
Abstract
A sensitive and rapid method for the simultaneous analysis of artificial sweeteners, aspartame, acesulfame, cyclamate, saccharine, and phenylalanine in water samples using solid-phase and large-volume sample stacking-capillary electrophoresis (SPE-LVSS-CE) has been developed. Under optimal conditions, the proposed method had a linear range of 0.08 to 2.0 mg L−1, with limits of detection ranging from 0.03 to 0.18 mg L−1 with inter- and intraday repeatabilities < 10% (as a relative standard deviation) in all cases. The enrichment factor obtained was in a range from 20 to 89 times for each artificial sweetener compared with a conventional capillary zone electrophoresis (CZE). The method is adequate to analyze artificial sweeteners in water samples with different ionic strengths. The proposed method was employed in the analysis of 20 samples including drinking water such as mineral water, distilled water, spring water, and tap water. Nine of the tested samples were positive for the presence of aspartame, saccharine, and acesulfame with concentrations between 0.19 and 0.75 mg L−1, 0.08 mg L−1, and 0.08 mg L−1, respectively. The SPE-LVSS-CE is a robust, easy, fast, and efficient strategy for online preconcentration of artificial sweeteners in complex matrices.

Graphical Abstract
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In the last decade, the study, analysis, and determination of different emerging contaminants have been of high relevance. These contaminants are originated from pharmaceuticals, personal care products, and pesticides. In recent years, as a result of changes in the food industry, food additives have been developed to improve the overall quality, safety, nutritive value, appeal, convenience, and cost of food, involving the use of several chemical compounds, including dyes, preservatives, antioxidant agents, thickeners, flavorings, and artificial sweeteners (Tran et al. 2014; Yang et al. 2018).
Artificial sweeteners are chemical compounds widely used as sugar substitutes, and are much sweeter than table sugar (sucrose); therefore, smaller amounts of high-intensity sweeteners are needed to achieve the same level of sweetness. Their use has increased to enhance or modify the flavor in food and pharmaceutical products, contributing only a few calories to the diet in comparison to traditional sugar, without inducing a rise in blood sugar levels (Ordoñez et al. 2013; Tran et al. 2014; Yang et al. 2015). Their excessive use and consumption has been the center of controversy due to possible human and environmental toxic effects such as dermatological problems, headaches, mood variations, alterations on human microbiota, and carcinogenic effects (Tran et al. 2014; Yang et al. 2015; Murray et al. 2016; Roca-Saavedra et al. 2018), and contamination of municipal effluents, surface water, and potable water at concentrations up to a microgram per liter or milligram per liter level (Ordoñez et al. 2013; Tran et al. 2014; Yang et al. 2018).
The European Union (EU) has approved the use of the following artificial sweeteners as food additives: acesulfame (ACE), cyclamate (CYC), saccharine (SAC), sucralose (SUC), and aspartame (ASP), as shown in Fig. 1 (Ordoñez et al. 2013).

Chemical structures of the artificial sweeteners phenylalanine (PHE), aspartame (ASP), cyclamate (CYC), saccharine (SAC), and acesulfame-K (ACE)
Their excessive use, high persistence, environmental distribution, and ecotoxicological impact have become regarded as emerging contaminants. In recent years, possible toxicological problems caused by artificial sweeteners at environmental and human health have incentivized the development of analytical methodologies for determination of artificial sweeteners in complex matrices at milligram per liter and microgram per liter levels; these include high-performance liquid chromatography (HPLC) (Zygler et al. 2009; Ordoñez et al. 2013; Salas et al. 2015), gas chromatography (Hashemi et al. 2011), spectrophotometry (Ni et al. 2009), and electrochemical (Lin et al. 2017) and capillary electrophoresis (CE) (Bergamo et al. 2011; Stojkovic et al. 2013; Yang et al. 2018). These techniques can be used individually or sequentially according to the complexity of the samples, the nature of the matrix, and the type of analyte. Sample treatment, including isolation, clean-up, and pre-concentration, are critical for the analysis of sweeteners at low concentrations. Traditional techniques, such as solid-phase extraction (SPE) (Zygler et al. 2010; Salas et al. 2015; Yang et al. 2018), liquid-liquid extraction (LLE) (Ordoñez et al. 2013; Núñez et al. 2017), and headspace single-drop microextraction (Hashemi et al. 2011), are the most commonly applied strategies to water samples. However, in some cases, these techniques require the use of high volumes of organic solvent, increasing the cost, sample contamination, and the generation of waste products (Yang et al. 2015).
In contrast, CE has received considerable attention as an alternative to traditional methods in recent years due to its potential application in on-line pre-concentration systems, such as transient isotachophoresis (tITP) (Crevillén et al. 2017), dynamic pH junction (Xu et al. 2015), sweeping (Xu et al. 2015), and field-amplified stacking (Li et al. 2012). The main advantages of these methods over traditional techniques are their higher efficiency, shorter analysis time, and lower reagent and sample consumption (Puig et al. 2008; Li et al. 2012).
On-line pre-concentration is performed in the capillary as part of the separation process. It is based on the injection of a larger-than-normal-volume sample into the capillary via hydrodynamic or electrokinetic methods between the sample zone and the background electrolyte due to the electric field, and is known as stacking (Puig et al. 2008; Lee et al. 2012). There are several approaches to stacking that have been studied, including field amplified sample stacking (FASS) and large-volume sample stacking (LVSS) (Lee et al. 2012). LVSS with polarity switching involves injecting a large volume sample into a capillary and removing the sample matrix plug from the capillary by reversing the polarity (Wang et al. 2016). In this technique, the analytes in the long sample zone can be focused into a narrow zone before separation and detection (Chen et al. 2012). LVSS has already been applied to the analysis of several compounds such as metals (Cheng et al. 2016), quinolones (Wang et al. 2016), sulfonamides (Wang et al. 2016), flavonoids (Lee et al. 2012), drugs (Li et al. 2012), amines (Malinina et al. 2018), chiral (Kawai et al. 2012), tetracyclines (Moreno-González et al. 2018), and water (Li et al. 2012), among others.
In this study, a novel method for the analysis of artificial sweeteners in drinking water samples was proposed. This method involved the sample clean-up by SPE, pre-concentration, and separation by LVSS-CE with photodiode array detection (SPE-LVSS-CE). The advantages of the developed method and its application in the determination of artificial sweetener residues in drinking water samples have also been discussed.
Experimental
Reagents and Standards
All solutions were prepared by dissolving the respective analytical-grade reagent in deionized water with one specific resistivity less than 18.0 MΩ cm, which was provided by a Milli-Q system (Millipore, Bedford, MA, USA). Sodium tetraborate, picric acid, sodium hydroxide, and hydrochloric acid were obtained from J.T.Baker (Phillipsburg, NJ, USA), sodium benzoate from Sigma-Aldrich (St. Louis, MO, USA), and methanol obtained from Mallinckrodt Baker (Xalostoc, Mexico).
Standard solutions were prepared by dilution of a stock solution of phenylalanine (PHE), saccharin (SAC), cyclamate (CYC), aspartame (ASP), and acesulfame-K (ACE) at concentrations of 500 and 100 mg L−1 (Sigma-Aldrich; St. Louis, MO, USA). These solutions were stored at 4 °C. Mixed standard working solutions were prepared by diluting the standard stock solution immediately prior to use. The background electrolyte (BGE) solution consisted of 5.0 mM sodium tetraborate and 0.5 mM sodium benzoate, adjusted to pH 9. Picric acid concentrations of 5.00 and 0.15 μg mL−1 were used as internal standards for CZE and LVSS, respectively.
Instrumentation
Electrophoretic experiments were performed using a Beckman Coulter P/ACE 5500 CE system, equipped with diode array detector (DAD). Data were acquired and analyzed using P/ACE MDQ version 2.3 software. A fused silica capillary (41.7 cm × 75 μm i.d.) was used for the separation. A pH/ion analyzer (model 450; Corning Science Products, NY, USA) was used to accurately adjust the pH of the electrolyte solution to within 0.01 pH units.
Capillary Zone Electrophoresis Procedure
At the beginning of each working day, the capillary was activated with 1.0 M NaOH for 15 min, followed by 0.1 M NaOH for 10 min, deionized water for 10 min, and finally BGE solution for 10 min. The capillary was washed between successive analyses using 1.0 M NaOH for 4 min, 0.1 M NaOH for 2 min, deionized water for 2 min, and BGE for 4 min at 20 psi. Peaks were identified by migration time and co-injection of standard solutions.
The conditions used for the separation were a fused silica capillary (41.7 cm × 75 μm i.d.), wavelength (λ) of 215 nm, hydrodynamic injection mode of 0.5 psi for 5 s, capillary temperature of 25 °C, and a separation voltage of 18 kV at normal polarity.
Sample Treatment and Analysis
A 1.0 mL sample was spiked with an internal standard (0.15 mg L−1) in polypropylene tubes and diluted to 10 mL with deionized water. The solution was then passed through a cartridge (Discovery DSC-18 cartridges, 500 mg) previously activated with 5 mL of methanol followed by 5 mL of deionized water at a maximum flow rate of 1 mL min−1. Analytes retained on the SPE cartridge were washed with 2.0 mL of methanol 5.0% (v/v). Retained artificial sweeteners were eluted with 2.0 mL of methanol. The eluted solution was evaporated to dryness, and the residue was dissolved in 1 mL of deionized water.
Samples treated were injected into the capillary system by hydrodynamic injection at 5 psi for 60 s, 3.84 nL of a total volume from the capillary of 1.84 μL according to the Hagen-Poiseuille equation. where L is the total length of the capillary, V is the injection volume, Ƞ is the viscosity of the solvent, △P is the pressure difference between the capillary ends, and d is the capillary I.D. (Vuorensola et al. 2002).
where L is the total length of the capillary, V is the injection volume, Ƞ is the viscosity of the solvent, △P is the pressure difference between the capillary ends, and d is the capillary I.D. (Vuorensola et al. 2002).
The capillary was then placed in BGE vials, and a potential of 8 kV was applied for 60 s (reverse polarity) to pre-concentrate the artificial sweeteners at the inlet, while water and other ions were removed from the capillary. Polarity was then returned at 18 kV, and the CE separation was carried out.
Results and Discussion
Optimization of the Capillary Zone Electrophoresis Procedure
Electrophoretic Separation
The BGE composition is an important factor in the separation of artificial sweeteners by capillary electrophoresis. As previously reported and according to the pKa values, an adequate separation between each artificial sweetener and the internal standard can be achieved with basic media adjusted with electrolyte solutions with pH values from 7.0 to 12.0. Na2B4O7 and NaH2PO4 (25 mM) solutions were studied; according to the results obtained, a better resolution and separation was define in the use of an electrolyte solution with Na2B4O7 (pH 9.0). Using this solution, a systematic study of the effect of pH was carried out between pH 8.0 and 10.0. Finally, pH 9.0 was chosen, as the best resolution of four of the five analytes was obtained. There was a presence of four signals in the electropherograms, attributable to ACE, SAC, PHE, and ASP, and this result was in accordance with the literature, in which the pH used for separation was also 9.0 (Oliveira et al. 2013). At this pH, all the analytes are found in their ionic form (Vistuba et al. 2015).
The effects of electrolyte concentrations on migration time and peak area were further tested from 2.5 to 40 mM under a separation voltage of 18 kV and temperature of 25 °C. The migration time and peak area of the analytes increased with increasing buffer concentration. This is due to the decreasing thickness of the electric double layer between the capillary walls and the buffer solution with increasing buffer concentrations.
Conversely, a higher buffer concentration could increase the coverage of sites on the silica surface, resulting in a decrease in migration speed due to a decrease in the number of silanol groups. Based on the above results, a 5 mM borate buffer (pH 9.0) was selected for higher sensitivity and faster analyses. The separation voltage was also optimized from 12 to 18 kV; as a result of this evaluation, 18 kV was selected as the optimum voltage in the analytical separation. The voltage regulates the current, and therefore, the migration speed and resolution are improved.
The addition of organic modifiers, such as acetonitrile and methanol, did not improve the resolution between the artificial sweeteners; therefore, they were not used. Picric acid, sodium benzoate, and aspartic acid were evaluated as internal standard (IS) for quantification. Picric acid was selected as its migration time did not affect the resolution of the artificial sweeteners studied.
However, the absence of chromophore groups in the chemical structure of CYC indicates that complete identification may be difficult. The use of a chromophore agent was therefore employed for indirect detection (Oliveira et al. 2013).
Optimization of Indirect UV Detection
Indirect UV detection is an important approach in CE to detect weak or non-UV-absorbing substances, which requires the addition of an appropriate chromophore agent to the BGE. In order to obtain favorable indirect detection, a close match in electrophoretic mobility between the chromophore used in the electrolyte and the analytes is necessary. The use of these compounds produces a negative analytical signal in the electropherogram. According to the literature on CE, two principal chromophores have been used for indirect UV detection of CYS, potassium di(2-ethylhexyl)-phthalate, and sodium benzoate. In this study, we investigated these two potential chomophore agents at levels of concentration ranging from 5 to 20 mM added to the electrolyte solution (Oliveira et al. 2013; Moreno-González et al. 2018). The concentrations of the chromophore agents are important to determine the limit of detection (LOD) in indirect UV detection (Malinina et al. 2018). However, for potassium di(2-ethylhexyl)-phthalate, the separation efficiency of the artificial sweeteners was affected considerably at all concentration levels evaluated. For sodium benzoate, separation significantly deteriorated at concentrations above 5.0 mM, affecting the detection of each analyte. Therefore, it was necessary to further investigate sodium benzoate at lower concentrations from 0.5 to 5 mM. From the results, the best separation and identification were achieved using 0.5 mM of benzoate in the electrolyte solution, maximizing the resolution and CYC analytical response.
Finally, picric acid was used as IS. The electropherograms correspond to the analysis of a sample containing the five analytes and the IS. For the identification of each signal, standard solutions were used.
Optimization of the Large-Volume Sample Stacking Procedure
Considering the low sensitivity of CE using DAD detection, we proposed an on-line pre-concentration system to improve the enrichment factors of CE using a simple methodology at low cost. In LVSS, a large-volume sample with low conductivity was hydrodynamically injected into the capillary, and a voltage was applied (reverse polarity). The analytes were stacked at the boundary between the sample and the BGE; after that, the separation was carried out using the CZE procedure (Fig. 2). Therefore, to optimize the system, it was necessary to consider variables such as the injection time, and pre-concentration voltage and time (Malinina et al. 2018).

Diagram of the preconcentration mechanism with LVSS. 1, low-conductivity sample is injected; 2, a negative polarity voltage is applied and the analytes are stacked and the sample matrix is removed out of the capillary; 3, the polarity in the system is switched to normal mode; and 4, the analytes are separated by CZE
Hydrodynamic Sample Injection
The sample injection was carried out using a hydrodynamic technique. In this pre-concentration modality, the injection time is crucial to improving the enrichment factors. In our study, different injection times ranging from 30 to 240 s, with injection pressures from 0.5 to 1.5 psi were evaluated. According to the literature, the analytical signal and peak area increment with the injection time. In our results, at a superior time of 60 s and 0.5 psi, deterioration in analytical signals was observed, and therefore, the on-line pre-concentration technique deteriorated with longer injection times. A remarkable broadening of peaks was observed, leading to a decrease in separation efficiency. As a result, an injection time and injection pressure of 60 s and 0.5 psi, respectively, were selected as adequate conditions for the LVSS.
Optimization of Inversion Electric Current
The selection of an adequate voltage for the LVSS procedure was crucial to ensure sufficient stacking time in the analytical pre-concentration to remove the sample matrix from the capillary without losing analytes (Cheng et al. 2016). In this study, we evaluated stacking voltages from − 6 to − 16 kV at 30 to 240 s. Experimental results showed several differences. At superior voltages greater than 10 kV, the analytical signals for PHE and ASP decreased. This may be explained by the loss of each analyte with time after 60 s. On the other hand, at − 6 kV, the efficiency of separation was affected at baseline, and at − 8 kV, an adequate enrichment factor was obtained without affecting the analytical separation. Finally, the stacking voltage was selected as − 8 kV, which produced a stable current of 2.90 μA. Optimal conditions for analyte stacking were − 8 kV for 60 s. From the results, a clear interaction was observed between the variables in pre-concentration systems using LVSS-CE.
Optimal conditions determined in this study were injection time of 60 s, stacking voltage of − 8 kV, and a pre-concentration time of 60 s.
The proposed methodology (LVSS-CE) was used to determine the artificial sweeteners in water samples. However, the composition of the analyte produces different electrophoretic mobilities, which can be attributed to the ionic strength of the sample. Ionic strength significantly increases the electrophoretic mobility of the analytes, thereby affecting LVSS pre-concentration and causing loss of the analyte if care is not taken when applying the negative polarity (Puig et al. 2008; Chen et al. 2012; Cheng et al. 2016; Wang et al. 2016).
According to the matrix complexity, one of the most important steps in LVSS-CE analysis is sample clean-up. Consequently, the clean-up step was used prior to the analysis by LVSS-CE. SPE was employed as a clean-up procedure in the water samples. This technique decreases the ionic strength effects, making samples suitable for analysis by LVSS-CE. For sample pre-treatment, 1.0 mL of sample was diluted to 10 mL with deionized water, and then passed through an activated C18 SPE cartridge. Analytes retained on the SPE cartridge were washed with two portions of 5.0 mL of deionized water. Retained artificial sweeteners were eluted with 2.0 mL of methanol, according to the methodology reported by Ordoñez et al. (2013). The eluted solution was evaporated to dryness and re-dissolved in 1.0 mL of deionized water containing 0.15 mg L−1 picric acid.
Analytical Parameters
Under optimal conditions, analytical parameters of the LVSS-CE method were evaluated at concentrations of 0.01–2.0 mg L−1 in a ratio of 1:5 for each artificial sweetener. Each standard was prepared and analyzed in triplicate using the proposed methodology. Peak areas were measured, and calibration curves were constructed from the peak area ratios (analyte/internal standard). Calibration curves showed a linear dependence on artificial sweetener concentration. Calibration regression parameters are shown in Table 1. LODs were calculated for a signal-to-noise ratio of 3.29, according to IUPAC recommendations (Currie 1995). The accuracy and precision of the method were measured in terms of intra- and interday repeatabilities for migration times and peak areas. Results were determined as the relative standard deviations (%RSD) obtained in the analysis of the artificial sweeteners at two concentrations. Based on these results, LVSS was adequate for the analysis of artificial sweeteners in water samples.
According to the results, CE-UV provides higher LODs (0.68–8.31 mg L−1) with respect to the LVSS-CE (0.03–0.18 mg L−1). These differences were evidenced by the t-test of two paired samples (tcalc 2.51 > tcrit 2.13). In addition, the analytical sensibility of each regression line (CE-UV and LVSS-CE) was statistically different (p < 0.05) in all the cases, which is evidenced in the enrichment factors obtained for PHE, CYC, ASP, SAC, and ACE of 89.44, 46.36, 27.92, 26.42, and 20.51, respectively.
Application to Real Samples
The proposed methodology SPE-LVSS-CE was developed and applied to the analysis of artificial sweeteners in 20 different water samples, including bottle water (BW), drinking water (DW), spring water (SW), and tap water (TW), obtained from different locations around Pachuca City. The samples consisted of seven BW, seven TW, two SW, and four DW. Three replicate determinations of each analyte in the selected samples were performed. Eight samples were determined to be positive for ASP in different ranges of concentrations depending of the type of sample: three samples were positive for BW with a concentration between 0.38 and 0.75 mg L−1, two samples for TW, 0.23–0.76 mg L−1; two samples for DW, 0.19–0.33 mg L−1; and one sample for SW, 0.49 mg L−1. On the other hand, SAC was found only in BW samples, positive in one sample with a concentration of 0.08 mg L−1. Finally, ACE was positive in one DW sample with a concentration of 0.08 mg L−1; with RSD < 10% in all the cases, as shown in Table 2. The residue concentrations of the artificial sweeteners in the water samples were identified by their migration times. In order to confirm the presence of each analyte, a standard addition was made to the sample extract. An increase in the peak area confirmed the presence of the artificial sweetener residues.
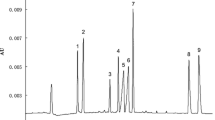
The presence of artificial sweeteners in water samples such as SW indicates that as Ordoñez et al. found, these substances reach aquatic environments as contaminants. The presence of these compounds in TW samples is the result of incomplete water treatment, and is important because this is the water that reaches homes for daily consumption. Finally, the fact that artificial sweeteners were observed in BW and DW samples indicates that these compounds can be found as a possible effect of contamination, and in some cases employed as additives in some industries, though there is no current regulation on their use. The electropherograms obtained are shown in Fig. 3; according to other works, the concentrations found in the water samples are superior than those found in other countries. It can be attributed to the fact that Mexico does not possess norms that regulate their elimination or the correct manipulation in water treatment process, or their use in bottle and drinking water as additives (Buerge et al. 2009).

Electropherograms of (a) standard solution of the artificial sweeteners and IS in concentrations of 1 mg L−1 (SAC, ACE), 20 mg L−1 (IS), and 5 mg L−1 (ASP, PHE, CYC) analyzed by CE procedure; (b) standard solution in concentrations 1 mg L−1 (SAC, ACE, IS) and 5 mg L−1 (ASP, PHE, CYC) analyzed by LVSS-CE procedure; (c) real water sample analyzed by the LVSS-CE procedure, without a cleaning step, dilution system (1:10); (d) real water sample analyzed by the LVSS-CE procedure, with a cleaning step, dilution system (1:10). Experimental conditions: CE procedure (pH = 9 borate buffer, separation voltage 18 kV at 215 nm; LVSS procedure) pH = 9.0 borate buffer; injection by pressure, 0.5 psi; injection time, 1 min; stacking voltage, − 8 kV; separation voltage, + 18 kV
Conclusions
A sensitive, fast, simple, and efficient on-line pre-concentration SPE-LVSS-CE method was developed for the simultaneous analysis of artificial sweeteners in the complex matrices of water samples. The proposed methodology required only 1.0 mL of water sample, whereas traditional methods require at least 10.0 mL. The use of SPE-LVSS-CE improved sensitivity and accuracy compared with CZE, with acceptable LODs for the analysis of water samples with artificial sweetener residues or pollutants. The method achieved enrichment factors ranging from 20 until 89 compared with the conventional injection mode. SPE-LVSS-CE was used to pre-concentrate, identify, and quantify artificial sweeteners in real water samples with satisfactory results, highlighting the great potential of this method.
References
Bergamo AB, Fracassi da Silva JA, Pereira de Jesus D (2011) Simultaneous determination of aspartame, cyclamate, saccharin and acesulfame-K in soft drinks and tabletop sweetener formulations by capillary electrophoresis with capacitively coupled contactless conductivity detection. J Food Chem 124:1714–1717. https://doi.org/10.1016/j.foodchem.2010.07.107
Buerge IJ, Buser H-R, Kahle M, Müller MD, Poiger T (2009) Ubiquitous occurrence of the artificial sweetener Acesulfame in the aquatic environment: an ideal chemical marker of domestic wastewater in groundwater. Environ Sci Technol 43(12):4381–4385. https://doi.org/10.1021/es900126x
Chen Y, Lü W, Chen X, Teng M (2012) Review of recent developments of on-line sample stacking techniques and their application in capillary electrophoresis. Cent Eur J Chem 10(3):611–638. https://doi.org/10.2478/s11532-012-0007-4
Cheng C, Li X, Xie X, Chang F, Li M, Zhu Z (2016) Highly sensitive detection of cooper (I) and cooper (II) in cells specimens by CE-UV with a large-volume sample stacking. Anal Methods 8:4272–4276. https://doi.org/10.1039/C6AY00933F
Crevillén AG, De Frutos M, Diez-Masa JC (2017) On-chip single column transient isotachophoresis with free zone electrophoresis for preconcentration and separation of α-lactalbumin and β-lactoglobulin. Microchem J 133:600–606. https://doi.org/10.1016/j.microc.2017.04.040
Currie LA (1995) Nomencature in evaluation of analytical methods including detection and quantification capabilities (IUPAC Recommendations 1995). Pure Appl Chem 67:1699–1723. https://doi.org/10.1351/pac199567101699
Hashemi M, Habibi A, Jahanshahi N (2011) Determination of cyclamate in artificial sweeteners and beverages using headspace single-drop microextraction and gas chromatography flame-ionisation detection. Food Chem 124:1258–1263. https://doi.org/10.1016/j.foodchem.2010.07.057
Kawai T, Koino H, Sueyoshi K, Kitagawa F, Otsuka K (2012) Highly sensitive chiral analysis in capillary electrophoresis with large-volume sample stacking with an electroosmotic flow pump. J Chromatogr A 1246:28–34. https://doi.org/10.1016/j.chroma.2012.02.001
Lee ISL, Boyce MC, Breadmore MC (2012) Extraction and on-line concentration of flavonoids in Brassica oleracea by capillary electrophoresis using large volume sample stacking. Food Chem 133:205–211. https://doi.org/10.1016/j.foodchem.2012.01.006
Li J, Huang Y, Huang L, Ye L, Zhou Z, Xiang G, Xu L (2012) Determination of imatinib mesylate and related compounds by field amplified sample stacking with large volume sample injection capillary electrophoresis. J Pharm Biomed Anal 70:26–31. https://doi.org/10.1016/j.jpba.2012.05.010
Lin H, Oturan N, Wu J, Sharma VK, Zhang H, Oturan MA (2017) Removal of artificial sweetener aspartame from aqueous media by electrochemical advanced oxidation processes. Chemosphere 167:220–227. https://doi.org/10.1016/j.chemosphere.2016.09.143
Malinina J, Kamencev M, Tkach K, Yakimova N, Kuchumova I, Moskvin N (2018) Large-volume sample stacking for the analysis of low molecular mass amines in steam water by CE using novel highly absorbing probe for indirect UV detection. Microchem J 137:208–213. https://doi.org/10.1016/j.microc.2017.10.018
Moreno-González D, Krulišová M, Gámiz-Gracia L, García-Campaña AM (2018) Determination of tetracyclines in human urine samples by capillary electrophoresis in combination with field amplified sample injection. Electrophoresis 39:608–615. https://doi.org/10.1002/elps.201700288
Murray S, Tulloch A, Criscitelli K, Avena NM (2016) Recent studies of the effects of sugar on brain systems involved in energy balance and reward: relevance to low calorie sweeteners. Physiol Behav 164:504–508. https://doi.org/10.1016/j.physbeh.2016.04.004
Ni Y, Xiao W, Kokot S (2009) A differential kinetic spectrophotometric method for determination of three sulphanilamide artificial sweeteners with the aid of chemometrics. Food Chem 113:1339–1345. https://doi.org/10.1016/j.foodchem.2008.08.068
Núñez M, Borrul F, Pocurull E, Fontanals N (2017) Pressurised liquid extraction and liquid chromatography-high resolution mass spectrometry to determine high-intensity sweeteners in fish samples. J Chromatogr A 1479:32–39. https://doi.org/10.1016/j.chroma.2016.12.006
Oliveira VN, Bellozi L, Pereira J, Vincenzi A, Tonin FG, Leal MA (2013) Simultaneous analysis of aspartame, cyclamate, saccharin and acesulfame-K by CZE under UV detection. Anal Methods 5:1524–1532. https://doi.org/10.1039/C3AY26187E
Ordoñez EY, Quintana JB, Rodil R, Cela R (2013) Determination of artificial sweeteners in sewage sludge samples using pressurised liquid extraction and liquid chromatography-tandem mass spectrometry. J Chromatogr A 1320:10–16. https://doi.org/10.1016/j.chroma.2013.10.049
Puig P, Tempels FWA, Somsen GW, de Jong JG, Borrull F, Aguilar C, Calull M (2008) Use of large-volume sample stacking in on-line solid-phase extraction-capillary electrophoresis for improved sensitivity. Electrophoresis 29:1339–1346. https://doi.org/10.1002/elps.200700636.
Roca-Saavedra P, Mendez-Vilabrille V, Miranda JM, Nebot C, Cardelle-Cobas A, Franco CM, Cepeda A (2018) Food additives, contaminants and other minor components: effects on human gut microbiota-a review. J Physiol Biochem 74:69–83. https://doi.org/10.1007/s13105-017-0564-2
Salas D, Borrul F, Fontanals N, Marcé RM (2015) Hydrophilic interaction liquid chromatography coupled to high-resolution mass spectrometry to determine artificial sweeteners in environmental waters. Anal Bioanal Chem 407:4277–4285. https://doi.org/10.1007/s00216-014-8330-6
Stojkovic M, Mai TD, Hauser PC (2013) Determination of artificial sweeteners by capillary electrophoresis with contactless conductivity detection optimized by hydrodynamic pumping. Anal Chim Acta 787:254–259. https://doi.org/10.1016/j.aca.2013.05.039
Tran NH, Hu J, Li J, Ong SL (2014) Suitability of artificial sweeteners as indicators of raw wastewater contamination in surface water and groundwater. Wat Res 48:443–456. https://doi.org/10.1016/j.watres.2013.09.053
Vistuba JP, Dolzan MD, Vitali L, Leal de Oliveira MA, Micke GA (2015) Sub-minute method for simultaneous determination of aspartame, cyclamate, acesulfame-K and saccharin in food and pharmaceutical samples by capillary zone electrophoresis. J Chromatogr A 1396:148–152. https://doi.org/10.1016/j.chroma.2015.03.070
Vuorensola K, Sirén H, Kostiainen R, Kotiaho T (2002) Analysis of catecholamines by capillary electrophoresis and capillary electrophoresis-nanospray mass spectrometry Use of aqueous and non-aqueous solutions compared with physical parameters. J Chromatogr A 979:179–189. https://doi.org/10.1016/S0021-9673(02)01256-6
Wang N, Su M, Liang S, Sun H (2016) Sensitive residue analysis of quinolones and sulfonamides in aquatic product by capillary zone electrophoresis using large-volume sample stacking with polarity switching combined with accelerated solvent extraction. Food Anal Methods 9:1020–1028. https://doi.org/10.1007/s12161-015-0269-5
Xu X, Jia Z, Shu Y, Liu L (2015) Dynamic pH junction–sweeping technique for on-line concentration of acidic amino acids in human serum by capillary electrophoresis with indirect UV detection. J Chromatogr B 980:20–27. https://doi.org/10.1016/j.jchromb.2014.12.009
Yang L, Zhou S, Xiao Y, Tang Y, Xie T (2015) Sensitive simultaneous determination of three sulfanilamide artificial sweeters by capillary electrophoresis with on-line preconcentration and contact less conductivity detection. Food Chem 188:446–451. https://doi.org/10.1016/j.foodchem.2015.04.060
Yang Y-Y, Zhao J-L, Liu Y-S, Liu W-R, Zhang Q-Q, Yao L, Hu L-X, Zhang J-N, Jiang Y-X, Ying G-G (2018) Pharmaceuticals and personal care products (PPCPs) and artificial sweeteners (ASs) in surface and ground waters and their applications as indication of wastewater contamination. Sci Total Environ 616–617:816–823. https://doi.org/10.1016/j.scitotenv.2017.10.241
Zygler A, Wasik A, Namiésnik J (2009) Analytical methodologies for determination of artificial sweeteners in foodstuffs. Trac-Trend Anal Chem 28:1082–1102. https://doi.org/10.1016/j.trac.2009.06.008
Zygler A, Wasik A, Namiésnik J (2010) Retention behaviour of some high-intensity sweeteners on different SPE sorbents. Talanta 82:1742–1748. https://doi.org/10.1016/j.talanta.2010.07.070
Acknowledgments
The authors wish to thank Programa para el Desarrollo Profesional Docente, para el Tipo Superior (PRODEP) for the approved project in the incorporation of new PTC (Profesores de Tiempo Completo)
Funding
This study was financially supported by Consejo Nacional de Ciencia y Tecnología (CONACyT) (Project INFR-2014-227999 and Retention Grant no. 251112).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of Interest
Lorena Camargo Medrano declares that she has no conflict of interest. Juan Francisco Flores-Aguilar declares that he has no conflict of interest. Gabriela Islas declares that she has no conflict of interest. José Antonio Rodríguez declares that he has no conflict of interest. Israel Samuel Ibarra declares that he has no conflict of interest.
Research Involving Human Participants and/or Animals
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Not applicable.
Rights and permissions
About this article

Cite this article
Medrano, L.C., Flores-Aguilar, J.F., Islas, G. et al. Solid-Phase Extraction and Large-Volume Sample Stacking-Capillary Electrophoresis for Determination of Artificial Sweeteners in Water Samples. Food Anal. Methods 12, 526–533 (2019). https://doi.org/10.1007/s12161-018-1383-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-018-1383-y