
Abstract
Polycyclic aromatic hydrocarbon (PAH) residue concentrations have been measured in honey samples collected on the Italian market. An ultrasound-vortex-assisted dispersive liquid-liquid micro-extraction (UVALLME) procedure coupled with a gas chromatography flame ionization detector or ion trap mass spectrometry (GC-IT/MS) is proposed for fast analysis of fluorene, phenanthrene, anthracene, fluoranthene, pyrene, chrysene, benzo(b)fluoranthene, benzo(a)pyrene, and benzoperylene. Different analytical parameters such as extraction solvent and relative volume, best extraction time, pH, NaCl concentration, and reproducibility at low and high concentrations were optimized. Under optimal conditions, the recoveries range from 95 to 107% and correlation coefficients range from 0.893 to 0.995 whereas the limits of detection (LODs) and limits of quantification (LOQs) are ≥36 and ≥41 ng g−1 in GC-FID and 0.030 and 0.069 ng g−1 in GC-IT/MS, respectively. The precision, expressed as relative standard deviations (RSDs), is ≤7.4 and ≤5.2% for low and high PAH concentration levels, respectively. The whole proposed methodology, demonstrated to be simple, reproducible, and sensible, has been applied to the determination of trace PAHs in five honey samples.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are well-known compounds containing three or more fused benzene rings. Such compounds may be formed and released during combustion and/or pyrolysis processes. Because of combustion of fossil fuels and organic waste, PAHs are ubiquitous in the environment. Differences in the configuration of rings may lead to differences in properties. From a toxicological point of view, the primary human health risk associated to PAH exposure is the cancer (Bostrom et al. 2002), but the relationship between PAH exposure and cardiovascular disease (Kenneth and Moorthy 2005) or poor fetal development (Sram et al. 2005; Suades-González et al. 2015) is also relevant. In any case, the PAH structure influences whether and how the individual compound is carcinogenic (Rubin 2001; Baird et al. 2005). Some carcinogenic PAHs are genotoxic and induce mutations that initiate cancer; others are not genotoxic and instead affect cancer promotion or progression (Ramesh et al. 2004; ATSDR, Environmental Medicine; Environmental Health Education 2011). The exposure can occur through different routes. Basically, the major routes of exposure are from inhaled air and food (Alexander et al. 2008). It is documented that, in non-occupational settings, up to 70% of PAH exposure for a non-smoking person can be associated with diet (Skupinska et al. 2004). Food can be contaminated by both environmental PAHs present in air, soil, or water and industrial food processing methods (e.g., heating, drying, and smoking processes) and home food preparation (e.g., grilling and roasting processes).
Among different highly nutritional foods, honey has valuable compounds; it is valued for its healing and prophylactic properties (Batelková et al. 2012) that result from its composition: the physicochemical and chemical indicators are the image of such characteristics (Lachman et al. 2010). Recent studies have indicated honey as environmental marker for trace element (Conti and Botre 2001), pesticides (Blasco et al. 2003), antibiotic residues (Hammel et al. 2008; Giannetti et al. 2010), and PAH (Dobrinas et al. 2008) contamination. Data on the PAH content in honey are very rare; nevertheless, some authors reported high concentrations of PAHs in honey (Dobrinas et al. 2008). Further, it should be added that PAHs are one of the major factors contributing to the onset of cancer in humans; in this way, it becomes fundamental to develop a quick, simple, and accurate protocol for their determination in such foods (Wenzl et al. 2006). Among the analytical methods available in literature for determining PAHs in honey matrix (Dobrinas et al. 2008; Albero et al. 2003; Perugini et al. 2009; Moret et al. 2010; Lambert et al. 2012; Ciemniak et al. 2013) and according to the large experience of the authors in such field (Russo 2000; Russo and Neri 2002; Russo et al. 2012a; Russo et al. 2012b; Russo et al. 2014a, 2014b; Notardonato et al. 2016), a modified method based on the dispersive liquid-liquid micro-extraction (Cinelli et al. 2014a; Cinelli et al. 2014b; Russo et al. 2014b; Russo et al. 2016) coupled with gas chromatography-flame ionization detector (GC-FID) and ion trap mass spectrometry (GC-IT/MS) has been developed.
Materials and Methods
Materials
Nine PAHs have been investigated: fluorene (abbreviation F; CAS number 86-73-7; chemical formula C13H10; molecular weight 166.222; pKa 22.6; Log K ow, octanol/water partition coefficient, 4.18; median lethal dose, DL50, N/A), phenanthrene (P; 85-01-8; C14H10; 178.233; >15; 4.46; 700 mg kg−1 oral), anthracene (Ant; 120-12-7; C14H10; 178.233; >15; 4.50; 3200 mg kg−1 oral), fluoranthene (Fl; 206-44-0; C16H10; 202.255; >15; 4.90; 2000 mg kg−1 oral), pyrene (Pyr; 129-00-0; C16H10; 202.255; >15; 5.63; >16,000 mg kg−1 oral), chrysene (Chr; 218-01-9; C18H12; >15; 5.63; 228.1928; -), benzo(b)fluoranthene (BbFl; 205-99-2; C20H12; 252.315; >15; 6.04; -), benzo(a)pyrene (BaPyr; 50-32-8; C20H12; 252.3148; >15; 6.06; 50 mg kg−1 subcutanea), and benzoperylene (BghiPer; 191-24-2; C22H12; 276.337; >15; 6.78; -). The PAHs are furnished by Sigma-Aldrich, Milan, Italy. Each PAH standard solution (concentration of 5 mg mL−1) was prepared in acetone: further, each solution was diluted with acetone to prepare final solutions (400 and 20 μg mL−1) for spiking the real samples. Five mix standard solutions (1, 5, 10, 15, and 20 μg mL−1 with the addition of 5 μL of I.S.) were prepared for studying the analytical parameters. Octacosane (C28H58) was used as internal standard (I.S.): 5 mg was dissolved in acetone/iso-octane (9 + 1 v/v) and after the solution was diluted ten times by acetone (0.5 mg mL−1).
The honey samples (no. 5) were purchased in the Italian market: the production year is 2015 whereas the products were produced in Central Italy (Latium and Molise region).
USVADLLME Procedure
A 0.1 g aliquot of each honey sample was transferred into a 10-mL Pyrex tube with a conical bottom and well dissolved in 10 mL of warm hydroalcoholic solution (5% ethanol). After addition of 0.1 g of NaCl (concentration 10 g L−1) and 5 μL of octacosane (0.5 mg mL−1), the extraction procedure is based on 150 μL chloroform as extraction solvent and vortex for 2 min: this step was repeated three times to obtain a stable emulsion. In details, different extraction solvents at different volumes were tested. After 2 min in an ultrasound bath, the solution was further centrifuged at 4000 rpm for 30 min: the micro-drop is formed and the supernatant transferred into a vial. Finally, after sodium sulfate addition for eliminating water residual, 1 μL was injected into GC-FID or GC-IT/MS for PAH determination.
GC-FID and GC-IT/MS Analysis and Quantification
The GC-FID analysis was carried out by means of a gas chromatograph DANI (Monza, Italy) equipped with an electronic flow control system, a programmed temperature vaporizer (PTV) injector, and a FID detector.
A fused-silica capillary column with chemically bonded phase (SE-54, 5% phenyl-95% dimethylpolysiloxane) was prepared in our laboratory (Russo et al. 1985; Cartoni et al. 1986; Russo et al. 1996) with the following characteristics: 30 m × 250-μm i.d., N (theoretical plate number) 125,000 for n-dodecane at 90 °C, K′ (capacity factor) 6.9, d f (film thickness) 0.24 μm, u opt (optimum linear velocity of carrier gas, hydrogen) 38.0 cm s−1, and utilization of theoretical efficiency (UTE%) 92%. The fused-silica capillary column used is very similar to commercial ones showing very good chromatographic efficiency and being more convenient from an economic point of view.
Helium was used as the carrier gas at a constant flow rate of 1 mL min−1. The oven temperature was programmed from 100 to 150 °C in 30 s (at 20.0 °C min−1) and from 150 to 290 °C in 180 s (at 20.0 °C min−1): finally, it was kept 7 min at 290 °C. The PTV injector was performed in splitless mode. Ten seconds after injection, the vaporizer was heated from 110 to 290 °C at 800 °C min−1 and cooled after 120 s; the splitter valve was opened for 120 s.
For the GC-IT/MS analysis, a gas chromatograph Finnigan Trace GC Ultra equipped with an ion trap mass spectrometry detector Polaris Q (Thermo Fisher Scientific, Waltham, MA), a PTV injector, and a PC with a chromatography station Xcalibur (Thermo Fisher Scientific) was used. The capillary column was the same used in the GC-FID analysis. The experimental conditions adopted were as follows: dumping gas in the ion trap at 0.3 mL min−1; transfer line and ion source held at 270 and 250 °C, respectively; PTV kept at 50 °C for 3 and after to 290 °C in 4 min at 14.5 °C min−1; and oven temperature kept for 30 s at 60 °C, after to 150 °C in 120 s (20 °C min−1) and 290 °C in 11 (20 °C min−1). Scan acquisition in positive chemical ionization was from m/z 100 up to 400 a.m.u. at 1.68 scan s−1 and 70 eV.
In both cases, the PAH concentrations were obtained by calibration graphs of the ratio area(PAH)/area(IS,C28) plotted versus each PAH concentration (pg μL−1). All the samples were quantified in triplicate.
Results and Discussion
Evaluation of the UVALLME Procedure
The PAHs investigated in this study, i.e., F, P, Ant, Fl, Pyr, Chr, BbFl, BaPyr, and BghiPer, are listed in the list of “priority pollutants” by US EPA (US EPA 1998): some of them are classified as probably carcinogenic to humans (group 2A) and others as possibly carcinogenic to humans (group 2B) according to the criteria established by the International Agency for Research on Cancer (IARC). According to the European Food Safety Authority (EFSA) CONTAM Panel conclusions (Alexander et al. 2008), it should be considered that BaPyr is not the only appropriate sign of the occurrence of carcinogenic and genotoxic PAHs in foods, but the sum of eight high molecular weight PAHs is important: so, our PAH choice is based on the need to analyze some PAHs at very low levels.
This study is focused to set up an analytical procedure for determining PAHs to be applied to real samples. Further, in the frame of the study, the authors evaluated the availability to use the analytical determination based on GC-FID, which is an equipment worldwide diffused: in this way, the methodology could be proposed as routine method to give accurate and rapid information about the PAH content in this kind of nutritionally high food.
About the cleanup procedure, the DLLME method (Cinelli et al. 2014c) is mainly based on the dispersive solvent: it promotes and helps the action of the extraction solvent finely dispersed in the sample solution. A key role in this procedure could be considered. Actually, in the proposed protocol, any dispersive solvent was not added. In fact, even if the ethanol presence is very low (hydroalcoholic solution 5%), it is sufficient for avoiding the use of dispersive solvent because the ethanol plays the co-surfactant effect (Cinelli et al. 2014b). Further, the dispersion is obtained, and increased as well, by means of endothermic energy furnished by vortex and ultrasounds, i.e., the an ultrasound-vortex-assisted dispersive liquid-liquid micro-extraction (UVALLME). The vortex is also used for dispersing the extraction solvent: the extraction solvent makes a biphasic system, where the phase with higher density is an emulsion. Finally, after the extraction solvent separation by centrifugation, the solution is injected in the GC instrument. All the experiments for optimizing the LLME procedure have been performed on real samples spiked (when it was necessary) with appropriate PAH amount (basically 20 μg mL−1 for each PAH) and using GC-FID analysis. For this aim, 0.1 g of honey (or similar samples) is solubilized in warm distilled water for every test.
For enhancing the extraction recovery, various analytical parameters, which might influence the experiment, were investigated.
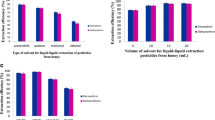
First, the study was focused on the extraction solvent choice based on some criteria such as higher density than water, low solubility in water, high extraction efficiency, and good gas chromatographic behavior (Rezaee et al. 2006). Following these characteristics, five solvents were tested: dichloromethane (CH2Cl2; d 1.33 g cm−3), chloroform (CHCl3; 1.47 g cm−3), carbon tetrachloride (CCl4; 1.5867 g cm−3), 1,1-dichloroethane (C2H4Cl2; 1.2 g cm−3), and 1,1,2,2-tetrachloroethane (C2H2Cl4; 1.59 g cm−3). Each solvent was tested at different volumes. Further, experiments using two different ISs, i.e., octacosane (C28) and dibenzothiophene (DBT), were carried out for evaluating the relative performance.
Table 1 shows the recoveries obtained using 150 μL of chloroform with both C28 and DBT as ISs whereas in Tables 1 and 2 of the Supplementary Material, all the detailed measurements are reported. The other four solvents do not show any significant recoveries; at lower and higher volumes than 150 μL, the recoveries show large variability or, at least, are very poor. In details, the recoveries determined using C28 as I.S. range between 96.8 and 103.9% whereas they vary between 81.7 and 106.6% using DBT.
Further, the reproducibility of the entire analytical method based on chloroform as the extraction solvent (volume 150 μL) has been investigated using both the ISs. In particular, for optimizing the method, two different spiking solutions were considered, i.e., solutions containing 0.1 and 1 μg mL−1 of each PAH, respectively. As reported in Table 2, the recoveries (five replicates) obtained on PAH-spiked real honey samples using C28 as I.S. are still better than those obtained on solutions using DBT as I.S., particularly that the relative standard deviations (RSDs) are very good: they range between 92.5 and 103.9% with a RSD ≤8.1 and between 94.7 and 102.3% with a RSD ≤ 4.9 for samples spiked with 0.1 and 1 μg mL−1 of each PAH, respectively.
As just reported in previous studies, a very critical point regards the salting-out effect: the possible PAH solubility variation in presence of different NaCl concentrations. According to our experience, we tested just three different NaCl concentrations (0, 10, 25 g L−1): the optimum recoveries are reached for addition of NaCl 10 g L−1; above this NaCl concentration, the PAH solubility slightly begins to decrease (salting out) as well as for no NaCl addition. This occurrence confirms the findings of previous studies (Cinelli et al. 2014b, c).
Similar considerations regard the pH influence. First, it should be evidenced that all honeys have an acid reaction, presenting pH values always less than 7, mostly between 3.5 and 4.5. This acidity is essentially due to the presence of numerous organic acids partly already contained in the nectar or honeydew, in part from the bees. The acidity increases with aging, with the fermentation, or is extracted from highly propolis honeycombs. Then, it is important to evaluate the pH value for obtaining best recoveries as possible: two different pH values, i.e., pH 4 (without any addition) and 9 (reached by addition of NaOH 1 M), were studied. Real honey samples were spiked with 1 μg mL−1 of each PAH and 5 μL of I.S. (500 μg mL−1), and the pH was adjusted according to the procedure. The recoveries at pH 9 are significantly lower than those found at pH 4: the gel obtained after addition of strong alkaline species adsorbs analytes and reduces strongly the recoveries. In fact, we would like to remember that PAHs are very weak acids (see pKa and Log K ow reported in “Materials” section): at acid pH, they are in molecular form and the extraction from aqueous solution to organic solvents is better.
So, under the optimized conditions (i.e., honey sample, 0.1 g, spiked with 1 μg mL−1 of each PAH; addition NaCl 10 g L−1; I.S. C28; extraction solvent 150 μL chloroform and 2-min vortex, repeated three times; 2 min of ultrasound bath at 25 °C; 30 min of stirring at 4000 rpm), the mean PAH recoveries range between 96 and 102% with a RSD below 6.3: this shows that the optimized extraction conditions are appropriate for PAH extraction and analysis in honey samples. Under nitrogen flow, the solution has been concentrated up to 10 μL achieving high enrichment factors: 1 μL is further injected for quantification into GC-FID and GC-IT/MS.
Quantification by Means of GC-FID
Table 3 reports the calibration curves with relative R 2: the results, obtained spiking honey samples with PAH at different increasing concentrations and adding 50 μL of I.S. in each, show a good linearity range, R 2 always above 0.89, in the range investigated, 80–1000 ng g−1. Further, the table shows the limits of detections (LODs) and limits of quantifications (LOQs) ranging between 36 and 63 and 41–74 ng g−1, respectively. These values were determined according to the Knoll’s definition (Knoll 1985), i.e., an analyte concentration that produces a chromatographic peak equal to three times (LOD) and ten times (LOQ) the standard deviation of the baseline noise.
Table 3 also shows the reproducibility obtained in spiking honey samples with different PAH standard solution concentrations, i.e., 100, 500, and 1000 ng mL−1 and 5 μL of I.S.: it ranges between 94 and 107% (RSD <12.1), 95–104% (RSD <9.5), and 96–102% (RSD <6.3), respectively: as expected, the parameter improves as the concentration increases, but it is also very good at low PAH concentrations. Finally, the inter- and intra-day precisions at two different concentrations (100 and 1000 ng g−1), evaluated as RSD, are below 8.2 and 6.0%, respectively.
Figure 1 shows the chromatograms of PAH standard solution 100 ng g−1 (Fig. 1a), honey sample (Fig. 1b), and the same honey sample spiked with 100 ng g−1 of each PAH (Fig. 1c).

GC-FID chromatograms of a standard solution (100 ng g−1 of each PAH), b honey sample with no PAH, and c same honey sample spiked with 100 ng g−1 of each PAH (for experimental conditions, see text). Peak list: 1: fluorene; 2: phenanthrene; 3: anthracene; 4: fluoranthene; 5: pyrene; 6: chrysene; IS: octacosane (C28); 7: benzo(b)fluoranthene; 8: benzo(a)pyrene; 9: benzoperylene
Quantification by Means of GC-IT/MS
For achieving better performance in the PAH determination, 1 μL of the final volume is also been injected into the GC-IT/MS instrument. In Table 4, the calibration curves are reported along with the relative R 2 >0.93: in this case, the linearity has been studied in the range 1–500 ng g−1. Taking into account the same definition of LOD and LOQ reported previously, they range between 0.030 and 0.199 ng g−1 (fluoranthene-benzo(a)pyrene) and 0.069 and 0.4656 ng g−1 (fluoranthene-benzo(a)pyrene), respectively: it means that they are much lower than those obtained by GC-FID from 67 to 481 times.
Table 4 also reports the reproducibility obtained in spiking honey samples with different PAH standard solution concentrations, i.e., 5, 50, and 100 ng g−1 and 5 μL of IS. The recoveries range between 91 and 105% (RSD <12.0), 93 and 104 (RSD <7.2), and 95 and 104% (RSD <5.6), respectively: even if they get better with the increase of the concentration, they are very good in any case. Finally, the inter- and intra-day precisions (as RSD) at two different concentrations (1 and 20 ng g−1) are below 6.2 and 7.4%, respectively.
Figures 2 and 3 show the chromatograms in total ion chromatogram (TIC) and selected ion chromatogram (SIM) modes, respectively, of PAH standard solution 5 ng g−1 (Figs. 2a and 3a), honey sample (Figs. 2b and 3b), and the same honey sample spiked with 5 ng g−1 of each PAH (Figs. 2c and 3c): the peaks are well solved and well separated. The chromatograms evidence no contamination problems.

GC-IT/MS chromatograms in TIC mode of a standard solution (5 ng g−1 of each PAH), b honey sample with no PAH, and c same honey sample spiked with 5 ng g−1 of each PAH (for experimental conditions: see text). Peak list: 1: fluorene; 2: phenanthrene; 3: anthracene; 4: fluoranthene; 5: pyrene; 6: chrysene; IS: octacosane (C28); 7: benzo(b)fluoranthene; 8: benzo(a)pyrene; 9: benzoperylene

GC-IT/MS chromatograms in SIM mode of a standard solution (5 ng g−1 of each PAH), b honey sample with no PAH, and c same honey sample spiked with 5 ng g−1 of each PAH (for experimental conditions, see text). Peak list: 1: fluorene; 2: phenanthrene; 3: anthracene; 4: fluoranthene; 5: pyrene; 6: chrysene; IS: octacosane (C28); 7: benzo(b)fluoranthene; 8: benzo(a)pyrene; 9: benzoperylene
Comparison with Similar Studies
Even if the studies regarding the PAH determination in such matrices are very few, some considerations can be drawn. Table 5 reports two important parameters such as the recoveries and LODs/LOQs for the papers present in literature: three studies use GC-MS as analytical methods (Giannetti et al. 2010; Wenzl et al. 2006; Moret et al. 2010) and two the HPLC with fluorescence or spectrofluorometer detection (Albero et al. 2003; Perugini et al. 2009). It can be noted that the UVALLME methodology (this paper) is able to investigate PAHs at levels similar to other methods with a good linear range. The main advantage regards the recoveries obtained in this study: they are very good if compared with the other, the RSD is good. The entire procedure is very easy, and it does not require particular technology such as hollow fiber, or disperser solvent and it takes few minutes.
Application to Different Real Honey Samples
Five different kinds of honey samples have been analyzed using the UVALLME-GC-IT/MS analytical procedure. Particularly, they are wildflower honey, chestnut honey, organic acacia honey, honey orange flowers, and Ambrosoli honey. For each kind of honey sample, different commercial brands were collected on the Italian market. Table 6 shows the levels found in each sample. The concentrations appear to be very low with BaPyr below the LOQ (0.465 ng g−1): the only PAHs detected are F, ranging between LOQ (0.18 ng g−1) and 17.9 ng g−1 and present in all the samples, and fluroranthene and Pyr, present in wildflower, chestnut (only Fl), and Ambrosoli samples.
Conclusions
The method developed allows to investigate PAHs at very low levels in rapid, efficient, and accurate way. The UVALLME-GC-IT/MS analytical procedure is demonstrated to be able to investigate such compounds in a difficult matrix such as honey, considered as an important alimentary food, especially for teenagers and sporty persons. In any case, it is necessary to regulate PAH levels in dietary supplements: for this, the development of highly accurate and precise analytical procedure is fundamental. In the samples analyzed in this study, mainly present on the Italian market, even if BaP is not mandatory for evaluating the carcinogenic characteristics of a food, its level is below LOQ (0.465 ng g−1) whereas the only PAHs detected show a high DL50 to be not so relevant for the human health issue.
References
Albero B, Sánchez-Brunete C, Tadeo JL (2003) Determination of polycyclic aromatic hydrocarbons in honey by matrix solid-phase dispersion and gas chromatography/mass spectrometry. J AOAC Inter 86:576–582
Alexander J, Benford D, Cockburn A, Cravedi J, Dogliotti E, Di Domenico A, Fernández-Cruz ML, Fink Gremmels J, Fürst P, Galli C, Grandjean P, Azyl J, Heinemeyer G, Johansson N, Mutti A, Schlatter J, Van Leeuwen R, Van Peteghem C, Verger P (2008) Scientific opinion of the panel on contaminants in the food chain on a request from the European Commission on polycyclic aromatic hydrocarbons in food 2008. EFSA J 724:1–114 . doi:10.2903/j.efsa.2008.724Available at: http://onlinelibrary.wiley.com/doi/10.2903/j.efsa.2008.724/epdf
ATSDR, Environmental Medicine; Environmental Health Education (2011). Toxicity of polycyclic aromatic hydrocarbons (PAHs): health effects associated with PAH exposure. Available at http://www.atsdr.cdc.gov/csem/pah/docs/pah.pdf; last access on August 2016
Baird WM, Hooven LA, Mahadevan B (2005) Carcinogenic polycyclic aromatic hydrocarbon-DNA adducts and mechanism of action. Environ Mol Mut 45:106–114. doi:10.1002/em.20095
Batelková P, Borkovcová I, Čelechovská O, Vorlová L, Bartáková K (2012) Polycyclic aromatic hydrocarbons and risk elements in honey from the South Moravian region (Czech Republic). Acta Vet Brno 81:169–174. doi:10.2754/avb201281020169
Blasco C, Fernández M, Pena A, Lino C, Silveira MI, Font G, Picó Y (2003) Assessment of pesticide residues in honey samples from Portugal and Spain. J Agr Food Chem 51:8132–8138. doi:10.1021/jf034870m
Bostrom C-E, Gerde P, Hanberg A, Jernstrom B, Johansson C, Kyrklund T, Rannug A, Tornqvist M, Victorin K, Westerholm R (2002) Cancer risk assessment, indicators, and guidelines for polycyclic aromatic hydrocarbons in the ambient air. Environ Health Perspect 110:451–488
Cartoni GP, Goretti G, Monticelli B, Russo MV (1986) Evaluation of capillary gas chromatographic columns in series: analytical application to lemon oil. J Chromatogr 370:93–101. doi:10.1016/S0021-9673(00)94677-6
Ciemniak A, Witczak A, Mocek K (2013) Assessment of honey contamination with polycyclic aromatic hydrocarbons. J Environ Sci Health B 48:993–998. doi:10.1080/03601234.2013.816609
Cinelli G, Avino P, Notardonato I, Centola A, Russo MV (2014a) Study of XAD-2 adsorbent for the enrichment of trace levels of phthalate esters in hydroalcoholic food beverages and analysis by gas chromatography coupled with flame ionization and ion-trap mass spectrometry detectors. Food Chem 146:181–187. doi:10.1016/j.foodchem.2013.09.064
Cinelli G, Notardonato I, Avino P, Russo MV (2014b) Rapid analysis of six phthalate esters in wine by ultrasound-vortex-assisted dispersive liquid-liquid micro-extraction coupled with gas chromatography-flame ionization detector or gas chromatography-ion trap mass spectrometry. Anal Chim Acta 769:72–78. doi:10.1080/03601234.2013.816609
Cinelli G, Avino P, Notardonato I, Russo MV (2014c) Ultrasound-vortex-assisted dispersive liquid-liquid microextraction coupled with gas chromatography with a nitrogen-phosphorus detector for simultaneous and rapid determination of organophosphorus pesticides and triazines in wine. Anal Meth 6:782–790. doi:10.1039/C3AY41641K
Conti ME, Botre F (2001) Honeybees and their product as potential bioindicators of heavy metals contamination. Environ Monit Assess 69:267–282. doi:10.1023/A:1010719107006
Dobrinas S, Birghila S, Coatu V (2008) Assessment of polycyclic aromatic hydrocarbons in honey and propolis produced from various flowering trees and plants in Romania. J Food Compos Anal 27:71–77. doi:10.1016/j.jfca.2007.07.003
Giannetti L, Longo F, Buiarelli F, Russo MV, Neri B (2010) Tetracycline residues in royal jelly and honey by liquid chromatography tandem mass spectrometry: validation study according to Commission Decision 2002/657/EC. Anal Bioanal Chem 398:1017–1023. doi:10.1007/s00216-010-3943-x
Hammel YA, Mohamed R, Gremaud E, LeBreton MH, Guy PA (2008) Multi-screening approach to monitor and quantify 42 antibiotic residues in honey by liquid chromatography-tandem mass spectrometry. J Chromatogr A 1177:58–76. doi:10.1016/j.chroma.2007.10.112
Kenneth RS, Moorthy B (2005) Bioactivation of polycyclic aromatic hydrocarbon carcinogens within the vascular wall: implications for human atherogenesis. Drug Met Rev 37:595–610. doi:10.1080/03602530500251253
Knoll JK (1985) Estimation of the limit of detection in chromatography. J Chromatogr Sci 23:422–425. doi:10.1093/chromsci/23.9.422
Lachman J, Orsák M, Hejtmánková A, Kovářová E (2010) Evaluation of antioxidant activity and total phenolics of selected Czech honeys. LWT Food Sci Technol 43:52–58. doi:10.1016/j.lwt.2009.06.008
Lambert O, Veyrand B, Durand S, Marchand P, Le Bizec B, Piroux M, Puyo S, Thorin C, Delbac F, Pouliquen H (2012) Polycyclic aromatic hydrocarbons: bees, honey and pollen as sentinels for environmental chemical contaminants. Chemosphere 86:98–104. doi:10.1016/j.chemosphere.2011.09.025
Moret S, Purcaro G, Conte LS (2010) Polycyclic aromatic hydrocarbons (PAHs) levels in propolis and propolis-based dietary supplements from the Italian market. Food Chem 122:333–338. doi:10.1016/j.foodchem.2010.02.041
Notardonato I, Avino P, Cinelli G, Russo MV (2016) Rapid and reliable method for analyzing acaricides in honey-based products. Food Anal Meth 9:1675–1685. doi:10.1007/s12161-015-0344-y
Perugini M, Di Serafino G, Giacomelli A, Medrzycki P, Sabatini AG, Persano Oddo L, Marinelli E, Amorena M (2009) Monitoring of polycyclic aromatic hydrocarbons in bees (Apis mellifera) and honey in urban areas and wildlife reserves. J Agric Food Chem 57:7440–7444. doi:10.1021/jf9011054
Ramesh A, Walker SA, Hood DB, Guillen MD, Schneider K, Weyand EH (2004) Bioavailability and risk assessment of orally ingested polycyclic aromatic hydrocarbons. Int J Toxicol 23:301–333. doi:10.1080/10915810490517063
Rezaee M, Assadi Y, Hosseini MRM, Aghaee E, Ahmadi F, Berijani S (2006) Determination of organic compounds in water using dispersive liquid-liquid microextraction. J Chromatogr A 1116:–9. doi:10.1016/j.chroma.2006.03.007
Rubin H (2001) Synergistic mechanisms in carcinogenesis by polycyclic aromatic hydrocarbons and by tobacco smoke: a bio-historical perspective with updates. Carcinogenesis 22:1903–1939. doi:10.1093/carcin/22.12.1903
Russo MV, Goretti G, Liberti A (1985) A fast procedure to immobilize polyethylene glycols in glass capillary columns. J High Resolut Chromatogr 8:535–538. doi:10.1002/jhrc.1240080911
Russo MV, Goretti G, Soriero A (1996) Preparation and application of fused-silica capillary microcolumns (25-50 μm ID) in gas chromatography. Ann Chim (Rome) 86:115–124
Russo MV (2000) Fast solid phase extraction of polychlorobiphenyls and chlorinated pesticide residues from mussels using Sep-Pak cartridges. Chromatographia 51:71–76. doi:10.1007/BF02490698
Russo MV, Neri B (2002) Fluvalinate residues in honey by capillary gas chromatography-electron capture detection-mass spectrometry. Chromatographia 55:607–610. doi:10.1007/BF02492909
Russo MV, Notardonato I, Cinelli G, Avino P (2012a) Evaluation of an analytical method for determining phthalate esters in wine samples by solid-phase extraction and gas chromatography coupled with ion-trap mass spectrometer detector. Anal Bioanal Chem 402:1373–1381. doi:10.1007/s00216-011-5551-9
Russo MV, Avino P, Cinelli G, Notardonato I (2012b) Sampling of organophosphorus pesticides at trace levels in the atmosphere using XAD-2 adsorbent and analysis by gas chromatography coupled with nitrogen-phosphorus and ion-trap mass spectrometry detectors. Anal Bioanal Chem 404:1517–1527. doi:10.1007/s00216-012-6205-2
Russo MV, Notardonato I, Avino P, Cinelli G (2014a) Determination of phthalate esters at trace levels in light alcoholic drinks and soft drinks by XAD-2 adsorbent and gas chromatography coupled with ion trap-mass spectrometry detection. Anal Meth 6:7030–7037. doi:10.1039/C4AY00926F
Russo MV, Notardonato I, Avino P, Cinelli G (2014b) Fast determination of phthalate ester residues in soft drinks and light alcoholic beverages by ultrasound/vortex assisted dispersive liquid-liquid microextraction followed by gas chromatography-ion trap mass spectrometry. RSC Adv 4:59655–59663. doi:10.1039/C4RA08574D
Russo MV, Avino P, Perugini L, Notardonato I (2016) Fast analysis of nine PAHs in beer by ultrasound-vortex-assisted dispersive liquid-liquid micro-extraction coupled with gas chromatography-ion trap mass spectrometry. RSC Adv 6:13920–13927. doi:10.1039/C5RA24873F
Skupinska K, Misiewicz I, Kasprzycka-Guttman T (2004) Polycyclic aromatic hydrocarbons: physiochemical properties, environmental appearance and impact on living organisms. Acta Pol Pharm 61:233–240
Sram RJ, Binkova B, Dejmek J, Bobak M (2005) Ambient air pollution and pregnancy outcomes: a review of the literature. Environ Health Perspect 113:375–382
Suades-González E, Gascon M, Guxens M, Sunyer J (2015) Air pollution and neuropsychological development: a review of the latest evidence. Endocrinol 156:3473–3482. doi:10.1210/en.2015-1403
US EPA (1998) EPA Methods 550.1/610/8100/8270C/8310. Polynuclear aromatic hydrocarbons (PAH) mixture
Wenzl T, Simon R, Kleiner J, Anklam E (2006) Analytical methods for polycyclic aromatic hydrocarbons (PAHs) in food and the environment needed for new food legislation in the European Union. TRAC-Trend Anal Chem 25:716–725. doi:10.1016/j.trac.2006.05.010
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The study was performed with no funds.
Conflict of Interest
Mario Vincenzo Russo declares that he has no conflict of interest. Pasquale Avino declares that he has no conflict of interest. Ivan Notardonato declares that he has no conflict of interest.
Ethical Approval
This article does not contain any studies with human or animal subjects, and so, the ethical approval is not necessary and not required.
Informed Consent
Not applicable. This article does not contain any studies with human or animal subjects.
Electronic supplementary material
ESM 1
(DOCX 24 kb)
Rights and permissions
About this article

Cite this article
Russo, M.V., Avino, P. & Notardonato, I. PAH Residues in Honey by Ultrasound-Vortex-Assisted Liquid-Liquid Micro-Extraction Followed by GC-FID/IT-MS. Food Anal. Methods 10, 2132–2142 (2017). https://doi.org/10.1007/s12161-016-0783-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-016-0783-0