
Abstract
In the present study, an extraction, preconcentration, and determination method has been reported for some synthetic phenolic antioxidants and bisphenol A in honey samples using dispersive liquid–liquid microextraction technique followed by gas chromatography-flame ionization detection. The main factors influencing the extraction efficiency including extractive solvent type and volume as well as the volume of dispersive solvent, salt addition, and pH are evaluated in this study. Under the optimum extraction conditions, limits of detection and quantification for all target analytes were obtained in the ranges of 0.4–4.7 and 1.3–14 ng g−1, respectively. Enrichment factors and extraction recoveries were in the ranges of 144–186 and 72–93 %, respectively. The method precision was evaluated at 100 ng g−1 of each analyte, and the relative standard deviations were found to be less than 7.6 % for intra-day (n = 6) and less than 8.3 % for inter-days (n = 4). The proposed method has been successfully applied to the analysis of different honey samples and two analytes, butylated hydroxytoluene and butylated hydroxyanisole, were determined at nanogram per gram level in one honey sample.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Phenolic compounds are a wide range of substances that can be found in different samples due to their use in several industrial processes, such as the manufacture of plastics, dyes, drugs, antioxidants, etc. Synthetic phenolic antioxidants (SPAs), mainly butylated hydroxytoluene (BHT) and butylated hydroxyanisole (BHA) are widely added to different samples. Also bisphenol A (BPA) is used to produce epoxy resins and polycarbonates. Therefore, these compounds are typically found in domestic and industrial products, waters, and foods (Rodil et al. 2010; Lin et al. 2013; Richard Prabakar and Sriman Narayanan 2010; Sun et al. 2012). The SPAs provide a high level of protection in maintaining food product quality, but an excess of antioxidants is a health risk (Williams 1994), and SPAs’ usage is strictly monitored in most countries. It is important to determine their contents in those materials that are used by people. Analytical methods must be able to determine the presence of these synthetic compounds at very low concentrations in different samples. Several analytical methods for the determination of these synthetic compounds have been reported. High-performance liquid chromatography (HPLC) (Huang et al. 2008; Li et al. 2009; Bahruddin et al. 2007) and gas chromatography (GC) methods (Gonzalez et al. 1999; Yang et al. 2002) are common in determination of BHT, BHA, and BPA. Several approaches have been reported for the extraction of SPAs from different matrices which liquid–liquid extraction (LLE) (Soliman et al. 2007) and solid-phase extraction (SPE) (Trenholm et al. 2008; Remberger et al. 2003) are the most commonly used techniques. LLE and SPE need large amounts of sample and/or organic solvents and they are time-consuming and expensive, and the materials used are not reusable. Therefore, many efforts have been directed towards miniaturizing the extraction procedure by greatly reducing the extraction solvent to aqueous phase volume ratio leading to the development of solvent microextraction methods. As a result, solid-phase microextraction (SPME) (Hernández et al. 2007; Arthur and Pawliszyn 1990; Lord and Pawliszyn 2000) and liquid-phase microextraction (LPME) (Jeannot and Cantwell 1996; Shen and Lee 2002; Ahmadi et al. 2006) have been developed. LPME as a solvent-minimized sample preparation procedure is inexpensive, and several microliters of an extracting solvent are used. One of the main approaches of LPME is single-drop microextraction (SDME) (Liu and Dasgupta 1996). However, the application of SDME is limited by the relatively long extraction time and is significantly affected by the stirring rate. Hollow-fiber liquid-phase microextraction (HF-LPME) (Rasmussen and Pedersen-Bjergaard 2004; Psillakis and Kalogerakis 2003; Melwanki et al. 2005) method overcomes some of these drawbacks. Typically, the extraction efficiency obtained with HF-LPME is higher than SDME due to the larger contact area between the analyte aqueous solution and extraction phase. However, in some cases, higher extraction time, carryover, and contaminations of the HF can be drawbacks of the method.
In 2006, Assadi and co-workers developed a new LPME technique as a highly efficient and powerful preconcentration method termed as dispersive liquid–liquid microextraction (DLLME) (Rezaee et al. 2006). This method is based on a ternary component solvent system including an extraction solvent, a disperser solvent, and an aqueous sample containing the compounds of interest. In this method, an extraction solvent is mixed with a water-miscible polar solvent (as a dispersive solvent) with a suitable proportion, and the mixture is rapidly injected into an aqueous solution by a syringe. A cloudy solution containing fine droplets of the extraction solvent dispersed entirely into the aqueous phase is formed. The analytes in the aqueous phase are extracted into the dispersed fine droplets of the extraction solvent, which is further separated by centrifugation, and the enriched analytes in the sedimented phase are determined by conventional analytical techniques. DLLME provides many advantages such as relatively high recoveries, high enrichment factors (EFs), simplicity, rapidity, easy to operate, low consumption of organic solvents, and so on. It has been used in extraction and preconcentration of different compounds from various samples (Jofre et al. 2010; Bidari et al. 2011; Rusnakova et al. 2011; Yan et al. 2010; Khodadoust and Hadjmohammadi 2011; Farajzadeh et al. 2009, 2012a, b, 2014a, b; Farajzadeh and Nouri 2012).
The goal of this study was to develop a simple and sensitive procedure for the analysis of BHT, BHA, BPA, and methyl-tert-butyl phenol (MTBP) (an intermediate in synthesis of BHT) in honey using DLLME combined with gas chromatography-flame ionization detection (GC-FID). It is noted that those antioxidants are extensively used in food industry. They are added to a wide variety of foods in order to improve their stability and especially to prevent rancidity in products. On the other hand, food products may also be contaminated by migration from polymers containing synthetic phenolic antioxidants and BPA coming into content with the food migration during processing and storage. The proposed analytical method consists of two steps: (i) extraction of the analytes from honey sample and (ii) performing DLLME for enrichment of them. In the first step, the selected analytes are extracted into acetonitrile which is used as a disperser in the following DLLME method. The effects of different experimental parameters on the extraction procedure performance are studied, and the optimal conditions are selected. Finally, the proposed method will be applied to investigate concentrations of the target analytes in honey samples prepared from different sources. To the best our knowledge, up to now, no study about using DLLME for the extraction and preconcentration of SPAs and BPA in honey has been reported.
Experimental
Reagents and Solutions
All analytes used (BHT, BHA, BPA, and MTBP) were purchased from Sigma (St. Louis, Mo, USA). The tested extraction solvents were supplied from the following sources: chloroform, 1,2-dibromoethane (1,2-DBE), 1,1,1-trichloroethane (1,1,1-TCE), and carbon tetrachloride were from Merck (Darmstadt, Germany), and 1,1,2,2-tetrachloroethane (1,1,2,2-TCE) was from Janssen Chimica (Beerse, Belgium). HPLC-grade acetonitrile, methanol, dimethyl sulfoxide (DMSO), dimethyl formamide (DMF), and acetone tested as extraction/disperser solvents for the analytes from honey samples and DLLME step were obtained from Merck. De-ionized water (Ghazi Company, Tabriz, Iran) was used for preparation of aqueous solutions. Analytical reagent grade sodium chloride, hydrochloric acid, and sodium hydroxide were purchased from Merck. A mixture stock solution of the selected analytes was prepared in methanol at a concentration of 1,000 μg mL−1 (each analyte). Two other standard solutions (100 and 10 μg mL−1) were prepared by dilution of the above mentioned solution with methanol. Working standard solutions were daily prepared by appropriate dilutions of the stock solution with de-ionized water. A standard solution of analytes (500 mg L−1 of each analyte) in 1,1,1-TCE was injected into GC-FID (three times in a day), and the obtained analytical signals (peak areas) were used for the calculation of enrichment factors and extraction recoveries. Calibration graphs were prepared by spiking a honey sample (analyte-free) with the analytes at eight levels (5–20,000 ng g−1 for MTBP and BHT, 12–20,000 ng g−1 for BHA, and 48–20,000 ng g−1 for BPA).
Samples
Four honey samples of different floral origins from the following sources were selected: two honey samples were directly obtained from beehives located in virgin mountainous lands (Kaleybar, East Azarbaijan Province, Iran), and two honey samples packed in polyethylene packages and refined by different companies were purchased from local supermarkets. A 2.0-g honey was mixed with 1 mL de-ionized water and manually shaken to obtain a homogeneous solution. This solution was left to equilibrate for at least 15 min prior to performing the proposed extraction method. Then, it was directly subjected to the microextraction procedure without filtration or any other pretreatment.
Procedure for Extraction of the Analytes from Honey Samples
Diluted honey sample (see Section 2.2) spiked with 1 μg g−1 (of each analyte, and 20 μL of a standard solution prepared in methanol at a concentration of 100 μg mL−1 was added) was transferred into a glass test tube. Then, 1.5 mL acetonitrile was added to the tube, and after manually shaking for 1 min, the mixture was centrifuged at 4,000 rpm for 5 min. After centrifuging, 1.1 mL organic phase (acetonitrile) was collected at the upper of aqueous phase, and the analytes were extracted into it. After that, 1.0 mL of organic phase was removed and applied in the following DLLME stage.
DLLME Procedure
To perform DLLME method, 5.0 mL de-ionized water was placed in a 10-mL glass test tube with conical bottom. To 1.0 mL of the organic phase obtained from the previous step, 40 μL 1,1,1-TCE (as an extraction solvent) was added, and the obtained solution was filled into a 5-mL glass syringe and injected rapidly into the de-ionized water. In this step, a cloudy solution resulting from dispersion of fine droplets of 1,1,1-TCE by acetonitrile into aqueous solution was formed, and the analytes were extracted into 1,1,1-TCE. In order to separate the organic phase from the aqueous phase, the mixture was centrifuged at 4,000 rpm for 5 min. After this process, the dispersed fine droplets of the extraction solvent (1,1,1-TCE) were sedimented at bottom of the conical test tube. Volume of the sedimented phase was measured using a 25-μL HPLC syringe and was 10 ± 1 μL. Finally, 1 μL of the sedimented phase was removed and injected into GC system for analysis.
Instrumentation
A gas chromatograph (GC-2014, Shimadzu, Kyoto, Japan) equipped with a split/splitless injector system and an FID was used for separation and determination of the selected analytes. Helium (99.999 %, Gulf Cryo, United Arabic Emirates) was used as the carrier gas at a constant linear velocity of 30 cm s−1 and make up gas (40 mL min−1). Chromatographic separation was achieved on a DB-1 capillary column (100 % poly dimethyl siloxane, 30 m × 0.25 mm i.d. with a 0.25-μm stationary film thickness) (Agilent Technologies, USA). The oven temperature was programmed as follows: initial temperature 80 °C (held 2 min), then ramped to 300 °C at a rate of 10 °C min−1 and held at 300 °C for 5 min. The FID temperature was maintained at 300 °C. Hydrogen gas was generated with a hydrogen generator (OPGU-1500S, Shimadzu, Japan) for FID at a flow rate of 30 mL min−1. Air flow rate for FID was 300 mL min−1. A Metrohm pH meter model 654 (Herisau, Switzerland) was used for pH measurements. Gas chromatography-mass spectrometry (GC-MS) analysis was carried out on an Agilent 7890A gas chromatograph with a 5975C mass-selective detector (Agilent Technologies, CA, USA). Helium was used as carrier gas at a flow rate of 1.0 mL min−1. The oven temperature programming and capillary column were the same as GC-FID analysis mentioned above. The Hettich centrifuge model D-7200 (Germany) was used for accelerating phase separation.
Analytical Parameters
Two main parameters, namely EF and extraction recovery (ER), have been employed for evaluation of the proposed method. EF is defined as the ratio between the analyte concentration in the sedimented phase (C sed) and the initial concentration of analyte (C 0) within the sample:
C sed is obtained by comparison with peak areas of the analytes obtained by directly injecting the standard solution prepared in the extraction solvent (500 mg L−1 in 1,1,1-TCE). ER is defined as the percentage of the total analyte amount (n 0) which is extracted into the sedimented phase (n sed):
where V sed and V aq are volumes of the sedimented phase and aqueous solution, respectively. Calibration curves were plotted using eight concentrations (5, 10, 100, 500, 1,000, 5,000, 10,000, and 20,000 ng g−1 in honey). These levels were obtained by spiking the honey with appropriate volumes of the standard solutions prepared in methanol (10, 100, or 1,000 μg mL−1 of each analyte). To study repeatability of the method, six (intra-days precision) or four (inter-day precision) repeated determinations were performed on the honey spiked with each analyte at a concentration of 100 ng g−1. In preparing calibration curves and calculating repeatability of the method, the honey sample obtained from virgin mountainous lands (see Section 2.2) which was free of the analytes was used.
Results and Discussion
In the present study, a liquid partitioning extraction method combined with DLLME is used for the extraction, cleanup, and preconcentration of some SPAs and BPA from honey. In this method, the selected analytes are initially extracted into an organic solvent from honey samples. Then, the extractant is used as a disperser solvent in the following DLLME step. Different experimental parameters such as selection of a suitable extraction solvent for extraction of the analytes from honey sample (also the disperser solvent for the following DLLME step) and its volume, type, and volume of extraction solvent in DLLME step, salt addition, pH, and centrifugation time and speed which may affect the process are systematically investigated.
Optimization of Parameters in Extraction of Some SPAs and BPA from Honey sample
Selection of Extraction Solvent
In this study, an extraction solvent should be used for extraction of the analytes from honey sample. On the other hand, it will act as a disperser solvent in the following DLLME procedure. Therefore, the extraction solvent has to meet the following requirements: extraction capability of the compounds of interest from honey, miscibility with both aqueous phase and organic phase (extraction solvent used in DLLME step), and formation of a two-phase system when it is added to the diluted honey sample. On the basis of the mentioned considerations, five solvents, DMF, DMSO, methanol, acetone, and acetonitrile, were examined. The results showed that among the tested solvents, only acetonitrile formed a two-phase system when it was added to the honey sample. Therefore, it was selected as the extraction solvent for the subsequent experiments.
Optimization of Acetonitrile Volume
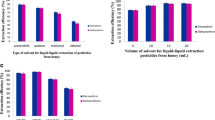
To assess the effect of acetonitrile volume on the extraction efficiency, different volumes of acetonitrile (from 0.5 to 2 mL at 0.5-mL intervals) were tested. Volume of the acetonitrile was effective on the volume of final upper phase after extraction. The obtained results showed that at the volume of 0.5 mL acetonitrile, no organic phase was collected and the method became useless. In the cases of 1.0, 1.5, and 2.0 mL acetonitrile, volume of the collected phase was 0.7, 1.1, and 1.4 mL, respectively. In all cases, 1 mL of organic phase was removed and used in DLLME step, except in the case of 1.0 mL acetonitrile, in which volume of the collected phase was 0.7 mL. In that case, whole of the organic phase was removed and mixed with 0.3 mL pure acetonitrile and used in the following DLLME step. As it can be seen from Fig. 1, the high analytical signals are obtained for all analytes in the case of 1.5 mL acetonitrile. Therefore, it was selected for the further experiments.

Effect of acetonitrile volume on performance of the developed method in extraction of SPAs and BPA from honey. Extraction conditions: sample, 2 g honey diluted with 1 mL de-ionized water spiked with the analytes (each analyte, 1 ng g−1); centrifuge rate, 4,000 rpm; centrifuge time, 5 min; volume of acetonitrile in DLLME step, 1 mL; extraction solvent, 1,1,1-TCE (40 μL); volume of aqueous phase (de-ionized water) in DLLME step, 5 mL; centrifuge rate in DLLME step, 4,000 rpm; and centrifuge time in DLLME step, 5 min. The error bars indicate the minimum and maximum of three determinations
Optimization of Centrifuging Time and Speed
The effect of time and speed of centrifuging were examined in the ranges of 3–8 min and 2,000–6,000 rpm, respectively. According to the obtained results in centrifuging time and speed of 5–8 min and 4,000–6,000 rpm, respectively, phase separation has occurred efficiently, and a clear upper phase was obtained. Therefore, 5 min and 4,000 rpm were selected as the centrifuging time and speed, respectively, in the following studies.
Optimization of Parameters in DLLME Process
Selection of Extraction Solvent
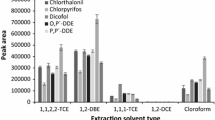
Selection of an appropriate extraction solvent has a primary importance on the performance of a DLLME process. Generally, the extraction solvent used in DLLME must fulfill some characteristics such as low solubility in water, extraction capability of the interested analytes, good chromatographic behavior, and formation of a two-phase system (cloudy solution) when injected into an aqueous solution. Based on these facts, some organic solvents named chloroform, carbon tetrachloride, 1,1,2,2-TCE, 1,2-DBE, and 1,1,1-TCE were examined. Experiments were performed using 1.0 mL of acetonitrile (obtained from the first step) containing different volumes of the extraction solvents to reach 10 μL of the sedimented organic phase volume. The needed volumes for each solvent were 45 μL chloroform, 40 μL carbon tetrachloride, 30 μL 1,1,2,2-TCE, 34 μL 1,2-DBE, and 40 μL 1,1,1-TCE. Figure 2 shows the effect of extraction solvent type on the extraction efficiency of the target analytes. The results reveal that 1,1,1-TCE and 1,2-DBE give better extraction efficiencies than the other tested solvents. However, by comparing peak areas for 1,1,1-TCE and 1,2-DBE shows that the former gives higher efficiency towards BHA. Therefore, 1,1,1-TCE was selected for the subsequent experiments.

Selection of extraction solvent kind in DLLME. Extraction conditions: extraction solvent in DLLME step, chloroform (45 μL), 1,2-DBE (34 μL), 1,1,1-TCE (40 μL), carbon tetrachloride (40 μL), and 1,1,2,2-TCE (30 μL). Other conditions are the same as in Fig. 1, except 1.5 mL acetonitrile was used in the first step. The error bars indicate the minimum and maximum of three determinations
Optimization of Extraction Solvent Volume in DLLME
Volume of the extraction solvent is another important factor that can affect volume of the sedimented organic phase, repeatability of the results, and extraction efficiencies. By increasing extraction solvent volume, the extracted amounts of analytes would increase, whereas their concentrations in the sedimented phase will decrease due to dilution effect. To evaluate the effect of the extraction solvent volume on the extraction performance, 1.0 mL acetonitrile containing different volumes of 1,1,1-TCE (30, 40, 50, 60, and 70 μL) subjected to the same DLLME procedures by keeping other experimental conditions remained constant. Maximum peak areas were obtained when 40 μL of the extraction solvent was added. The obtained results reveal that by increasing the extraction solvent volume from 40 to 70 μL, the analytical signals decrease due to the increase in volume of the sedimented phase from 10 to 40 μL and its dilution effect on concentrations of the analytes in the extractant. It is noted that in the case of 30 μL extraction solvent volume or less, no sedimented phase was formed, and the method has failed to work. Therefore, 40 μL was selected as the optimal volume of 1,1,1-TCE, which leads to obtain 10 ± 1 μL sedimented phase volume.
Effect of Salt Addition
Generally, adding a salt decreases the solubility of analytes in aqueous phase with increasing its ionic strength due to salting out effect. On the other hand, salt addition leads to an increase in volume of the sedimented phase by decreasing the solubility of extraction solvent into aqueous phase. For investigating the influence of ionic strength of aqueous phase on the extraction efficiency of the developed DLLME procedure, various experiments were performed by adding NaCl in the range of 0–7.5 %, w/v, to aqueous phase, while the other experimental conditions were kept constant. The experiments were performed using different volumes of the extraction solvent to achieve 10 μL of the sedimented phase volume (40, 37, 34, and 30 μL for 0, 2.5, 5.0, and 7.5 % NaCl, w/v, respectively). The results (not shown) indicated that by increasing NaCl concentration, the analytical signals are nearly constant. Therefore, further experiments were performed without the addition of the salt. It is noted that in the presence of NaCl 10 %, w/v, or higher, no organic phase has settled down due to the increasing density of the aqueous phase.
Effect of pH
The pH of aqueous phase affects the extraction performance for acidic and basic compounds. Therefore, the effect of pH was studied within the range of 2–12 adjusted with 1 M HCl or NaOH solutions. The obtained results indicated that the analytical signals remained constant up to pH 10 and decreased noticeably at higher pHs. Decreasing in extraction efficiencies of the target compounds can be attributed to ionization of the selected phenolic compounds in alkaline pHs. It is noted that the pK a of the studied compounds are as follows: BHA, 10.75; BHT, 12.75; BPA, 9.9 and 11.3; and MTBP, 11.72. The pH of aqueous phase was 6.5; therefore, there is no need for pH adjustment in the following studies.
Optimization of Centrifugation Time and Speed
In DLLME, centrifugation is a mandatory process to achieve a rapid separation of the extractant droplets from the aqueous phase. In order to obtain the optimum values of centrifugation time and speed, several experiments were performed in the ranges of 3–8 min and 2,000–6,000 rpm, respectively. Except for the centrifugation time of 3 min, separation of two phases was performed completely and the analytical signals were nearly constant. Also, the centrifuge speed had a little influence on the extraction efficiency. Therefore, 4,000 rpm and 5 min were selected as the optimal centrifuge rate and time, respectively, in the following studies.
Quantitative Aspects
Analytical characteristics of the method were evaluated in determination of the target analytes according to the recommended procedure for estimating figures of merits of the method under the optimized experimental conditions. Some important quantitative features of the method such as linear range (LR), correlation coefficient (r), relative standard deviation (RSD %), EF, ER, limit of detection (LOD), and limit of quantification (LOQ) based on the signal-to-noise ratio (S/N) of 3 and 10, respectively, were calculated and are summarized in Table 1. The calibration graphs were constructed by analyzing the honey sample spiked with the analytes at eight concentration levels in the ranges of 5–20,000 ng g−1 for MTBP and BHT; 12–20,000 for BHA; and 48–20,000 ng g−1 for BPA. Good linearity was obtained for all analytes with r higher than 0.997. The intra-day and inter-day precisions of the proposed method, expressed as RSD %, were calculated by performing the method on six repeated samples in a day (for intra-day) and four repeated samples in different days (for inter-days) at a concentration of 100 ng g−1 of each analyte. The obtained RSDs varied between 1.8 and 7.6 and 2.4–8.3 %, respectively. LODs and LOQs in honey were in the ranges of 0.4–4.7 and 1.3–14 ng g−1, respectively, and in solution 1.4–16 and 4.5–48 ng mL−1, respectively, which are significantly low values. High EFs and ERs ranging from 144 to 186 and 72–93 %, respectively, were obtained. These results show that the proposed method can be considered as a sensitive and repeatable method with high EFs for the analysis of such compounds at trace levels in honey.
Honey Sample Analysis
In order to investigate applicability of the developed method, four honey samples from different producers being commercially available from local supermarkets were analyzed. The typical GC-FID chromatographic data for the selected samples, along with direct injection of standard solution (100 mg L−1 of each analyte in methanol), are shown in Fig. 3. According to the chromatograms in one honey sample (packed in polyethene package), two peaks were observed in the retention times belonging to BHT and BHA, and other samples were free of analytes. For more identification, that honey sample was also analyzed by GC-MS after performing the proposed extraction method. Total ion current (TIC) chromatogram and mass data for BHT and BHA are given in Fig. 4. The presence of BHT and BHA in the studied honey sample was confirmed by comparison of mass data for scans 1786 and 1722 (retention times 15.71 and 15.28 min, respectively) with those of the studied analytes. The obtained concentrations for BHA (33 ng g−1) and BHT (42 ng g−1) in honey sample (sample 1) are given in Table 2. In order to evaluate the method accuracy and matrix effect, samples were spiked with the analytes at three concentration levels (100, 250, and 500 ng g−1 of each analyte). The obtained data expressed as recoveries are summarized in Table 2. The results show that the recoveries, defined as the percentage ratio between the found concentration of the selected analytes and the added concentration of them, are from 91 to 104 %. This indicates that there is no significant matrix effect in the analyzed samples. So the developed method is acceptable and applicable for the quantitative analysis of the studied phenolic compounds in real samples and could be used in routine analysis.

GC-FID chromatograms of a honey, b honey spiked with 250 ng g−1 of each analyte, and c standard solution (100 mg L−1) prepared in methanol. Chromatogram (c) was obtained by direct injection, whereas in the cases of other chromatograms, the proposed method was carried out on the samples, and 1 μL of the sedimented phase was injected into the separation system

a Total ion current (TIC) chromatogram of honey sample and mass spectra of b BHA and scan 1722 (retention time 15.28 min) and c BHT and scan 1786 (retention time 15.71 min)
Comparison of the Proposed Method with Other Methods
Table 3 summarizes the values of LR, RSD, LOD, LOQ, and ER of some analytical methods along with the proposed method for the extraction and determination of the selected analytes in different samples. The repeatability of the method is good, and RSDs % for the proposed method are lower than or comparable with those of the mentioned methods. In comparison with other methods, the proposed method provides wider LRs and lower LODs except solid-phase extraction–ultra high-performance liquid chromatography–tandem mass spectrometry (SPE–UPLC–MS/MS) and micro liquid–liquid extraction–GC–MS. It should be noted that in those methods, a high sensitive detection system (mass spectrometry) was used which is naturally more sensitive than FID. These results reveal that the presented DLLME–GC–FID method is a sensitive, simple, rapid, and repeatable technique and can be used for the preconcentration and determination of synthetic phenolic antioxidants and bisphenol A from honey samples.
Conclusion
In this study, a microextraction method based on DLLME has been reported for the extraction and preconcentration of BHT, BHA, BPA, and MTBP from honey samples followed by GC-FID determination. The results reveal that the developed method exhibits many merits such as high ERs and EFs, low LODs and LOQs, shorter extraction time, simplicity, low cost, and better repeatability. The experimental results demonstrate that the present method is a suitable procedure for analysis of the target analytes at nanogram per gram level in honey.
Abbreviations
- BHA:
-
Butylated hydroxyanisole
- BHT:
-
Butylated hydroxytoluene
- BPA:
-
Bisphenol A
- DLLME:
-
Dispersive liquid–liquid microextraction
- EF:
-
Enrichment factor
- FID:
-
Flame ionization detection
- ER:
-
Extraction recovery
- GC:
-
Gas chromatography
- LLE:
-
Liquid–liquid extraction
- LOD:
-
Limit of detection
- LOQ:
-
Limit of quantification
- LPME:
-
Liquid-phase microextraction
- r :
-
Correlation coefficient
- RSD:
-
Relative standard deviation
- SPE:
-
Solid-phase extraction
- SPME:
-
Solid-phase microextraction
References
Ahmadi F, Assadi Y, Hosseini MRM, Rezaee M (2006) J Chromatogr A 1101:307–312
Arthur CL, Pawliszyn J (1990) Anal Chem 62:2145–2148
Bahruddin S, Yong YS, Mohd AN, NoorHasani H, Abdussalam SMA, Muhammad IS, Shaida FS, Khairuddin MT, Kamarudzaman A (2007) Food Chem 105:389–394
Bidari A, Ganjali MR, Norouzi P, Hosseini MRMM, Assadi Y (2011) Food Chem 126:1840–1844
Cariot A, Dupuis A, Albouy LM, Legube B, Rabouan S, Migeot V (2012) Talanta 100:175–182
Farajzadeh MA, Nouri N (2012) Talanta 99:1004–1010
Farajzadeh MA, Bahram M, Vardast MR (2009) J Sep Sci 32:4200–4212
Farajzadeh MA, Djozan D, Afshar Mogaddam MR, Norouzi J (2012a) J Sep Sci 35:742–749
Farajzadeh MA, Djozan D, Khorram P (2012b) Anal Chim Acta 713:70–78
Farajzadeh MA, Afshar Mogaddam MR, Ghorbanpour H (2014a) J Chromatogr A 1347:8–16
Farajzadeh MA, Feriduni B, Afshar Mogaddam MR (2014b) J Food Sci 79:2140–2148
Gonzalez M, Gallego M, Valcarcel M (1999) J Chromatogr A 848:529–536
González-Casado A, Navas N, Del Olmo M, Vílchez JL (1998) J Chromatogr Sci 36:565–570
Hernández F, Portoles T, Pitarch E, López FJ (2007) Anal Chem 79:9494–9504
Huang W, Niu H, Shi B, Yang M, Jiang J (2008) J Food Lipids 15:1–12
Jeannot MA, Cantwell FF (1996) Anal Chem 68:2236–2240
Jofre VP, Assof MV, Fanzone ML, Goicoechea HC, Martinez LD, Silva MF (2010) Anal Chim Acta 683:126–135
Khodadoust S, Hadjmohammadi M (2011) Anal Chim Acta 699:113–119
Li XQ, Ji C, Sun YY, Yang ML, Chu XG (2009) Food Chem 113:692–700
Lin X, Ni Y, Kokot S (2013) Anal Chim Acta 765:54–62
Liu H, Dasgupta PK (1996) Anal Chem 68:1817–1821
Lord H, Pawliszyn J (2000) J Chromatogr A 885:153–193
Melwanki MB, Hsu WH, Huang SD (2005) Anal Chim Acta 552:67–75
Mingzhen D, Jiankai Z (2012) Food Chem 131:1051–1055
Olmo M, Gonzalez-Casado A, Navas NA, Vilchez JL (1997) Anal Chim Acta 346:87–92
Psillakis E, Kalogerakis N (2003) J Chromatogr A 999:145–153
Rasmussen KE, Pedersen–Bjergaard S (2004) Trends Anal Chem 23:1–10
Remberger M, Kaj L, Palm A, Sternbeck J, Kvernes E, Brorström–Lundén E (2003) IVL Swedish Environ Res Inst, Stockholm
Rezaee M, Assadi Y, Milani Hosseini MR, Aghaee E, Ahmadi F, Berijani S (2006) J Chromatogr A 1116:1–9
Ricardo PC, Adriana GFA, João BGS, Tatiane AA, Luiz HV, Magno AGT, Valdir SF (2013) Fuel 105:306–313
Richard Prabakar SJ, Sriman Narayanan S (2010) Food Chem 118:449–455
Rodil R, Quintana JB, Basaglia G, Pietrogrande MC, Cela R (2010) J Chromatogr A 1217:6428–6435
Rusnakova L, Andruch V, Balogh IS, Skrlikova J (2011) Talanta 85:541–545
Shen G, Lee HK (2002) Anal Chem 74:648–654
Soliman MA, Pedersen JA, Park H, Castaneda–Jimenez A, Stenstrom MK, Suffet IH (2007) Water Environ Res 79:156–167
Sun YY, Xie YF, Wang HY, Qian H, Yao WR (2012) Appl Surf Sci 261:431–435
Trenholm RA, Vanderford BJ, Drewes JE, Snyder SA (2008) J Chromatogr A 1190:253–262
Williams GM (1994) Eur J Cancer Prev 3:89–99
Yan HY, Liu B, Du JJ, Yang GL, Row KH (2010) J Chromatogr A 1217:5152–5157
Yang MH, Lin HJ, Choong YM (2002) Food Res Int 35:627–633
Acknowledgments
The authors thank the Research Council of Tabriz University for financial support.
Conflict of Interest
Mir Ali Farajzadeh has received research grants from University of Tabriz. Maryam Abbaspour declares that she has no conflict of interest. Mohammad Reza Afshar Mogaddam declares that she has no conflict of interest. Houshang Ghorbanpour declares that she has no conflict of interest. This article does not contain any studies with human or animal subjects.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Farajzadeh, M.A., Abbaspour, M., Mogaddam, M.R.A. et al. Determination of Some Synthetic Phenolic Antioxidants and Bisphenol A in Honey Using Dispersive Liquid–Liquid Microextraction Followed by Gas Chromatography-Flame Ionization Detection. Food Anal. Methods 8, 2035–2043 (2015). https://doi.org/10.1007/s12161-015-0087-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-015-0087-9