
Abstract
The electrochemical behavior of caffeine was investigated by using a simple, cheap, and highly selective pseudo carbon paste electrode. The electrode was characterized using scanning electron microscope (SEM). Cyclic voltammetry of caffeine showed only one oxidation peak at 1.46 V (vs. SCE) in phosphate buffer pH 7. Chemical calculations were performed using PM3 method to prove the electrochemical oxidation mechanism of caffeine studied by cyclic voltammetry. Square wave voltammetry was used for the direct electrochemical determination of caffeine. The effect of different experimental parameters was investigated on the peak height of caffeine. Caffeine showed a linear calibration curve ranged from 1 × 10−6 to 1 × 10−3 M, with 3.03 × 10−7 standard deviation and a correlation coefficient of 0.999. A lower detection limit of 3.48 × 10−7 M was obtained. The effect of ascorbic acid and paracetamol on the peak height of caffeine was studied. The proposed method has been successfully applied for the direct electrochemical determination of caffeine in different real samples such as tea, coffee, cola, and pharmaceutical formulations.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Caffeine, (1,3,7-Trimethylxanthine) (I), is the most widely consumed drugs in the world. It naturally occurs in coffee, tea, chocolate products, and in many soft drinks. Caffeine has different physiological effects such as relaxation of bronchial muscle, stimulation of the central nervous system, gastric acid secretion, dieresis (Spatarua et al. 2002), and cardiovascular systems (Zen et al. 1998). Caffeine is considered to be a risk factor for cardiovascular diseases and may cause depression and hyperactivity (Aklilu et al. 2008).
For these reasons, it is very important to control the concentration of caffeine in its different sources. Different methods have been developed for the determination of caffeine including spectrophotometric (Ding and Zou 2008; Zhang et al. 2008; Ito et al. 2008; Khoshayand et al. 2008; Shrivas and Wu 2007; Khanchi et al. 2007; Zou and Li 2006; Belay et al. 2008; Singh and Sahu 2006; Yamauchi et al. 2008) and chromatographic methods (Evans and Siittonen 2008; Emre and Ozaltın 2007; Tzanavaras and Themelis 2007; Brunetto et al. 2007; Aranda and Morlock 2006). The mentioned methods have many disadvantages such as highly expensive, time consuming, and more complicated than the electroanalytical methods (Spatarua et al. 2002; Zen et al. 1998; Aklilu et al. 2008; Lau et al. 1989). The electrochemical determination of caffeine depends on the oxidation of caffeine which occurs at high positive potentials and leads to overlapping with the discharge of the background electrolyte (Spatarua et al. 2002). To avoid the overlapping of the oxidation peak of caffeine with the electrolyte, several types of electrodes have been examined using electrochemical techniques. From these electrodes: glassy carbon electrode (Lau et al. 1989), glassy carbon electrode modified with Fullerene-60 (Goyal et al. 2005a, b), Nafion-covered glassy carbon electrode (Brunetti et al. 2007), graphite pencil electrode (Ly et al. 2004), boron-doped diamond electrode (Lourennao et al. 2009), pyrolytic graphite electrode (Goyal et al. 2008a, b), pyrolytic graphite electrode modified with single-wall carbon nanotubes (Goyal et al. 2009a, b), Fullerene-60 modified electrodes (Goyal et al. 2007a, b, c; Goyal et al. 2008a, b; Goyal et al. 2009a, b), gold nanoparticles modified indium tin oxides electrodes (Goyal et al. 2005a, b; Goyal et al. 2006; Goyal et al. 2007a, b, c), modified polyvinyl chloride electrodes (Srivastava et al. 1995; Jain et al. 1995a, b; Srivastava et al. 1996; Gupta and Kumar 1999; Prasad et al. 2004; Gupta et al. 2005; Gupta et al. 2006; Singh et al. 2007), and enzymatic electrodes (Ly et al. 2009; Pizzariello et al. 1999; Babu et al. 2007).
For the electrochemical determination of caffeine, Nafion-ruthenium oxide pyrochlore chemically modified electrodes have been used (Zen et al. 1998; Dryhurst and Hansen 1971). Indirect voltammetric method was used for the determination of caffeine in coffee using 1, 4 benzoquinone modified carbon paste electrode (Aklilu et al. 2008).
Carbon paste electrodes represent one on the most types of working electrode. Carbon paste prepared by mixing graphite powder with insulating liquids such as paraffin oil, silicon oil, bromonaphthalene, tricresyl phosphate, and others. Carbon paste electrodes have many advantages such as easily renewable surface, low cost, and have very low background currents especially in the anodic region (Svancara et al. 2009; Svancara et al. 2001; Safavi et al. 2006). One of the disadvantages of carbon paste electrodes is the tendency of organic binder to dissolve in solutions containing organic solvents. The main objective of the present work is to prepare unmodified pseudo carbon paste microelectrode by mixing graphite powder with paraffin wax and to study the direct electrochemical behavior of caffeine and to detect caffeine concentration using this electrode. Measurements were conducted under various operating conditions using cyclic voltammetry and square wave voltammetric techniques.
Experimental
Chemicals and Reagents
All chemicals used were of analytical grade and used without further purifications. Caffeine, paracetamol, ascorbic acid graphite powder, and paraffin wax were obtained from Sigma-Aldrich (Germany), while H3BO3, H3PO4, CH3COOH, and NaOH were obtained from Merck (Germany). Stock solutions of 0.01 M caffeine were freshly prepared daily in phosphate buffer. Britton–Robinson buffer by mixing 0.04 M H3BO3, 0.04 M H3PO4, and 0.04 M CH3COOH. The desired pH was adjusted by the addition of 0.2 M NaOH. Double-distilled water was used for the preparation of solutions.
Preparation of Pseudo Carbon Paste Electrode and Electrochemical Measurements
Pseudo carbon paste electrode was prepared by mixing 65% graphite powder and 35% paraffin wax. Paraffin wax was heated till melting and then mixed very well with graphite powder to produce a homogeneous paste. The resulted paste was then packed into the end of an insulin syringe. External electrical contact was established by forcing a copper wire down the syringe. The surface of the electrode was polished with a piece of weighting paper and then rinsed with distilled water thoroughly.
Preparation of Real Samples
Caffeine in Coffee Samples
Coffee sample was treated as described by (Belay et al. 2008; Aklilu et al. 2008.). In this method 1.0 g of coffee samples was milled and ground with mortar. The ground sample was transferred into a 250-mL Erlenmeyer flask fitted with a stopper, then 50.0 mL of distilled water and 2.0 g of sodium carbonate were added to the flask and boiled for 1 h with continuous stirring. The hot solution was allowed to settle and filtered to get rid of particles. Coffee was extracted from the solution by liquid–liquid extraction using dichloromethane. Twenty-five milliliters of dichloromethane was added to the filtered solution and mixed very well. Using separatory funnel, coffee was extracted from solution. The extraction was preceded three times with dichloromethane. The extracted solution was passed through 0.25 g of anhydrous sodium sulfate for drying. Dichloromethane was left to evaporate and the residue was dissolved in 10 mL of supporting electrolyte.
Caffeine in Tea Samples
For tea samples, into a 250-mL Erlenmeyer flask, 100 mL distilled water and 2 g of sodium carbonate was added to 5 g of tea sample. The mixture was boiled in a hot plate with stirring till sodium carbonate dissolved. The mixture was allowed to cool for 5 min, and then decant into another beaker and continued cooling to about 20 C. The extraction process was the same as in coffee samples and residue was dissolved in 10 mL of supporting electrolyte.
Caffeine in Coca Cola Samples
The cola samples were used without treatment, just diluted with supporting electrolyte.
Caffeine in Drug Formulation Samples
Ten tablets were weighted accurately and finely powdered in a mortar and 0.2 g of the powder was weighted accurately and transferred to 100 mL calibrated flask, which then was completed to the volume with the supporting electrolyte and then the solution was filtered to get the sample solution.
Apparatus
Cyclic voltammetry and square wave voltammetry were performed using an Autolab potentiostat PGSTAT 302 (Eco Chemie, Utrecht, The Netherlands) driven by the General purpose electrochemical systems data processing software (GPES, software version 4.9, Eco Chemie). Electrochemical cell with three electrodes was used; carbon paste electrode was used as a working electrode, SCE was used as a reference electrode while platinum wire was used as a counter electrode. The pH values were measured using a Metrohom pH-meter (Germany) with a combined glass electrode. Scanning electron microscopy (SEM; JEOL JSM-6390) was used to characterize the morphology of pseudo carbon paste electrode.
Computational Methods
Molecular modeling and quantum semi-empirical calculations were carried out using PM3 method. The geometry optimizations of caffeine were obtained by the application of the Polak–Ribiere algorithm with convergence limit of 0.01 kcal/mol and RMS gradient of 0.01 kcal/mol.
Results and Discussion
Characterization of Carbon Paste Electrode
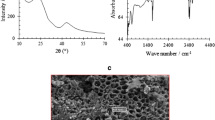
The SEM images for the carbon past electrode showed microstructures with a discontinuous grain growth with a large unclear crystal structure (Fig. 1). The surface structure of the carbon paste electrode showed that the graphite particles are covered by a very thin film of paraffin wax. The microstructure appeared hardly compact and this will increase the electrical conductivity of the carbon paste.

SEM micrographs of carbon paste electrode surface at (A) low magnification and (B) at high magnification
Cyclic Voltammetry and Square Wave Voltammetric Measurements
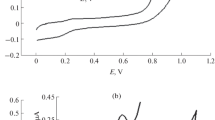
The electrochemical behavior of caffeine on carbon paste electrode was studied by cyclic voltammetry (CV) using a potential range from 0.0 to +2.0 V with a scan rate of 50 mV s1. Various supporting electrolytes were investigated such as: sodium phosphate buffer, Britton–Robinson buffer, sodium acetate buffer, sodium perchlorate buffer, borate buffer, and sodium sulfate. Figure 2a shows the results obtained with 1.0 mM caffeine in different supporting electrolytes at pH 7. Caffeine showed only one oxidation peak in phosphate, sodium acetate, Britton–Robinson, borate, and sodium sulfate buffers, while no peaks appeared in case of perchlorate buffer. The best results were obtained using phosphate buffer, where caffeine showed a sharp oxidation peak, while in the other buffers broad peaks were obtained.

a Cyclic voltammetric response for 1 mM caffeine in different supporting electrolytes: (a) phosphate, (b) acetate, (c) BR, (d) borate, (e) perchlorate, and (e) sodium sulfate buffers, pH 7 at carbon paste electrode with scan rate of 50 mV/s. b Square wave voltammetric response for 1.0 mM caffeine in different supporting electrolytes: (a) phosphate, (b) acetate, (c) BR, (d) borate, (e) perchlorate, and (f) sodium sulfate buffers, pH 7 at carbon paste electrode
On the other hand, the effect of the same supporting electrolytes at pH 7 on the peak current of 1.0 mM caffeine was studied using square wave voltammetry. In the absence of caffeine no signals were observed, while in the presence of caffeine (Fig. 2b), only one sharp oxidation peak was observed in phosphate and borate buffers. So phosphate buffer was selected for further studies.
In cyclic voltammetry, with the addition of 1.0 mM caffeine to phosphate buffer, an oxidation peak appeared during the anodic scan at 1.48 V (vs. SCE), and no cathodic peaks were observed on the reverse scan indicating that the oxidation of caffeine is an irreversible process (Spatarua et al. 2002). Using square wave voltammetry, caffeine showed only one oxidation peak at 1.46 V (vs. SCE). The resulting oxidation peak position of caffeine is compared with the obtained by using the different mentioned electrodes in Table 1. It has been shown that the electrochemical oxidation of caffeine is due to the oxidation at the C8 position on the purine ring through 4e−, 4H+ process (Brunetti et al. 2007). The first step is a 2e−, 2H+ oxidation of the C-8 to N-9 bond to give the substituted uric acid, followed by an intermediate 2e−, 2H+ oxidation to the 4,5-diol analogue of uric acid, and that rapidly fragments.
Computational Studies
In order to characterize redox orbitals for caffeine and to prove the results obtained from the cyclic voltammetric experiments quantum chemical calculations were performed. The optimized structure for caffeine was obtained through molecular mechanics calculation by applying semi-empirical PM3 method. Figure 3a shows the ball and stick model for the optimized geometries of caffeine. The energy values of the highest energy occupied orbital (HOMO) and the lowest energy unoccupied orbital (LUMO) and their energy gaps reflect the chemical activity of the molecules. HOMO represents the ability of the molecule to donate electrons (oxidation) and LUMO represents the ability of the molecule to obtain electrons (reduction). The electronic density in the HOMO and LUMO for caffeine is mainly located in C8, N9, and 4,5 diol as shown in Fig. 3b, c, indicating that C8, N9, and 4,5 diol in caffeine are the active sites. Localization of electronic densities on C8, N9, and 4,5 diol confirms that the oxidation of caffeine occurs in C8, N9, and 4,5 diol as mentioned by the pervious mechanism from cyclic voltammetric techniques. The energy gap between the energy of LUMO (−7.952 eV) and the energy of HOMO (−9.494 eV) equals +1.542 eV, this lower energy gap enables electrons to transfer from HOMO to LUMO orbital’s easily.

a Ball and stick model for the optimized geometry of caffeine, b the electronic density in the HOMO orbitals, and c the LUMO orbitals obtained from semi-empirical PM3 for caffeine
Effect of pH
The influence of pH on the oxidation peak current and peak potential of caffeine was investigated in the pH range of 2.7 to 10.3 using 1.0 mM caffeine in 0.1 M phosphate buffer. The results showed that the dependence of the oxidation peak on the pH of the supporting electrolyte. This is clearly seen from the sharp decrease of the caffeine oxidation peak with pH. The highest peak current was observed at pH 2.7, which was chosen for the present analytical applications. The influence of pH on the peak potentials of caffeine was also examined. The peak potentials showed a little shift to the negative direction with the increase in pH of the supporting electrolyte.
Effect of Scan Rate
The effect of scan rate on the cyclic voltammograms for 1.0 mM caffeine on the carbon paste electrode in phosphate buffer at pH 2.7 was studied; see Fig. 4a. The oxidation peak currents were proportional to the square root of the scan rate (ν1/2; Fig. 4b), which indicates that the electron transfer reaction is diffusion controlled. The linear regression equation was Ip (A) = 6.62 × 10−6 + 4.158 × 10−6 v with a 0.999 correlation coefficient.

a Cyclic voltammograms of 1.0 mM caffeine at carbon paste electrode in phosphate buffer pH 7 at various scan rates: 10, 25, 50, 75, 100, 150, 200, 250, 300, 350, 400, 450, and 500 mV s−1. b Plot of anodic peak current Ip vs. ν1/2, c plot of peak current Ep vs. log ν and d plot of log Ip vs. Ep
A linear relation between the peak potential (Ep) and log ν was obtained, Fig. 4b. Such behavior reveals the irreversible nature of the electrochemical process for caffeine (Zhang and Wang 2000). This irreversible behavior can be also observed from the presence of one oxidation peak for caffeine in anodic scan and no reduction peaks appeared in the cathodic scan in the used scan rate values from 10 to 500 mV/s.
To get some information on the rate-determining step, two methods were used. The first one obtained from the following equation (Harison and Khan 1970):
where α is the transfer coefficient, n a is the number of electrons transferred, ν is the potential scan rate, F is the Faraday's constant, R the rate gas constant, T is the absolute temperature, and b is the Tafel slope. Plotting the relation between the peak potential (Ep) and log ν for 1 mM caffeine displayed a straight line (Fig. 4c). The linear regression equation was calculated as Ep (V) = 0.07866 log ν + 1.27686 (r = 0.9968). The slope of the above equation was found to be 0.07866 V, so b = 0.1573 V. This slope indicates that a one-electron-transfer process is the rate-determining step. Assuming that the number of transferred electrons in the rate-determining step equals one, a transfer coefficient (α = 0.62) is obtained. If we assumed two electrons in the rate-determining step, α should be 0.81, which is not a common value, because for most electrode processes α ranges from 0.7 to 0.3 (Bard and Faulkner 1890).
The second method was used to estimate the value of α from Tafel plot by plotting log Ip versus Ep for 1 mM caffeine. The slope of this plot can be used to estimate the number of transferred electrons in the rate-determining step. A linear relation obtained using a scan rate range from 10 to 500 mV s−1 (Fig. 4d). The linear regression equation was calculated as Ep (V) = 5.498 log ν − 12.032 (r = 0.991). The Tafel slope b for the linear relation was found 5.498. This value of b indicates that one electron transfer process is the rate-determining step, assuming a transfer coefficient of α = 0.67. The results obtained from the two different methods were in good agreement.
Effect of Accumulation Time and Accumulation Potential
The influence of accumulation time on the peak currents of 1.0 mM caffeine was investigated over the range from 0 to 600 s using the same conditions mentioned in Fig. 4. By increasing the accumulation time, the peak current increases (Fig. 5a). The peak currents for caffeine increased linearly using accumulation time from 0 to 120 s (insert of Fig. 5a). When the accumulation time exceeds 120 s, the oxidation peak current increases slightly. For further analytical work accumulation time of 120 s was selected.

a Square wave voltammetric peak current at different accumulation times from 0 to 600 s (from 0 to 120 inserted plot) for 1.0 mM caffeine with initial potential 0.0 V, final potential 1.7 V, step potential 0.002 V, and amplitude of 0.002 V and frequency of 20 Hz. b Square wave voltammetric peak current different accumulation potentials from −1.0 to +0.6 V for 1.0 mM caffeine and other conditions as in Fig. 4a
The influence of accumulation potential on the peak current of caffeine was investigated using accumulation potential range of −1.0 to +0.6 V. It follows from Fig. 4b that shifting the peak potential to more positive values increased the peak current reaching a maximum value at about 0.1 V (SCE). At higher accumulation potentials, the peak current decreased sharply. Based on these results, an accumulation potential of 0.1 V was selected for further work.
Effect of Square Wave Voltammetric Parameters
The effect of different square wave voltammetric parameters was examined on the peak height of caffeine and is shown in Fig. 6. The influence of square wave frequency on the peak current of 1.0 mM caffeine using 120 s accumulation time and 0.1 V accumulation potential is shown in Fig. 6a. The values of 8, 9, 10, 11, 12, 13, and 15 Hz were chosen in this study. It can be observed that the peak current increases linearly as the frequency increases up to 11 Hz, and at higher frequencies the peak current sharply decreased. Thus for further works, 11 Hz frequency was used where it gave the highest peak current value.

a Square wave voltammetric peak current at different square wave frequencies from 8 to 15 Hz for 1.0 mM caffeine with initial potential 0.1 V, final potential 1.7 V, square wave step potential 0.002 V, square wave amplitude of 0.002 V, and 120 s accumulation time. b Square wave voltammetric peak current at different square wave amplitudes from 5 to 100 mV for 1.0 mM caffeine with initial potential 0.1 V, final potential 1.7 V, step potential 0.002 V, 11 Hz square wave frequency, and 120 s accumulation time. c Square wave voltammetric peak current at different square wave step potentials from 1 to 10 mV for 1.0 mM caffeine with initial potential 0.1 V, final potential 1.7 V, square wave amplitude of 60 mV, 11 Hz square wave frequency, and 120 s accumulation time
Figure 6b illustrates the effect of square wave pulse amplitude on the peak current of 1.0 mM caffeine using 120 s accumulation time, 0.1 V accumulation potential, and 11 Hz square wave frequency. The pulse amplitude ranged from 5 to 100 mV. The peak current increased linearly from 5 to 45 mV with 0.998 correlation coefficient and 1.16 × 10−6 standard deviation. At higher square wave pulse amplitude values, the linearity decreased (Fig. 6b). So, a 60 mV will be the optimum square wave pulse amplitude height and will be used in the further studies.
The last parameter investigated for square wave voltammetric measurements was the step potential. The effect of step potential (1–10 mV) on the peak height of 1.0 mM caffeine is depicted using 60 mV square wave pulse amplitude and the other experimental parameters as in Fig. 6b. As the step potential increases, the peak height increases linearly up to 7 mV, after that the increase in the peak height is not pronounced (Fig. 6c). So for further work a step potential of 7 was selected.
Calibration Curve and Detection Limit
Using the following optimum conditions: 0.1 M phosphate buffer pH 2.7, 120 s accumulation time, 11 Hz square wave frequency, 60 mV square wave pulse amplitude, and 7 mV step potential, caffeine was determined using square wave voltammetry. Caffeine showed a linear range from 2 × 10−6–1 × 10−3 M (Fig. 7) with a correlation coefficient of 0.9994 and a relative standard deviation (RSD) of 3.03 × 10−7. The detection limit for caffeine was calculated based on a three signal to noise ration and it was found of 3.5 × 10−7 M.

Calibration plots for different caffeine concentrations from 2 × 10−6 to 1 × 10−3 M obtained at the following optimum conditions: 0.1 V accumulation potential, final potential 1.7 V, square wave amplitude of 60 mV, 11 Hz square wave frequency, 7 mV square wave step potential, and 120 s accumulation time
Reproducibility
The reproducibility of the results was examined by successive ten measurements of 1 × 10−4 M caffeine using the optimum conditions mentioned above. The RSD was calculated and it was found to be 0.3%, which indicated that this method give a good reproducibility for the obtained results.
Interferences
The effect of ascorbic acid and paracetamol as the most interfering substances in the electrochemical determination of some organic compounds was examined. Ascorbic acid showed one anodic oxidation peak at 0.285 V (vs. SCE) under the used conditions mentioned above (Fig. 7). The influence of ascorbic acid was examined over the range from 1 × 10−5 to 1 × 10−3 to 1 × 10−3 M caffeine, where no marked effect on the peak current was observed (Fig. 8a). Paracetamol showed also an oxidation peak at 0.7 V (vs. SCE) and this peak separated from the oxidation peak of caffeine. Figure 8b showed the effect of different concentrations from paracetamol on the peak height of 1 × 10−3 M caffeine. Addition of paracetamol concentrations from 1 × 10−5 to 1 × 10−3 M has no effect peak current of caffeine.

Effect of different concentrations ranged from 1×10-5 to 1×10-3 M from (A) ascorbic acid and (B) paracetamol on 1 mM caffeine, other conditions as in Fig. 7
Analytical Applications
The applicability of the proposed method was checked by analyzing synthetic samples of caffeine. Using the optimum conditions mentioned above and calibration curve (analytical equation: \( y = {9}.{83} \times {1}{0^{{ - {6}}}} + 0.0{3223}x \)). The results obtained are shown in Table 2. As can be seen from the Table 2, the concentrations added and found were generally in good agreement, with a recovery between 92.7% and 106%. The proposed method was also applied for the determination of caffeine in several real samples including coffee, tea, Coca Cola, and pharmaceutical formulations. Table 3 shows the results obtained in a study of different samples as caffeine concentration (milligrams per liter).
Conclusion
Pseudo carbon paste microelectrode prepared by mixing graphite powder with paraffin wax showed a good sensitivity and high selectivity for the direct electrochemical determination of caffeine using square wave voltammetric technique. The prepared sensor was used for the analysis of caffeine at different real samples such as tea, coffee, cola, and pharmaceutical formulations.
References
Aklilu M, Tessema M, Redi-AbshiroRr M (2008) Talanta 76:742
Aranda M, Morlock G (2006) J Chromatogr A 1131:253
Babu GVRS, Patra S, Karanth NG, Kumar MA, Thakur MS (2007) Anal Chim Acta 582:329
Bard AJ, Faulkner LR (1890) Electrochemical methods fundamentals and applications. Wiley, New York
Belay A, Ture K, Redi M, Asfaw A (2008) Food Chem 108:310
Brunetti B, Desimoni E, Casati P (2007) Electroanalysis 19:385
Brunetto MR, Gutiérrez L, Delgado Y, Gallignani M, Zambrano A, Gomez A, Ramos G, Romero C (2007) Food Chem 100:459
Ding MZ, Zou JK (2008) Chin J Anal Chem 36:381
Dryhurst G, Hansen BH (1971) J Electroanal Chem 30:407
Emre D, Ozaltın N (2007) J Chromatogr B 47:126
Evans RL, Siittonen PH (2008) J Chromatogr Sci 46:61
Goyal RN, Gupta VK, Sangal A, Bachheti N (2005a) Electroanalysis 17:2223
Goyal RN, Gupta VK, Munetaka O, Bachheti N (2005b) Electrochem Commun 7:803
Goyal RN, Gupta VK, Munetaka O, Bachheti N (2006) Electrochem Commun 8:65
Goyal RN, Gupta VK, Munetaka O, Bachheti N (2007a) Talanta 71:1110
Goyal RN, Gupta VK, Munetaka O, Bachheti N (2007b) Talanta 72:976
Goyal RN, Gupta VK, Bachheti N (2007c) Anal Chim Acta 597:82
Goyal RN, Gupta VK, Bachheti N, Chatterjee S (2008a) Electrochim Acta 53:5360
Goyal RN, Gupta VK, Bachheti N, Sharma RA (2008b) Electroanalysis 20:757
Goyal RN, Gupta VK, Chatterjee S (2009a) Biosens Bioelectron 24:3562
Goyal RN, Gupta VK, Chatterjee S (2009b) Biosens Bioelectron 24:1649
Gupta VK, Kumar P (1999) Anal Chim Acta 389:205
Gupta VK, Ludwing R, Agarwal S (2005) Anal Chim Acta 538:213
Gupta VK, Singh AK, Gupta B (2006) Anal Chim Acta 575:198
Harison RJA, Khan ZA (1970) Electroanal Chem 28:153
Ito M, Suzuki T, Yada S, Kusai A, Nakagami H, Yonemochi E, Terada K (2008) J Pharm Biomed Anal 47:819
Jain AK, Gupta VK, Sahoo BB, Singh LP (1995a) Anal Proc Including Anal Comm 32:21
Jain AK, Gupta VK, Singh LP (1995b) Anal Proc Including Anal Comm 32:263
Khanchi AR, Mahani MK, Hajihosseini M, Maragheh MG, Chaloosi M, Bani F (2007) Food Chem 103:1062
Khoshayand MR, Abdollahi H, Shariatpanahi M, Saadatfard A, Mohammadi A (2008) Spectrochim Acta Part A: Molecul Biomol Spectr 70:491
Lau O, Luk S, Cheung Y (1989) Analyst 114:1047
Lourennao BC, Medeiros RA, Rocha-Filho RC, Mazo LH, Fatibello-Filho O (2009) Talanta 78:748
Ly SY, Lee CHN, Jung YS (2009) Neuromol Med 11:20
Ly SY, Jung YS, Kim MH, Han IK, Jung WW, Kim HS (2004) Microchim Acta 146:207
Pizzariello AJS, vorc Stred’ansky M, Miertus S (1999) J Sci Food Agric 79:1136
Prasad R, Gupta VK, Kumar A (2004) Anal Chim Acta 508:61
Safavi A, Maleki N, Moradlou O, Tajabadi F (2006) Anal Biochem 359:224
Shrivas K, Wu H (2007) J Chromatogr A 1170:9
Singh AK, Gupta VK, Gupta B (2007) Anal Chim Acta 585:171
Singh DK, Sahu A (2006) Anal Biochem 349:176
Spatarua N, Sarada BV, Tryk DA, Fujishima A (2002) Electroanalysis 14:721
Srivastava SK, Gupta VK, Dwivedi MK, Jain S (1996) Electroanalysis 8:938
Srivastava SK, Gupta VK, Jain S (1995) Anal Proc Including Anal Comm 32:21
Svancara I, Walcarius A, Kalcher K, Vytras K (2009) Central European J Electrochem 7:598
Svancara I, Vytras K, Barek J, Zima J (2001) Critical Reviews in Anal Chem 31:311
Tzanavaras PD, Themelis DG (2007) Anal Chim Acta 581:89
Yamauchi Y, Nakamura A, Kohno I, Kitai M, Hatanaka K, Tanimoto T (2008) Chem Pharm Bull 56:185
Zen J, Ting Y, Shih Y (1998) Analyst 123:1145
Zhang Y, Mehrotra N, Budha NR, Christensen ML, Meibohm B (2008) Clin Chim Acta 398:105
Zhang Z, Wang E (2000) Electrochemical principles and methods. Science, Beijing, p 242
Zou J, Li N (2006) J Chromatogr A 1136:106
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mersal, G.A.M. Experimental and Computational Studies on the Electrochemical Oxidation of Caffeine at Pseudo Carbon Paste Electrode and Its Voltammetric Determination in Different Real Samples. Food Anal. Methods 5, 520–529 (2012). https://doi.org/10.1007/s12161-011-9269-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-011-9269-2