
Abstract
GLI1 fusions involving ACTB, MALAT1, PTCH1 and FOXO4 genes have been reported in a subset of malignant mesenchymal tumors with a characteristic nested epithelioid morphology and frequent S100 positivity. Typically, these multilobulated tumors consist of uniform epithelioid cells with bland nuclei and are organized into distinct nests and cords with conspicuously rich vasculature. We herein expand earlier findings by reporting a case of a 34-year-old female with an epithelioid mesenchymal tumor of the palate. The neoplastic cells stained positive for S100 protein and D2-40, whereas multiple other markers were negative. Genetic alterations were investigated by targeted RNA sequencing, and a PTCH1-GLI1 fusion was detected. Epithelioid mesenchymal tumors harboring a PTCH1-GLI1 fusion are vanishingly rare with only three cases reported so far. Due to the unique location in the mucosa of the soft palate adjacent to minor salivary glands, multilobulated growth, nested epithelioid morphology, focal clearing of the cytoplasm, and immunopositivity for S100 protein and D2-40, the differential diagnoses include primary salivary gland epithelial tumors, in particular myoepithelioma and myoepithelial carcinoma. Another differential diagnostic possibility is the ectomesenchymal chondromyxoid tumor. Useful diagnostic clues for tumors with a GLI1 rearrangement include a rich vascular network between the nests of neoplastic cells, tumor tissue bulging into vascular spaces, and absence of SOX10, GFAP and cytokeratin immunopositivity. Identifying areas with features of GLI1-rearranged tumors should trigger subsequent molecular confirmation. This is important for appropriate treatment measures as PTCH1-GLI1 positive mesenchymal epithelioid neoplasms have a propensity for locoregional lymph node and distant lung metastases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fusions or amplifications of the GLI1 gene have been discovered in soft tissue tumors of variable morphology, immunoprofile and clinical behavior. GLI1-fusion positive tumors were initially reported by Dahlen et al. in 2004 as a group of distinctive smooth muscle actin positive mesenchymal neoplasms harboring an ACTB-GLI1 gene fusion, and they were designated as “pericytoma with t(7;12) translocation” [1]. Three of their five cases were located in the tongue and one each in the stomach and the calf [1]. Subsequently, other GLI1-rearranged tumors have been reported to arise in diverse tissues [2, 3], including the ovary [4, 10], the bones [5], the gastrointestinal tract [6,7,8,9], the soft tissue of the extremities [10], the retroperitoneum [2], the lung [3] or the kidney [15]. A broad spectrum of malignant mesenchymal neoplasms with relatively monomorphic round cell or epithelioid morphology, with a frequent S100 protein expression, and with recurrent GLI1 gene fusions with ACTB, MALAT1, PTCH1 or FOXO4 have been characterized [2, 3, 15]. Although sharing some histologic features with pericytoma, including monomorphic and occasionally round cell phenotype, nested growth pattern and an intricate capillary network, these lesions display morphologic variability, lack of actin immunoreactivity, and propensity for locoregional lymph node and distant lung metastases [2].
Even though in the first published series of tumors harboring ACTB-GLI1 fusions there was a marked predilection for occurrence in the tongue [1], other GLI1-rearranged tumors have been later reported to arise in other locations of the head and neck, including the submandibular gland and soft tissues of the neck [11]. However, a mucosal location in the palate has never been reported. Herein, we present a case of a 34-year-old female patient with an epithelioid mesenchymal tumor of the soft palate harboring a PTCH1-GLI1 fusion. Several cases with GLI1 rearrangements in the head and neck have been described [11] but to date, only two of them harbored the PTCH1-GLI1 fusion. They arose in soft tissues of the submandibular region and the neck [2, 11] and the tongue [11]. One additional case with this fusion arose in soft tissues of the thorax [12]. To the best of our knowledge, ours is only the fourth case with the PTCH1-GLI1 fusion reported so far.
All reported tumors with the PTCH1-GLI1 rearrangement (including our case) showed prominent epithelioid features with a multinodular or plexiform growth pattern at low magnification and a distinctive nested architecture with a rich, delicate arborizing vascular network. The tumor cells displayed a monomorphic cytomorphology with uniform round to ovoid nuclei, multiple clear-cell areas, a focal spindled morphology, and S100 protein immunopositivity. Conversely, smooth muscle actin and wide-spectrum cytokeratin (AE1/AE3) expressions were negative on immunohistochemical examination. The unusual intraoral mucosal localization of the tumor in the palate and overlapping multilobulated growth, nested epithelioid morphology, clear cytoplasm and S100 protein immunopositivity represent possible differential diagnostic features shared with some primary salivary gland neoplasms, such as myoepithelioma, cellular pleomorphic adenoma, and myoepithelial carcinoma. Useful diagnostic clues for tumors with a GLI1 rearrangement include a rich vascular network, nested architecture, tumor tissue bulging into vascular spaces, and the absence of cytokeratin, SOX10, GFAP and calponin immunopositivity. Identification of areas with classic histopathologic features of GLI1-rearranged tumors should trigger efforts for molecular confirmation of a GLI1 rearrangement. This aberration has not been reported in the myoepithelial carcinoma of salivary glands [13, 14].
Materials and Methods
The study was approved by the institutional review board of the Faculty of Medicine in Pilsen, Charles University. The case described in this study was retrieved from the consultation files of AS. The clinical follow-up information was obtained from the treating clinicians (RP, ZD) and the referring pathologist (JM).
For conventional microscopy, excised tissues were fixed in formalin, processed routinely, embedded in paraffin (FFPE), cut, and stained with hematoxylin and eosin. For immunohistochemical analysis, 4-μm-thick sections were cut from paraffin blocks and mounted on positively charged slides (TOMO; Matsunami Glass IND, Japan). Sections were processed on a BenchMark ULTRA (Ventana Medical System, Tucson, AZ), deparaffinized, and then subjected to heat-induced epitope retrieval by immersion in CC1 solution at 95 °C and pH 8.6. All primary antibodies used are summarized in Table 1. Bound antibodies were visualized using the UltraView Universal DAB Detection Kit (Roche, Basel, Switzerland) and the UltraView Universal Alkaline Phosphatase Red Detection Kit (Roche). The slides were counterstained with Mayer’s hematoxylin. Appropriate positive and negative controls were employed.
Targeted Next-Generation Sequencing
For next-generation sequencing (NGS) RNA was extracted using Total NA Maxwell RSC DNA FFPE kit (automated on Maxwell RSC 48 Instrument, Promega, Madison, WI). Purified RNA was quantified using the Qubit Broad Range RNA Assay (Thermo Fisher Scientific, Waltham, MA). Fusion transcript detection and mutational analysis were performed using the customized FusionPlex Sarcoma kit No.6 (Archer DX, Boulder, CO). The process of library preparation and sequencing was done as described previously [14] and data analysis was performed using the Archer Analysis software version 6.2.1. The complete list of genes is available in Online Resource.
Case Presentation
The patient was a 34-year-old female who had observed a stiff lesion in her soft palate 2 months prior to first contact with a maxillofacial surgeon. The lesion was initially considered to be an abscess with partial regression of its size after dental work and an antibiotic treatment. However, a CT angiography revealed a persistent non-homogenous tumorous lesion in the soft palate and lateral nasopharyngeal wall. A preliminary biopsy was inconclusive, but suggestive of a low-grade round-cell sarcoma with immunohistochemical positivity for D2-40 and neuroendocrine markers (a diffuse positivity for synaptophysin and NSE, and a focal positivity for S-100 protein). At the time of excision, the tumor measured 34 × 21 × 13 mm. Oral surface above the tumor was ulcerated in an area of 18 × 13 mm and to a depth of 4 mm (Fig. 1A). The tumor consisted of soft tan-brown tissue on cut surface, and there was no discernible capsule.

Macroscopic features of the tumor. A The tumor grew on the left side of the soft palate with a superficial ulceration. B Residual tumor tissue was resected during second surgery, together with the left palatine tonsil
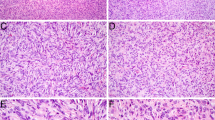
Microscopically, the excision was covered by a superficially ulcerated stratified squamous epithelium with reactive changes and an extensive pseudoepitheliomatous hyperplasia. Underneath, there was inflamed granulation tissue with occasional giant cells attached to the multilobular tumor (Fig. 2A). Invasive tumor front extended into the surrounding small salivary gland (Fig. 2B) and the striated muscle tissue (Fig. 2C), and perineural invasion was noted (Fig. 2D). Tumor cell nests were separated by thick or delicate fibrous septae (Fig. 3A) and occasionally they protruded into vascular spaces (not shown). An abundant arborizing capillary network was seen throughout the neoplastic tissue. The network was well pronounced especially in solid areas of the tumor (Fig. 3A, B). The tumor cells were organized either in a solid-trabecular fashion (Fig. 3B), a fascicular (myoepithelial carcinoma-like) pattern (Fig. 3C, D), or a reticular pattern with a myxoid stroma (Fig. 3E). A minor part of the tumor displayed a pseudoglandular architecture (Fig. 3F). Tumor cells appeared relatively uniform, round to oval, with bland centrally located basophilic-staining nuclei, inconspicuous nucleoli and a moderate amount of delicate granular eosinophilic cytoplasm. A spindled cell morphology was present in the fascicular areas. Occasional cells displayed clearing of the cytoplasm, sometimes with a perinuclear halo (Fig. 3G), escalating in some areas into a pseudolipoblast appearance with prominent cytoplasmic vacuolization (Fig. 3H). Tumor-infiltrating lymphocytes were abundant, especially in the solid-trabecular areas of the tumor (Fig. 3H). Cell proliferation was low, reaching 2 mitotic figures per 10 HPF. No tumor necrosis nor any atypical mitoses were seen. The tumor was not completely resected.

Low-power microscopic view of the tumor (H&E*). A The multilobular tumor grew underneath an ulcerated stratified squamous epithelium of the oropharyngeal mucosa. B Tumor cells extended into adjacent minor salivary gland. C Tumor cells infiltrated adjacent striated muscle tissue. D Perineural infiltration was noted. *Hematoxylin and eosin

Histomorphologic features of the tumor (H&E*). Nests of tumor cells were surrounded by thick or delicate fibrous septae (A). Arborizing capillary network was interspersed among tumor cells throughout the lesion (A, B). Tumor cells were relatively uniform, round to oval, occasionally spindled, and with bland basophilic oval nuclei and inconspicuous nucleoli (B, C). Several architectural patterns were seen. Tumor cells grew predominantly in a solid-trabecular (B) and a fascicular, myoepithelial carcinoma-like fashion (C, D). Less commonly, a reticular pattern with abundant myxoid stroma (E) and a pseudoglandular microcystic pattern (F) were seen. Focally, tumor cells displayed clearing of the cytoplasm with an infrequent perinuclear halo (G). The clear cell change escalated in some areas into a pseudolipoblast-like appearance with marked vacuolization of the cytoplasm (H). A focal lymphocytic infiltrate was seen throughout the tumor (H). *Hematoxylin and eosin
Immunohistochemically, a focal and patchy S-100 protein (Fig. 4A) and CD56 (Fig. 4B) positivity, as well as a diffuse and strong D2-40 (Fig. 4C) expression were demonstrated in the tumor cells. A wide spectrum of various markers was negative, including markers of epithelial (AE1/AE3, OSCAR, CAM5.2, CK5/6, CK7, CK14), myogenic (myoD1, desmin), melanocytic (SOX10), and endothelial (CD31, CD34) differentiation, as well as various other markers such as CD99, p63, HHV-8, DDIT3, STAT6 and CDK4. Proliferative index was low to intermediate, in lymphocyte-poor areas Ki-67 stained up to 7% of nuclei, while in lymphocyte-rich areas Ki-67 was positive in 20–25% of tumor cells (Fig. 4D). As the immunohistochemical examination of the probatory biopsy at the referring institution showed diffuse positivity for synaptophysin and NSE, we conducted an additional analysis of the expression of neuroendocrine markers. While NSE was strongly diffusely positive, patchy synaptophysin expression was detected in up to 5% cells and chromogranin was positive in one focus. However, multiple blocks had to be examined to demonstrate the positivity of synaptophysin and chromogranin.

Immunohistochemical features of the tumor. A Immunohistochemical examination revealed focal and patchy S-100 protein expression (positive internal control in a nerve is seen in the upper right corner). B Similar pattern was seen with CD56 immunostain. C Diffuse and strong D2-40 positivity was present throughout the tumor. D Ki-67 stained up to 7% of cells in areas with sparse lymphocytes, while 20–25% of tumor cells were positive in lymphocyte-rich foci
Using the Archer FusionPlex custom panel, a PTCH1-GLI1 fusion involving exon 1 of the PTCH1 gene and exon 6 of the GLI1 gene was detected. The reference transcript sequences used for describing PTCH1 and GLI1 have accession numbers NM_001083602.1 and NM_005269, respectively. The chromosomal position is described using the reference genome GRCh37 (hg19), and breakpoints at chr9:98278751, and chr12:5785939 (Fig. 5).

PTCH1-GLI1 fusion transcript. Exon 1a of PTCH1 gene containing a Gli1 binding site (black arrow) is fused (red arrow) to exon 6 of GLI1 gene. Gli1 part of the fusion protein retains its FOXP coiled-coil domain (FOXP-CC) and three zinc fingers of C2H2 type (C2H2 Zn fingers), which may enable dimerization and binding to Gli1 target genes
Re-resection and lateral block neck dissection were performed 2 months after the primary excision (Fig. 1B). Small amounts of residual tumor were identified in the resection sample from the oropharynx, but the margins were clear. Twenty-two neck lymph nodes were identified, and they were free of tumor infiltration. A PET-CT scan was performed before the first surgery, and it did not reveal any suspected metastatic lesions. Subsequent adjuvant therapy was not recommended by an oncologist. The patient is currently in the second trimester of pregnancy. The case is recent and only a limited follow-up of 4 months is available with no evidence of disease.
Discussion
Genetic abnormalities of GLI1, including gene fusions and amplification, have been reported recently in a subset of soft tissue tumors with a distinctive epithelioid nested histomorphology and a propensity for locoregional and distant metastasis [2]. To the best of our knowledge, 45 cases of GLI1-altered tumors, including 14 cases (31% of all cases) in the head and neck region, have been reported to date. Rearrangements of GLI1 have been detected in tumors arising from various parts of the body, including soft tissues of extremities or trunk (11/31 cases) [2, 3, 10], gastrointestinal tract (10/31 cases) [6,7,8,9], bones (4/31 cases) [2, 5, 10], ovary (2/31 cases) [4, 10], and one case each in intrathoracic soft tissues [12], kidney [15], lung [3], and retroperitoneum [2]. In the head and neck region, the most common site of origin was tongue (11/14 cases), followed by submandibular and neck soft tissue (3/14 cases) [1,2,3, 11]. There are no previous reports of a tumor of the soft palate. The tumors were diagnosed either as “pericytoma with t(7,12) translocation” or mesenchymal neoplasm with GLI1 gene alterations (the name malignant epithelioid neoplasm with GLI1 gene rearrangements was also used for this entity in the past [2]). Additionally, two cases of gastroblastoma with a MALAT1-GLI1 fusion [7] and five cases of gastric plexiform fibromyxoma with the same fusion or GLI1 amplification were reported recently [8]. Strikingly, with the exception of the plexiform fibromyxoma cases, most tumors demonstrated similar histopathologic features and were described as vaguely lobular proliferations of bland spindled to ovoid epithelioid cells with pale eosinophilic or clear cytoplasm, often with some myxoid areas and delicate arborizing vasculature. Even though the first reported cases of GLI1-altered tumors stained positive for smooth muscle actin, leading to their diagnosis as pericytoma, much higher variability in the immunoprofile of these tumors was later noted. In addition to the actin, the tumors are frequently positive for CD56 (16/19 cases tested, 84%), S100 protein (17/43 cases tested, 39.5%) and, less commonly, wide spectrum cytokeratins (AE1/AE3) (8/39 cases tested, 20.5%). Although a wide age range has been observed in patients with GLI1-rearranged tumors (ranging from 1 to 79 years), patients in their 4th decade seem to be most commonly affected (median 32 years). The situation is very similar with regards specifically to the head and neck tumors, where the age of the patients has ranged from 1 to 65 years, with a median of 34.5 years. According to the obtainable data, local recurrence and/or distant metastasis occurred in 36% cases with follow-up information available, including two cases in the head and neck area. The tumor in the first case occurred in the submandibular soft tissue and harbored a PTCH1-GLI1 fusion (discussed further in the next paragraph), in the second case the tumor with a GLI1 amplification arose from the neck soft tissue. Both patients developed both local recurrence and distant metastasis.
Presumably, this is only the fourth neoplasm with a PTCH1-GLI1 fusion reported to date (Table 2). One of the previously described tumors with this fusion grew as an intrathoracic mass in a 13-year-old male [12]. Two tumors with the fusion described by Antonescu and Xu [2, 11] occurred in the head and neck, one appearing in the submandibular soft tissues of a 32-year-old female patient, and the other in the tongue of a 38-year-old male patient. Similarly, the 34-year-old female patient in our case presented with an ulcerated tumor of the soft palate. A stronger predilection for the head and neck can be noted in the PTCH1-GLI1 fused tumors than in other GLI1-rearranged neoplasms, although the number of cases known at present is limited. One of the three previously reported cases with a PTCH1-GLI1 fusion developed local recurrence and lung metastasis. The mitotic index in this case was high (6 mitoses per 10 HPF), and the tumor also contained areas of necrosis. Conversely, both of these features were lacking in the other cases that did not metastasize, including the case reported here. Even though mitotic activity in our case was rather low and the tumor was ultimately resected with tumor-free margins, a careful follow-up is mandatory.
Considering the histomorphology, tumors with the PTCH1-FLI1 fusion do not differ greatly from other tumors in the group. As in the other cases of GLI1-rearranged tumors reported in the head and neck, the present tumor consisted of monomorphic round cells with epithelioid features and a focal clear-cell change that were organized into nests interspersed with delicate capillary networks. Only limited information is available in the literature on histomorphology of the pediatric tumor occurring in the 13-year-old male. The authors describe a tumor consisting of uniform small round cells organized into cord-like structures [12]. In rough agreement with the immunoprofile of all GLI1 rearranged tumors, the PTCH1-GLI1 fused tumors reported to date were at least focally positive for CD56 and negative for AE1/3, while S100 protein, which is only positive in approximately 40% of all GLI1-rearranged tumors, was expressed at least in some cells in each tumor with the PTCH1-GLI1 fusion. At least focal expression of CD56 and S100 protein (observed in 3/3 and 4/4 analyzed cases, respectively) with consistently negative cytokeratins and smooth muscle actin, together with the typical histomorphology, seems to be a common denominator for PTCH1-GLI1 fused tumors [11].
The transcription factor Gli1 is a nuclear effector of the Hedgehog signaling pathway, which under normal conditions plays important roles in organogenesis during embryonic development. The pathway is deregulated in various cancers, for instance a GLI2 amplification has been demonstrated in some medulloblastomas, while a proportion of rhabdomyosarcomas reportedly harbor inactivating mutations in genes of tumor suppressor proteins Patched and Sufu [16]. The significance of the Hedgehog signaling pathway during embryonic development and in carcinogenesis is evident in patients with the nevoid basal cell carcinoma syndrome, who suffer from multiple basal cell carcinomas and various congenital abnormalities caused by an inactivating germline mutation in the PTCH1 gene. This gene encodes the Patched protein, a tumor suppressor localized in the cell membrane, where it binds and inhibits the Smoothened receptor. When any of the Hedgehog signaling proteins binds Patched, it is inactivated and Smoothened is released. Consequently, a signaling cascade leads to the activation of Gli1. In deregulated signaling during tumorigenesis, various proteins involved in cell proliferation, loss of cell adhesion, increased motility or cancer cell stemness belong to Gli1 targets [16, 17]. Importantly, the Patched-encoding gene PTCH1 contains a Gli1 binding site in its exon 1. The proposed gene product of the PTCH1-GLI1 fusion retains the Gli1 binding site in the PTCH1 part, while the proposed fusion protein contains zinc fingers in the Gli1 part that provide binding to specific regulatory sequences of genes downstream of the Hedgehog pathway, including PTCH1 (Fig. 5). In neoplastic cells, this might in turn create a positive feedback loop, in contrast to the opposing relationship between Gli1 and Patched in normal tissues. This would increase the expression of the fusion protein which remains an active transcription factor for other targets in the Hedgehog pathway.
The differential diagnosis of a GLI1-rearranged mesenchymal epithelioid neoplasm is challenging. Due to an overall bland morphology with monomorphic round to epithelioid cells, multilobulated growth, nested architecture, clear cytoplasm and S100 protein immunopositivity, these tumors are strongly reminiscent of primary epithelial and/or myoepithelial neoplasms of the salivary glands. In the present case, due to the corded, microglandular or reticular growth patterns within a myxoid stroma, the epithelioid and spindle-shaped uniform neoplastic cells, and the unique mucosal location in the palate in close proximity to minor salivary glands, the resemblance to myoepithelial neoplasms including myoepithelioma, cellular pleomorphic adenoma, and myoepithelial carcinoma, was particularly intriguing. Importantly, in contrast to the multinodular and infiltrative growth pattern of the GLI1-rearranged mesenchymal neoplasm, a benign myoepithelioma is well-circumscribed. Moreover, pleomorphic adenoma shows a biphasic phenotype with both ductal and myoepithelial differentiation, whereas all GLI1-rearranged tumors were composed of a single cell type. Myoepithelial carcinoma shows at least focally invasive features and is characterized by a multinodular architecture divided by fibrous bands in a zonal arrangement and a hypercellular peripheral rim surrounding a hypocellular, usually myxoid and/or necrotic center. Useful diagnostic clues for tumors with a GLI1 rearrangement include a rich vascular network among nests of tumor cells, tumor tissue bulging into vascular spaces, and absence of cytokeratin, SOX10, GFAP and calponin immunopositivity. Identification of areas with these classic features of GLI1-rearranged tumors should trigger efforts for molecular confirmation of a GLI1 rearrangement, which in contrast has not been reported in myoepithelial carcinomas of salivary glands [13, 14].
Ectomesenchymal chondromyxoid tumor is another tumor included in the differential diagnosis of GLI1-rearranged tumors. It is a rare mesenchymal neoplasm characterized by a striking predilection for the anterior dorsal tongue. It has a multilobulated growth pattern and is immunohistochemically positive for S100 protein, CD56, cytokeratin, and SMA [18, 19]. However, ectomesenchymal chondromyxoid tumor typically shows a more spindled cytomorphology and an abundant myxoid stroma, it lacks an arborizing capillary network and may express other myoepithelial markers such as calponin and GFAP [18]. At the molecular level, these tumors harbor a characteristic RREB1-MKL2 fusion. Thus, molecular testing can be used in differential diagnosis in challenging cases [19].
In a tumor with positive neuroendocrine markers and a prominent vascular network in between round to oval cells with moderate amounts of pale eosinophilic cytoplasm organized in a solid-trabecular pattern, the differential diagnosis of paraganglioma could be considered. Nonetheless, a more solidly nested growth pattern is usually present in paraganglioma. Furthermore, S100 protein immunohistochemistry displays a distinct pattern in paragangliomas, with only the sustentacular cells staining positive. Whereas CD56 immunopositivity is very frequent in epithelioid soft tissue tumors with GLI1 rearrangements, other neuroendocrine markers are typically negative. In fact, there have only been four other cases thus far with a reported positivity for any neuroendocrine markers. These include a case of a 57-year-old female with a tibial tumor with rare cells staining with an antibody against synaptophysin [10], two cases diffusely positive for NSE, including a 67-year-old male with a bone (talus) tumor [5] and a 48-year-old male with a jejunal tumor [6], and an additional case of a 28-year-old male with a gastroblastoma positive for NSE in the epithelial component of the tumor [6]. All but one of these patients later suffered distant metastases [6, 7, 10], possibly linking such neuroendocrine features to a more aggressive clinical behavior. This further highlights the importance of careful follow-up of the patient in the case presented here.
A correct diagnosis supported by the detection of a GLI1 gene rearrangement may have therapeutic implications, as tumors with GLI1 oncogenic activation and subsequent PTCH1 overexpression may be sensitive to SHH pathway inhibitors [20]. Identification of GLI1 alterations and oncogenic activation in this unique tumor entity may, therefore, allow patients, especially those with advanced disease, to gain access to Hedgehog or GLI1-targeted therapies.
In summary, the presented case improves our understanding of the emerging group of GLI1-rearranged tumors. The tumor consisted of nests of relatively uniform round to spindled cells with bland basophilic nuclei, an occasional clearing of tumor cell cytoplasm and focal immunohistochemical positivity for S-100 protein and CD56. A reticular, trabecular, pseudoglandular or fascicular architecture was present in different parts of the tumor. A delicate branching vasculature permeated the lesion. Molecular genetic testing revealed a PTCH1-GLI1 fusion. GLI1-rearranged tumors usually have an indolent behavior; however, propensity for locoregional recurrence and distant metastatic spread should not be underscored. Information about molecular genetic background of the tumor is of great clinical importance, especially for patients with advanced disease who could benefit from treatment with selective Hedgehog pathway inhibitors.
Data Availability
Data supporting the findings of this study are available within the article and its supplementary information files.
Code Availability
Not applicable.
References
Dahlén A, Fletcher CD, Mertens F, et al. Activation of the GLI oncogene through fusion with the beta-actin gene (ACTB) in a group of distinctive pericytic neoplasms: pericytoma with t(7;12). Am J Pathol. 2004;164(5):1645–53.
Antonescu CR, Agaram NP, Sung YS, Zhang L, Swanson D, Dickson BC. A distinct malignant epithelioid neoplasm with GLI1 gene rearrangements, frequent S100 protein expression, and metastatic potential: expanding the spectrum of pathologic entities with ACTB/MALAT1/PTCH1-GLI1 fusions. Am J Surg Pathol. 2018;42(4):553–60.
Agaram NP, Zhang L, Sung YS, et al. GLI1-amplifications expand the spectrum of soft tissue neoplasms defined by GLI1 gene fusions. Mod Pathol. 2019;32:1617–26.
Koh NWC, Seow WY, Lee YT, Lam JCM, Lian DWQ. Pericytoma with t(7;12): the first ovarian case reported and a review of the literature. Int J Gynecol Pathol. 2019;38(5):479–84.
Bridge JA, Sanders K, Huang D, et al. Pericytoma with t(7;12) and ACTB-GLI1 fusion arising in bone. Hum Pathol. 2012;43(9):1524–9.
Prall OWJ, McEvoy CRE, Byrne DJ, et al. A malignant neoplasm from the jejunum with a MALAT1-GLI1 fusion and 26-year survival history. Int J Surg Pathol. 2020;28(5):553–62.
Graham RP, Nair AA, Davila JI, et al. Gastroblastoma harbors a recurrent somatic MALAT1–GLI1 fusion gene. Mod Pathol. 2017;30(10):1443–52.
Spans L, Fletcher CDM, Antonescu CR, et al. Recurrent MALAT1–GLI1 oncogenic fusion and GLI1 up-regulation define a subset of plexiform fibromyxoma. J Pathol. 2016;239(3):335–43.
Castro E, Cortes-Santiago N, Suarez Ferguson LM, Rao PH, Venkatramani R, López-Terrada D. Translocation t(7;12) as the sole chromosomal abnormality resulting in ACTB-GLI1 fusion in pediatric gastric pericytoma. Hum Pathol. 2016;53:137–41.
Kerr DA, Pinto A, Subhawong TK, et al. Pericytoma with t(7;12) and ACTB-GLI1 fusion: reevaluation of an unusual entity and its relationship to the spectrum of GLI1 fusion-related neoplasms. Am J Surg Pathol. 2019;43(12):1682–92.
Xu B, Chang K, Fople AL, et al. Head and neck mesenchymal neoplasms with GLI1 gene alterations: a pathologic entity with distinct histologic features and potential for distant metastasis. Am J Surg Pathol. 2020;44(6):729–36.
Ichikawa D, Yamashita K, Okuno Y, et al. Integrated diagnosis based on transcriptome analysis in suspected pediatric sarcomas. NPJ Genomic Med. 2021;6:49.
Dalin MG, Katabi N, Persson M, et al. Multi-dimensional genomic analysis of myoepithelial carcinoma identifies prevalent oncogenic gene fusions. Nat Commun. 2017;8(1):1197.
Skálová A, Agaimy A, Vanecek T, et al. Molecular profiling of clear cell myoepithelial carcinoma of salivary glands with EWSR1 rearrangement identifies frequent PLAG1 gene fusions but no EWSR1 fusion transcripts. Am J Surg Pathol. 2021;45(1):1–13.
Pettus JR, Kerr DA, Stan RV, et al. Primary myxoid and epithelioid mesenchymal tumor of the kidney with a novel GLI1-FOXO4 fusion. Genes Chromosom Cancer. 2021;60(2):116–22.
Sari IN, Phi LTH, Jun N, Wijaya YT, Lee S, Kwon HY. Hedgehog signaling in cancer: a prospective therapeutic target for eradicating cancer stem cells. Cells. 2018;7(11):208.
Carpenter RL, Lo HW. Hedgehog pathway and GLI1 isoforms in human cancer. Discov Med. 2012;13(69):105–13.
Laco J, Mottl R, Höbling W, et al. Cyclin D1 expression in ectomesenchymal chondromyxoid tumor of the anterior tongue. Int J Surg Pathol. 2016;24(7):586–94.
Dickson BC, Antonescu CR, Argyris PP, et al. Ectomesenchymal chondromyxoid tumor: a neoplasm characterized by recurrent RREB1-MKL2 fusions. Am J Surg Pathol. 2018;42(10):1297–305.
Rimkus TK, Carpenter RL, Qasem S, Chan M, Lo HW. Targeting the sonic hedgehog signaling pathway: review of smoothened and GLI inhibitors. Cancers (Basel). 2016;8(2):22.
Acknowledgements
This study was supported by study grant SVV 22639 from the Ministry of Education, Czech Republic (NK).
Funding
This study was supported by study grant SVV 22639 from the Ministerstvo školství, mládeže a tělovýchovy, Czech Republic (NK).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by AS, JM and VH. The first draft of the manuscript was written by NK and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethical Approval
The study was approved by the institutional review board of the Faculty of Medicine in Pilsen, Charles University. The procedures used in this study adhere to the tenets of the Declaration of Helsinki.
Informed Consent
No patient consent was required for this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Klubíčková, N., Kinkor, Z., Michal, M. et al. Epithelioid Soft Tissue Neoplasm of the Soft Palate with a PTCH1-GLI1 Fusion: A Case Report and Review of the Literature. Head and Neck Pathol 16, 621–630 (2022). https://doi.org/10.1007/s12105-021-01388-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12105-021-01388-4