://doi.org/10.1038/s41586-020-2668-z
Preza GC, Ruchala P, Pinon R, Ramos E, Qiao B, Peralta MA, Sharma S, Waring A, Ganz T, Nemeth E (2011) Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J Clin Invest 121(12):4880–4888. https://doi.org/10.1172/JCI57693
Lescano MA, Tavares LC, Santos P (2017) Juvenile hemochromatosis: HAMP mutation and severe iron overload treated with phlebotomies and deferasirox. World J Clin Cases 5(10):381–383. https://doi.org/10.12998/wjcc.v5.i10.381
Han Y, Zhang X (2019) Genetic diagnosis of hereditary hemochromatosis. J Clin Hepatol 35(8):1673–1679
Funding
This study was supported by Grant 2022YFA1103301 of National Key R&D Program of China and Grant 81890992 of National Natural Science Foundation of China.
Author information
Authors and Affiliations
Contributions
JH investigated the pedigree and drafted manuscript; YL interpreted the imaging findings; LF and FZ served as the patient’s physicians; GP analyzed the sequencing data; XZ wrote and revised the manuscript, interpreted the data; all authors issued final approval for the version to be submitted.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Ethical approval
This study was approved by the Ethical Committee of the Institute of Hematology & Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College.
Informed consent
Informed consent was obtained from the patient following the Declaration of Helsinki.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hu, J., Li, Y., Zhang, L. et al. Iron overload due to SLC40A1 mutation of type 4 hereditary hemochromatosis. Med Mol Morphol 56, 233–238 (2023). https://doi.org/10.1007/s00795-023-00359-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00795-023-00359-8