
Abstract
An activated carbon was prepared using physical activation from date stone. This activated carbon was characterized by SEM, XRD, and FT-IR. This composite was used as modifier of carbon paste electrode (AC/CPE) for electrochemical determination of catechol (CC) and hydroquinone (HQ) using cyclic voltammetry. The electrochemical experiments indicated that the modified electrodes can simultaneously determinate HQ and CC at an oxidative and reductive peaks separation of about 125 and 120 mV, respectively. Furthermore, differential pulse voltammetry (DPV) of the sensing platform showed wide linear responses in the presence of 5.0 × 10−5 mol L−1 CC with the limit of detection (S/N = 3) of 7.1 × 10−8 mol L−1. At the same time, the oxidation peak current of CC was linear to its concentration with the limit of detection (S/N = 3) of 5.23 × 10−8 mol L−1 in the presence of 1.0 × 10−4 mol L−1 HQ.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Phenolic compounds and its substitute are important organic intermediates for the products of industrial raw and synthetic in cosmetic, pharmaceutical, tanning, and pesticide industries, etc. [1,2,3,4], for example, dihydroxybenzene compounds such as catechol (CC, 1,2-benzenediol) and hydroquinone (HQ, 1,4-benzenediol). Unfortunately, these two isomers are broadly distributed in soil and aquatic environment and are difficult to be degraded due to their high toxicity and high stability in the ecological environment [5,6,7]. It is highly toxic to human health even at very low concentrations [8], which are considered as environmental pollutants by the US Environmental Protection Agency (EPA) and the European Union (EU) [9]. Moreover, CC and HQ usually coexist in the environmental samples and have similar structures and properties, which make it difficult to determinate them simultaneously. Therefore, the development of a sensitive, simple, and rapid method for simultaneous determination of HQ and CC has become the most important study.
Up to now, many instrumental methods such as chromatography [10], chemiluminescence [11, 12], and spectrophotometry [13] have been commonly employed for quantification of CC and HQ. In fact, these techniques require expensive, complicated instruments and time consuming procedure. However, the electrochemical methods offer the practical advantages including easy operation, satisfactory sensitivity, and low cost of instrument [14,15,16,17,18,19,20,21]. The simultaneous determination of the two dihydroxybenzene isomers CC and HQ represents a serious problem, which arises from the adjacent potentials in the direct oxidation and reduction at most unmodified electrode surfaces. Therefore, it is very important to develop novel materials with excellent conductivity and catalytic activity for the simultaneous determination of CC and HQ.
Activated carbon (AC) is a carbonaceous material, and it is highly porous over a broad range of pore sizes and various functional groups on the surface [22, 23]. Due to their good chemical stability, large aspect ratio, excellent electrical conductivity, and being cost effective [24,25,26,27], AC was widely used in the development of high-performance electrochemical sensors [28]. The price of commercial activated carbon varies between 0.8 and 10 €/kg [29]. However, because the commercial production of activated carbon is not cost effective and together with the fact that the regeneration of used activated carbon is extremely difficult, much attention has been given to synthesizing amorphous activated carbon from renewable sources [30]. Many researchers turn their attention from fossil fuels to cheaper biomass, which is viewed as a veritable way to make full use of agricultural/industrial wastes and reduce carbon emission [31,32,33].
The novelty of this work is the simultaneous determination of the catechol and hydroquinone using an AC. This material having the advantages of very low cost and facilitates the charge transfer, which can enhance the electrocatalytic performance and anti-interference proprieties of the modified electrode.
In this study, we presented a simple and general method to prepare activated carbon from date stone through physical activation. The result was demonstrated to be an effective material for the fabrication of electrochemically modified paste carbon electrode (AC/CPE). The performance of the fabricated electrode (AC/CPE) and their catalytic effect on HQ and CC behaviors were investigated using cyclic voltammetry (CV). Moreover, the fabricated AC/CPE was successfully applied to the simultaneous detection of HQ and CC by differential pulse voltammetry (DPV) with good sensitivity, wide linear range, and low limit of detection.
Experimental
Reagents
All reagents employed were of analytical grade and were used without further purification. Hydroquinone (HQ) and catechol (CC), dibasic potassium phosphate, and potassium dihydrogen phosphate were obtained from Sigma-Aldrich. Its stock solution was prepared with distilled water. Phosphate buffer solutions were prepared by mixing the stock solutions of 0.1 mol L−1 K2HPO4 and 0.1 mol L−1 KH2PO4 at different rations to adjust the pH value. Carbon graphite powder (particle size < 100 μm) used for constructing electrodes was supplied from Carbone Lorraine (Lorraine, France; ref. 9900).
Apparatus
The electrochemical experiments, including cyclic voltammetry (CV) and differential pulse voltammetry (DPV), were performed with an eDAQecorder/potentiostat EA163 controlled by the eDAQEChem data acquisition software, with a three electrode system. The working electrode was unmodified carbon paste or that modified by activated carbon (AC/CPE) with cavity geometric surface of 0.1256 cm2. Reference electrode and counter electrode used were Ag/AgCl/saturated KCl and platinum, respectively. pH measurements were carried out using a pH-meter sensION™ (pH 31), with a combined pH glass electrode calibrated against standard phosphate buffer solutions.
The activated carbon was characterized by X-ray diffraction (D 2-PHASER of BRUKER-AXS, Cu-radiation, λKα1 = 1.54060 Å and λKα2 = 1.54439 Å) performed at room temperature. The external surface of the activated carbon was examined by a scanning electron microscope VEGA3 TESCAN. Fourier Transform Infrared (FT-IR) spectroscopy (FTIR-2000, Perkin Elmer) was used also to evaluate the chemical structure properties. The infrared spectrum was collected in a range of 4000–400 cm−1.
Preparation of activated carbon modified carbon paste electrode
The activated carbon was prepared according to the method reported in the literature [34]. Briefly, activated carbons were prepared from date stones by pyrolysis under nitrogen flow and activation under water vapor. Moreover, the carbon paste mixture was prepared by hand mixing of graphite powder and an appropriate amount of activated carbon to form a mixture well homogenized [34, 35].
Results and discussion
Characterization of the activated carbon
X-ray diffraction patterns for activated carbon were shown in Fig. 1a. The positions of the peaks due to d002 and d100 reflections are attributed to 2θ = 23.5° and 43.5°, respectively [36]. These diffraction peaks are evidence that the samples have an amorphous structure.

a X-ray diffraction (XRD). b Infrared spectra (IR). c SEM-EDX of activated carbon
FT-IR was used to determine the functional groups involved in activated carbon. In the FT-IR spectra (Fig. 1b), the presence of –OH group stretching at wave number of 3500 cm−1 (stretching vibration) with low signal was indicated. Similarly, at wave number of 2924 cm−1, it was attributed to C-H interaction with the surface of the carbon [37], with moderate intensity. Then, the wave numbers of 1645 cm−1–1678 cm−1 showed the C=C bonds of an alkene compound type, while 1516 cm−1–1541 cm−1 showed the C=C bonds of the aromatic ring compound type; this is supported by the presence of C–O at wave number of 1112 cm−1 [38].
Scanning electron microscope (SEM) picture of activated carbon is shown in Fig. 1c. The picture shows the highly porous characteristics of activated carbon with full of cavities. The porous surface morphology of the prepared carbon is a positive point for adsorption of hydroquinone and catechol.
Effect of activated carbon on the separation peak potential of HQ and CC
Figure 2a, b displays the individual electrochemical behaviors of HQ and CC at the CPE and AC/CPE in 0.1 M PBS (pH 7.0), respectively. From Fig. 2a, it can be observed that at AC/CPE, the potential differences between anodic and cathodic peaks of HQ were determined to be 0.233 and − 0.037 V, respectively. After the CPE modified with AC, the peak potential separation (ΔEP) decreased from 0.49 to 0.27 V. For CC (Fig. 2b), at CPE, the abroad anodic and cathodic peak potentials of CC appear at about 0.393 and 0.016 V, respectively. While at AC/CPE, the anodic and cathodic peaks appear at 0.312 and 0.054 V, respectively, and the ΔEP is extensively narrowed to 119 mV. The above results further confirm that the electrochemical reversibility of CC at the AC/CPE can be improved, similar to the observation in the case of HQ at the AC/CPE. It is evident that the AC could be an effective electrocatalyst for the redox reaction of dihydroxybenzene isomers.

a, b, and c are CVs of 1.0 × 10−3 mol L−1 HQ, 1.0 × 10−3 mol L−1 CC, and mixture of 1.0 × 10−3 mol L−1 HQ and 1.0 × 10−3 mol L−1 CC in 0.1 mol L−1 PBS (pH 7.0) at CPE and AC/CPE at 50 mV s−1, respectively
The cyclic voltammograms of a mixture containing HQ and CC (1.0 × 10−3 mol L−1) in PBS (pH 7.0) at CPE and AC/CPE are shown in Fig. 2c. At CPE, a large broad peak centered at + 0.314 V is observed for the oxidation reaction, while a narrower peak centered at 0.022 V is observed for the reduction reaction. These behaviors showed that the oxidation and reduction peaks of HQ and CC were characterized by the overlapping peaks, and it was impossible to separate these two compounds easily at the CPE. Also, the peak potential separation (ΔEp = Epa − Epc) of 292 mV indicates the irreversibility of the reactions. On the other hand, at AC/CPE, two pairs of well-defined peaks are apparent. The peaks at + 0.022 and + 0.147 V correspond to the oxidation of HQ and CC, respectively, and the reduction peaks are observed at − 0.016 V (HQ) and + 0.104 V (CC), with the values of ΔEp = 38 mV for HQ and ΔEp = 43 mV for CC. Additionally, a separation of 125 and 120 mV between the oxidation and reduction peaks, respectively, of HQ and CC is large enough for the simultaneous determination of the dihydroxybenzene isomers. The AC/CPE electrode exhibited an efficient electrocatalytic oxidation of HQ compared to CC (peak shift), due to high adsorption of the proposed modifier toward the molecules containing more than one groupment “OH.”. The variation of the adsorption capacity as a function of the “OH” position serves to promote the separation of the HQ and CC peaks. From the obtained results, it is found that the HQ adsorbs better than the CC. Consequently, the best adsorption capacity was observed for the molecule with “para” position compared to the ortho isomers. As well as the difference between the oxidation potentials facilitated the sensitive and accurate detection of the two compounds [39,40,41].
The excellent performance of AC/CPE may attribute to the following reasons. Firstly, the hydroxyl, carboxyl, and lactonic groups in activated carbon could interact with the hydroxyl groups in two isomers (CC and HQ) via H-bonding. Secondly, in the PBS of pH 7.0, the negatively charged AC (pHzpc = 7.6) could also interact with the positively charged HQ (pKa 9.96) and CC (pKa 9.48) through the favorable electrostatic attraction. These interactions may help to lower the activation energy of the redox reactions required. The overpotential of reactions was therefore decreased. Thirdly, the two isomers can be enriched on AC/CPE due to the large specific surface area of the AC.
Optimization of parameters
Effect of the mass ratio of AC
In order to obtain a better electrochemical performance of modified paste carbon electrode (AC/CPE) for the simultaneous detection of HQ and CC, the mass ratio of AC used in the fabrication of the modified electrode was investigated by CV. From Fig. 3, the modified electrodes fabricated with different mass ratios of AC behaved differently on the detection. When the mass ratio of AC was 2%, the resulting modified electrode exhibited the largest peak current, while the oxidation peak of HQ and CC could be completely separated. Thus, the optimum mass ratio of AC was selected as 2%.

CVs of AC/CPE with the mass ratios of AC at 1, 2, 3, 4, and 5% in 0.1 mol L−1 PBS (pH 7.0) containing 1.0 × 10−3 mol L−1 HQ and 1.0 × 10−3 mol L−1 CC; plots of variation of Ia and Ea with mass ratios of AC
Effect of solution pH
The electrochemical oxidation of hydroxyl aromatic compounds always involves proton transfer to form quinone [42]. So, to obtain the best electrochemical response of HQ and CC at the AC/CPE, the effect of the solution pH on the electrochemical responses was also investigated by the CV in 0.1 mol L−1 buffer phosphate solution, with a pH range of 5–8.5. As can be seen in Fig. 4, the anodic peak potentials of HQ and CC shifted negatively with the increase of pH from 5 to 8.5, revealing the participation of protons in the present electrochemical redox process [43]. The linear relationship of the formal potential of HQ and CC with pH can be expressed as the following equations (Eq. (1) and (2)):

CVs at AC/CPE in 0.1 mol L−1 PBS with different pH containing 1.0 × 10−3 mol L−1 HQ and 1.0× 10−3 mol L−1 CC; plot of Ea CC and Ea HQ vs. pH value; plot of Ia CC and Ia HQ vs. pH value
The slopes values of the two equations closed well with the theory value of 59 mV/pH that deduced from the Nernst equation (dEp/dpH = 2.303 mRT/nF, m and n are protons and electrons numbers). Based on these results, the redox reaction of HQ (or CC) at AC/CPE should be a two electron and two proton processes [44, 45]. Therefore, the probable reactions of HQ and CC on AC/CPE are described as Schema 1.

Redox reaction of HQ and CC
Figure 4 also shows that the oxidation peak current of HQ and CC increased with increasing the pH value until 7.0 and then decreased when the pH further increased. For the detection of HQ and CC simultaneously, pH 7.0 was selected as an optimal value of pH.
Effect of scan rate
The effect of scan rate on the redox of two isomers HQ and CC were investigated on the AC/CPE using CV. Figure 5 showed the CV plots with different scan rates (30–400 mV s−1) in 1.0 × 10−3 mol L−1 HQ and CC. The oxidation peaks current vs. scan rate for the HQ and CC which exhibited the linearity with the linear regression equation Ipa (μA) = 0.114 υ μA/mVs−1 + 3.63 μA (R2 = 0.9966) for HQ and Ipa (μA) = 0.133 υ μA/mVs−1 + 6.15 μA (R2 = 0.9959) for CC. From the slopes (0.8606 and 0.8613) found from the graph of log sweep rate (υ) versus the Log of anodic peak current (Log Ipa) for HQ and CC is nearly to the theoretically obtained value of 1.0 for an adsorption controlled electrode process [46, 47]. This study indicates that the electrocatalytic behavior of HQ and CC at the surface of AC/CPE was controlled by the adsorption controlled electrochemical process [48].

CVs of a mixture of 1.0 × 10−3 mol L−1 HQ and CC in 0.1 mol L−1 PBS on AC/CPE at different scan rates (30, 40, 50, 60, 80, 100, 153, 200, 250, 320, and 400 mV s−1); plot I vs. scan rate; plot Log Ia vs. Log υ
Simultaneous determination of HQ and CC
Differential pulse voltammetry technique (DPV) has been established to be very sensitive in the detection of micromolar amounts of chemical species compared with cyclic voltammetry. The effect of the chemical and DPV parameters on the response of the electrodes has been studied by means of seven factors. According to our experience, the optimal combination of chemical and DPV parameters chosen for these studies was as follows: accumulation time = 60 s, pH = 7, mass ratio of AC = 2%, step weight = 150 ms, step height = 5 mV, pulse weight = 50 ms, and step height = 80 mV.
The selective determination of HQ and CC at AC/CPE was studied by increasing the concentration of one isomer while keeping the concentration of the other isomer constant. Figure 6a shows that the DPV signals of CC oxidation increased remarkably with the increase of CC concentration, and the coexisted HQ had no effect on the detection of CC. As shown in the insert of Fig. 6a, the increase of Ipa fits the linear equation of Ipa (μA) = 0.2457 [CC] (μmol L−1) + 16.07 (R2 = 0.9965) when the concentration of CC increases from 1.0 × 10−6 to 1.0 × 10−3 mol L−1. The AC/CP exhibits good limit of detection (LOD) and limit of quantification (LOQ) for CC using Eq. (3) and (4) [49, 50] and found to be 1.83 × 10−8 mol L−1 and 5.5 × 10−8 mol L−1, respectively.
where, S is the standard deviation and M is the slope.

DPVs of AC/CPE in 0.1 mol L−1 PBS (pH 7.0) containing a various concentrations of HQ (a–p 1.0 × 10−7 to 1.0 × 10−3 mol L−1) in the presence of 5.0 × 10−5 mol L−1 CC, b various concentrations of CC (a–o 1.0 × 10−6 to 1.0 × 10−3 mol L−1) in the presence of 1.0 × 10−4 mol L−1 HQ, and c various concentrations of HQ and CC (a–m 1.0 × 10−6 to 1.0 × 10−3 mol L−1). The inserts show the relationships between the peak currents and target concentrations
Similarly, as shown in Fig. 6b, in the coexistence of CC, the linear range of HQ was from 1.0 × 10−7 to 1.0 × 10−3 mol L−1 with a regression equation of Ipa (μA) = 0.1907[HQ] (μmol L−1) + 8.945 (R2 = 0.9943). The calculated LOD and LOQ are 7.1 × 10−8 mol L−1 and 2.36 × 10−7 mol L−1, respectively. The above results indicate that the oxidation reactions of HQ and CC at AC/CPE take place independently.
The simultaneous determination of HQ and CC at AC/CPE was further studied by synchronously changing the concentration of HQ and CC in their binary mixture. The results are shown in Fig. 6c. The oxidation peak currents of HQ and CC were linearly correlated with their concentrations. The regression equations for CC and HQ were Ipa (μA) = 0.261 [CC] (μmol L−1) + 1.5128 (R2 = 0.9948) and Ipa (μA) = 0.1773 [HQ] (μmol L−1) + 3.1801 (R2 = 0.9948), and the limit of detection was estimated to be 5.23 × 10−8 mol L−1 and 7.7 × 10−8 mol L−1, respectively. These values are, respectively, in accordance with those obtained in their selective detection. It is confirmed that the fabricated AC/CPE is suitable for the simultaneous detection of HQ and CC in mixed systems.
A comparison of the proposed electrode AC/CPE with other electrodes for HQ and CC detection is listed in Table 1. Our proposed method achieved lower limits of detection, wider linear concentration ranges, and larger separation anodic potential (ΔEp) value toward the simultaneous determination of HQ and CC, which is superior to the reported in references [3, 8, 48, 51,52,53].
Interference of coexisting substances
The influence of various substances as compounds potentially interfering with the determination of HQ and CC was studied under optimum conditions. For this fact, the interferences of some common metal ions were evaluated. Experimental results which show the effect of some common metal ions including Na+, Ni2+, Zn2+, Cd2+, Ag+, Al3+, Mg2+, Pb2+, Co2+, and Fe2+ (each of 1.0 × 10−4 mol L−1) on the peak potential and intensity of 1.0 × 10−5 mol L−1 HQ and 1.0 × 10−5 mol L−1 CC were represented in Table 2. The variation in current and potential peak caused by the interference metal ions was less than 5%, which proved an excellent selectivity of AC/CPE. Additionally, for the common interference in biological samples, 5.0 × 10−5 mol L−1 of 4-nitrophenol, phenol, paracetamol, and dopamine were investigated in the presence of 5.0 × 10−5 mol L−1 HQ and CC. The use of the AC/CPE for the simultaneous determination was demonstrated by the clean separation of the potential peaks compared with CPE (Fig. 7). These results demonstrated that the AC-modified electrode exhibited excellent selectivity for determination of CC and HQ without interference of other coexisting substances.

DPV curves after exposure to a solution containing 5.0 × 10−5 mol L−1 4-nitrophenol (4-NP), phenol (Ph), paracetamol (PCT), dopamine (DA), hydroquinone (HQ) and catechol (CC) at CPE (a), and AC/CPE (b)
Stability and reproducibility of the AC/CPE
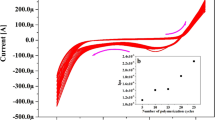
The stability and reproducibility of AC/CPE were also investigated by DPV and the modified electrodes exhibited nice properties in these two aspects. When the AC/CPE was stored at room temperature for about 1 month, the peak currents and peak potential of HQ and CC decreased merely 5%, indicating the high stability of AC/CPE (Fig. 8a). In addition, under the optimized conditions, the reproducibility was investigated comparing the peak currents and peak potential of the same concentration of HQ and CC (Fig. 8b). The relative standard deviations (RSDs) were less to 4%, which obtained for eight measurements in a solution of 1.0 × 10−5 mol L−1 CC and 1.0 × 10−5 mol L−1 HQ.

The stability (a) and reproducibility (b) of AC/CPE with repetitive measurements of DPV response for 1.0 × 10−5 mol L−1 CC and 1.0 × 10−5 mol L−1 HQ in 0.1 mol L−1 PBS with pH 7.0
Analytical application
In order to check the applicability and validity of the proposed method, the AC/CPE was applied for the simultaneous determination of HQ and CC in local tap water samples. Typically, the sample was prepared by adding 0.1 mol L−1 PBS (pH 7) to tap water and then spiked with appropriate amounts of CC and HQ. The electroanalytical curves were recorded using differential pulse voltammetry.
The results demonstrated that no analytes could be detected in the real samples, meaning that two isomers HQ and CC contents were below than the limits of detection. Then, the amount of hydroquinone in the presence of catechol, as well as catechol in the presence of hydroquinone in local tap water samples, was calculated from the calibration method using differential pulse voltammetry and the results are summarized in Table 3. The recoveries were found in the range from 95.50 to 97.85% for HQ and from 95.25 to 98.60% for CC, with the RSDs (below 5%). The obtained recovery results indicate that AC/CPE modified electrode can be successfully used for the determination of the concentration of two dihydroxybenzene isomers in real tap water samples.
Conclusion
In this work, a facile, effective and highly sensitive electrochemical method is devoted based on the activated carbon for the simultaneous and quantitative detection of HQ and CC. Therefore, due to the excellent electrocatalytic activity of AC, it showed two well-defined voltammetric peaks with greatly enhanced peak currents compared to that at bare CPE, which provided high sensitivity and good reversibility for the oxidation of HQ and CC. After the optimization of the experimental conditions, the measurable concentration ranges of HQ and CC using DPV at AC/CPE are expanded remarkably with lower limits of detection compared to previously reported electrochemical sensors. Additionally, the proposed modified electrode was applicable to determine HQ and CC in tap water with satisfying results. All the results showed that the proposed method is simple, low cost, and effective, which provides for the simultaneous detection of two diphenol isomers.
References
Du H, Ye J, Zhang J, Huang X, Yu C (2011) A voltammetric sensor based on graphene-modified electrode for simultaneous determination of catechol and hydroquinone. J Electroanal Chem 650:209–213
Blaut M, Braune A, Wunderlich S, Sauer P, Schneider H, Glatt H (2006) Mutagenicity of arbutin in mammalian cells after activation by human intestinal bacteria. Food Chem Toxicol 44:1940–1947
Yin H, Zhang Q, Zhou Y, Ma Q, liu T, Zhu L, Ai S (2011) Electrochemical behavior of catechol, resorcinol and hydroquinone at graphene–chitosan composite film modified glassy carbon electrode and their simultaneous determination in water samples. Electrochim Acta 56:2748–2753
Khodaei MM, Alizadeh A, Pakravan N (2008) Polyfunctional tetrazolic thioethers through electro-oxidative/Michael-type sequential reactions of 1,2- and 1,4- dihydroxybenzenes with 1-phenyl-5-mercaptotetrazole. J Org Chem 73:2527–2532
Jagetia GC, Aruna R (1997) Hydroquinone increases the frequency of micronuclei in a dose-dependent manner in mouse bone marrow. Toxicol Lett 93:205–213
Taysse L, Troutaud D, Khan NA, Deschaux P (1995) Structure-activity relationship of phenolic compounds (phenol, pyrocatechol and hydroquinone) on natural lymphocytotoxicity of carp (Cyprinus carpio). Toxicology 98:207–214
Hirakawa K, Oikawa S, Hiraku Y, Hirosawa I, Kawanishi S (2002) Catechol and hydroquinone have different redox properties responsible for their differential DNA-damaging ability. Chem Res Toxicol 15:76–82
Yuan X, Yuan D, Zeng F, Zou W, Tzorbatzogloub F, Tsiakaras P, Wang Y (2013) Preparation of graphitic mesoporous carbon for the simultaneous detection of hydroquinone and catechol. Appl Catal B Environ 129:367–374
Ahammad AJS, Sarker S, Rahman MA, Lee JJ (2010) Simultaneous determination of hydroquinone and catechol at an activated glassy carbon electrode. Electroanalysis 22:694–700
Asan A, Isildak I (2003) Determination of major phenolic compounds in water by reversed-phase liquid chromatography after pre-column derivatization with benzoyl chloride. J Chromatogr A 988:145–149
Li SF, Li XZ, Xu J, Wei XW (2008) Flow-injection chemiluminescence determination of polyphenols using luminol–NaIO4–gold nanoparticles system. Talanta 75:32–37
Cui H, Zhang QL, Myint A, Ge XW, Liu LJ (2006) Chemiluminescence of cerium(IV)–rhodamine 6G–phenolic compound system. J Photochem Photobiol A Chem 181:238–245
Nagaraja P, Vasantha RA, Sunitha KR (2001) A sensitive and selective spectrophotometric estimation of catechol derivatives in pharmaceutical preparations. Talanta 55:1039–1046
Unnikrishnan B, Ru PL, Chen SM (2012) Electrochemically synthesized Pt–MnO2 composite particles for simultaneous determination of catechol and hydroquinone. Sensors Actuators B Chem 169:235–242
Li DW, Li YT, Song W, Long YT (2010) Simultaneous determination of dihydroxybenzene isomers using disposable screen-printed electrode modified by multiwalled carbon nanotubes and gold nanoparticles. Anal Methods 2:837–843
Zhang X, Duan S, Xu XM, Xu S, Zhou CL (2011) Electrochemical behavior and simultaneous determination of dihydroxybenzene isomers at a functionalized SBA-15 mesoporous silica modified carbon paste electrode. Electrochim Acta 56:1981–1987
Shaikh AA, Saha SK, Bakshi PK, Hussain A, Ahammad AJS (2013) Poly(brilliant cresyl blue)-modified electrode for highly sensitive and simultaneous determination of hydroquinone and catechol. J Electrochem Soc 160:37–42
Ahammad AJS, Rahman MM, Xu GR, Kim S, Lee JJ (2011) Highly sensitive and simultaneous determination of hydroquinone and catechol at poly(thionine) modified glassy carbon electrode. Electrochim Acta 56:5266–5271
Vilian ATE, Chen SM, Huang LH, Ali MA, Al-Hemaid FMA (2014) Simultaneous determination of catechol and hydroquinone using a Pt/ZrO2-RGO/GCE composite modified glassy carbon electrode. Electrochim Acta 125:503–509
Guo HL, Peng S, Xu JH, Zhao YQ, Kang XF (2014) Highly stable pyridinic nitrogen doped graphene modified electrode in simultaneous determination of hydroquinone and catechol. Sensors Actuators B Chem 193:623–629
Yue XY, Pang SP, Han PX, Zhang CJ, Wang JL, Zhang LX (2013) Carbon nanotubes/carbon paper composite electrode for sensitive detection of catechol in the presence of hydroquinone. Electrochem Commun 34:356–359
Kumar A, Jena HM (2016) Preparation and characterization of high surface area activated carbon. Results Phys 6:651–658
Bachrun S, AyuRizka N, Annisa SH, Arif H (2016) Preparation and characterization of activated carbon from sugarcane bagasse by physical activation with CO2 gas. IOP Conf Ser Mater Sci Eng 105:12–27
Jain A, Xu C, Jayaraman S, Balasubramanian R, Lee JY, Srinivasan MP (2015) Mesoporous activated carbons with enhanced porosity by optimal hydrothermal pre-treatment of biomass for supercapacitor applications. Microporous Mesoporous Mater 218:55–61
Ghasemi M, Mashhadi S, Asif M, Tyagi I, Agarwal S, Gupta VK (2016) Microwave- assisted synthesis of tetraethylenepentamine functionalized activated carbon with high adsorption capacity for Malachite green dye. J Mol Liq 213:317–325
Saeidi N, Parvini M, Niavarani Z (2015) High surface area and mesoporous graphene/activated carbon composite for adsorption of Pb(II) from wastewater. J Environ Chem Eng 3:2697–2706
Zhu Z, Li A, Xia M, Wan J, Zhang Q (2008) Preparation and characterization of polymer based spherical activated carbons. Chin J Polym Sci 26:645–651
Park JH, Park OO, Shin KH, Jin CS, Kim JH (2002) An electrochemical capacitor based on a Ni (OH) 2/activated carbon composite electrode. Electrochem Solid State Lett 5:7–10
Stavropoulos GG, Zabaniotou AA (2009) Minimizing activated carbons production cost. Fuel Process Technol 90:952–957
Muniandy L, Adam F, Mohamed AR, Ng EP (2014) The synthesis and characterization of high purity mixed microporous/mesoporous activated carbon from rice husk using chemical activation with NaOH and KOH. Microporous Mesoporous Mater 197:316–323
Malik R, Ramteke DS, Wate SR (2006) Physico-chemical and surface characterization of adsorbent prepared from groundnut shell by ZnCl2 activation and its ability to adsorb colour. Indian J Chem Technol 13:319–328
Gratuito MKB, Panyathanmaporn T, Chumnanklang RA, Sirinuntawittaya N, Dutt A (2008) Production of activated carbon from coconut shell: optimization using response surface methodology. Bioresour Technol 99:4887–4895
Hussaro K (2014) Preparation of activated carbon from palm oil shell by chemical activation with Na2CO3 and ZnCl2 as imprenated agents for H2S adsorption. Am J Environ Sci 10:336–346
Hammani H, Boumya W, Laghrib F, Farahi A, Lahrich S, Aboulkas A, El Mhammedi MA (2017) Electro-catalytic effect of Al2O3 supported onto activated carbon in oxidizing phenol at graphite electrode. Mater Today Chem 3:27–36
Hammani H, Boumya W, Laghrib F, Farahi A, Lahrich S, Aboulkas A, El Mhammedi MA (2017) Electrocatalytic effect of NiO supported onto activated carbon in oxidizing phenol at graphite electrode: application in tap water and olive oil samples. J Assoc Arab Univ Basic Appl Sci 24:26–33
Kumar K, Saxena RK, Kothari R, Suri DK, Kaushik NK, Bohra JN (1997) Correlation between adsorption and x-ray diffraction studies on viscose rayon based activated carbon cloth. Carbon 35:1842–1844
Deraman M, Talib IA, Omar RM, Jumali HHJ, Taer E, Saman MM (2010) Microcrystallite dimension and total active surface area of carbon electrode from mixtures of PreCarbonized oil palm empty fruit bunches and green petroleum cokes. Sains Malaysiana 39:83–86
Yang T, Lua A (2003) Characteristics of activated carbons prepared from pistachio-nut shells by physical activation. J Colloid Interface Sci 267:408–417
Kuskur CM, Swamy BEK, Jayadevappa H (2017) Poly (naphthol green B) modified carbon paste electrode sensor for catechol and hydroquinone. J Anal Chem 804:99–106
Ganesh PS, Swamy BEK (2016) Voltammetric resolution of catechol and hydroquinone at eosin Y film modified carbon paste electrode. J Mol Liq 220:208–215
Naik TSSK, Swamy BEK (2017) Modification of carbon paste electrode by electrochemical polymerization of neutral red and its catalytic capability towards the simultaneous determination of catechol and hydroquinone: a voltammetric study. J Anal Chem 804:78–86
Yin H, Ma Q, Zhou Y, Ai S, Zhu L (2010) Electrochemical behavior and voltammetric determination of 4-aminophenol based on graphene–chitosan composite film modified glassy carbon electrode. Electrochim Acta 55:7102–7108
Liu Z, Wang Z, Cao Y, Jing Y, Liu Y (2011) High sensitive simultaneous determination of hydroquinone and catechol based on graphene/BMIMPF 6 nanocomposite modified electrode. Sensors Actuators B Chem 157:540–546
Song D, Xia J, Zhang F, Bi S, Xiang W, Wang Z, Xia L, Xia Y, Li Y, Xia L (2015) Multiwall carbon nanotubes-poly(diallyldimethylammonium chloride)-graphene hybrid composite film for simultaneous determination of catechol and hydroquinone. Sensors Actuators B Chem 206:111–118
Gan T, Sun J, Huang K, Song L, Li Y (2013) A graphene oxide–mesoporous MnO2 nanocomposite modified glassy carbon electrode as a novel and efficient voltammetric sensor for simultaneous determination of hydroquinone and catechol. Sensors Actuators B Chem 177:412–418
Kumar M, Swamy BEK, Chandra U, Gebisa AW (2017) Co3O4/CuO composite nanopowder/sodium dodecyl sulphate modified carbon paste electrode based voltammetric sensors for detection of dopamine. Int J Nanotechnol 14:930–944
Mohan K, Swamy BEK, Asif MHM, Viswanath CC (2017) Preparation of alanine and tyrosine functionalized graphene oxide nanoflakes and their modified carbon paste electrodes for the determination of dopamine. Appl Surf Sci 399:411–419
Si WM, Lei W, Zhang YH, Xia MZ, Wang FY, Hao QL (2012) Electrodeposition of graphene oxide doped poly (3,4-ethylenedioxythiophene) film and its electrochemical sensing of catechol and hydroquinone. Electrochim Acta 85:295–301
Shankar SS, Swamy BEK, Pandurangachar M, Chandra U, Chandrashekar BN, Manjunatha JG, Sherigara BS (2010) Electrocatalytic oxidation of dopamine on acrylamide modified carbon paste electrode: a voltammetric study. Int J Electrochem Sci 5:944–954
Chandra U, Swamy BEK, Mahanthesha KR, Vishwanath CC, Sherigara BS (2013) Poly (malachite green) film based carbon paste electrode sensor for the voltammetric investigation of dopamine. Chem Senses 3:1–6
Zheng LZ, Xiong LY, Li YD, Xu JP, Kang XW, Zou ZJ, Yang SM, Xia J (2013) Facile preparation of polydopamine-reduced graphene oxide nanocomposite and its electrochemical application in simultaneous determination of hydroquinone and catechol. Sensors Actuators B Chem 177:344–349
Wang LP, Meng Y, Chen Q, Deng JH, Zhang YY, Li HT, Yao SZ (2013) Simultaneous electrochemical determination of dihydroxybenzene isomers based on the hydrophilic carbon nanoparticles and ferrocene-derivative mediator dual sensitized graphene composite. Electrochim Acta 92:216–225
Wang L, Bai J, Wang H, Zhang L, Zhao Y (2007) Direct simultaneous electrochemical determination of hydroquinone and catechol at a poly(glutamic acid) modified glassy carbon electrode. Int J Electrochem Sci 2:123–132
Acknowledgements
The research described in this article has been funded wholly by the university Hassan 1, Morocco.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hammani, H., Laghrib, F., Lahrich, S. et al. Effect of activated carbon in distinguishing the electrochemical activity of hydroquinone and catechol at carbon paste electrode. Ionics 25, 2285–2295 (2019). https://doi.org/10.1007/s11581-018-2648-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-018-2648-6