
Abstract
Sarcoidosis is a complex disorder that often times involves the head and neck. Despite the presence of strong clinical evidence, tissue diagnosis and imaging is needed for confirmation of the disease. Although typically managed medically, when found in the sinonasal tract or intracranially, it may necessitate the intervention of a rhinologist-skull base surgeon. This article seeks to provide a comprehensive review of head and neck sarcoidosis, as this fascinating disorder often poses a diagnostic and therapeutic challenge. A brief discussion of surgical treatment for pituitary lesions is also provided. Articles from 1997 to 2013 were selected and reviewed by three researchers utilizing the most recent literature regarding sarcoidosis in the head and neck. PubMed searches were conducted using search terms such as “sarcoidosis”, “neurosarcoid”, and “extra-pulmonary sarcoid”, among many others. A large collection of articles was generated and reviewed by the team of authors, and appropriate information was extracted to compose a thorough and expansive review of the subject. 10–15 % of patients with sarcoidosis have head and neck manifestations. Sinonasal and pituitary sarcoidosis presents a diagnostic challenge owing to its non-specific symptoms. Although systemic steroid therapy is often the first time treatment, endoscopic surgery is commonly used to treat advanced pituitary sarcoidosis refractory to medical management. As tissue diagnosis and imaging is key, a multi-disciplinary team approach is advantageous. Our study collates the available literature on head and neck sarcoidosis to provide a comprehensive review of the subject. This provides helpful information to guide all practitioners involved in the care of these challenging patients, namely pathologists, radiologists, otolaryngologists, and skull base surgeons, in the workup and management of head and neck sarcoidosis.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Sarcoidosis is a multisystem disease that has been a point of clinical and research interest for many years. Our limited understanding and complex framework mandate continual and progressive assessment. Otolaryngologic manifestations of sarcoidosis have offered an opportunity to apply new advances and innovations in our field, further expanding the potential for discovery.
Most patients who are diagnosed with sarcoidosis, usually present with pulmonary findings, which often resolve spontaneously. Manifestations within the head and neck, along with other extrathoracic manifestations, are associated with more extensive disease requiring coordinated treatment. Therefore, it is important to understand the rare manifestations of sarcoidosis as they can quickly involve vital structures.
Diagnosis is initially suspected by clinical acumen, furthered by radiologic evidence, and finally confirmed by a histopathologic assessment. While pulmonary sarcoidosis is generally managed by an internal medicine specialist, once a patient begins having head and neck involvement, the otolaryngologist is often involved both for diagnostic and interventional purposes.
This article will provide a current background on sarcoidosis, describe the breadth of head and neck manifestations, discuss the mainstays and novel forms of treatment, and provide a brief discussion on the endoscopic approach to pituitary lesions.
Methods
An initial search was performed in PubMed, with the only search criteria being ‘review’ articles, using the search terms: “Sarcoidosis”, “Head and Neck Manifestations”, “Otolaryngology”, and “ENT”. Additional iterations of these terms were used to find multiple reviews on sarcoidosis, head and neck manifestations, and oromaxillofacial findings. After a comprehensive reading of major ENT reviews of sarcoidosis was done, further targeted searches were done for each respective section, with the major search terms being “Sarcoidosis” and “Background”, “Diagnosis”, “Etiology”, etc. Specific journals, such as The Laryngoscope, which comprised previous articles on head and neck sarcoidosis, were also searched for relevant additional reports. Finally, reference lists from each article were reviewed and cross-referenced to obtain an expansive literature base, accounting for various resources.
Background
Sarcoidosis is a chronic granulomatous disease, confirmed by the presence of non-caseating granulomas within a particular organ system. A large majority of the clinical manifestations are found within the pulmonary system and adjacent thoracic lymphatics. While these readily identifiable manifestations are common, the clinical spectrum of sarcoidosis may involve any organ [1]. Otolaryngologic manifestations are identified in 10–15 % of patients, the most common being cervical adenopathy. It is important to note that it is also rare for patients to present with regional signs in isolation [1, 2]. In addition, even if patients have extrathoracic involvement, many lack any associated symptoms [3].
Pathophysiology
Sarcoidosis is caused by a granulomatous reaction, an example of a cell-mediated immune response caused by an initial reaction to an unidentified antigen. Macrophages initially identify the antigen and in cohort with cytokine activity and T cell stimulation an immune response is mounted, leading to macrophage-giant cell creation. These giant cells mass together, walling off the offending antigen from the rest of the body, a classic feature seen in granulomas [3]. Figure 1a shows a well formed granuloma with an inflammatory background of lymphoid hyperplasia. Figure 1b shows a magnified giant cell histiocyte within a sarcoid granuloma. Further studies on granuloma formation have demonstrated key factors involved in the process. Cytokine mediators such as TNF-α, IF-ϒ, IL-12 and IL-18 have been found to be critical in the multifocal cell-activation [4]. T-cells initially have an accelerated immune response to antigens, but then maintain a suppressed response to destruction of the antigen. This creates a state of hyper- and hypo-antigen sensitivity, or anergy [5]. Compositional analysis of sarcoid granulomas have found that CD4 + T-cells exist within the middle of the encased granuloma, while CD8 + cells remain close to the epitheloid edge. HLA-DRB 1*0301 and HLA-DRB 1*1101 have been linked to this distinct separation of expression, showing that sarcoid risk and granuloma development can be linked to HLA-I and -II molecules [4, 6]. A novel theory proposes that after a mycobacterial infection, microbial antigens, which have been effectively killed by T-cell response, are then left in a poorly soluble state and never properly cleared from the body. An aberrant antigen removal system over time creates a nidus for secondary granuloma infection [4, 6].

a 10 × magnification of a nasopharyngeal specimen in a patient with sarcoidosis showing a well formed granuloma with lymphoid hyperplasia in an inflammatory background (Original image provided courtesy of Dr. Codrin Iacob, Department of Pathology at New York Eye and Ear Infirmary-Mount Sinai Health System). b 40 × magnification of a granuloma isolated from a nasopharyngeal specimen. Note the presence of a well developed giant cell histiocyte. (Original image provided courtesy of Dr. Codrin Iacob, Department of Pathology at New York Eye and Ear Infirmary-Mount Sinai Health System)
Etiology
Although unclear, the causes of sarcoidosis have been categorized into the following: genetic factors, infectious agents, and environmental exposures. While some advancement has been made by ways of HLA typing of patients and investigation of ACE, TNF-α and Vitamin D receptor genes, no clear etiology has been found [3]. The theory of an initial poorly soluble antigen being an instigating factor has become the primary avenue for novel research [6]. ACCESS (A case control etiologic study of sarcoidosis) was a cross-institutional study conducted with a primary goal of identifying the etiology and mechanism of sarcoidosis. ACCESS added to the previous categorization of sarcoid mechanisms, outlining them as genetic, environmental, infectious, and immune [7]. The study encompassed the assessment of all these factors in a total of 738 patients and 710 controls, including HLA typing and extensive exposure histories. At the conclusion of the study, reaffirmations were made on the previous associations of HLA-DRB1, yet no definitive etiology was discovered [7].
Epidemiology
The epidemiology of sarcoidosis is critical to the future understanding of the disease, since locally and globally there are particularly increased incidences in both African Americans and women. The ethnic distribution of sarcoidosis is vast and a variation has been noticed worldwide [3–5]. In the United States, African Americans have a nearly three times higher incidence when compared to Caucasians (Table 1). The lifetime risk of each group in the United States is 8.5 and 2.4 % respectively [5]. Table 2 shows a global slight higher incidence of sarcoidosis in women compared to men [4]. Similarly the incidence can rise as high as 50/100,000 in Scandinavian countries, demonstrating sarcoidosis as a global disease. In addition, when considering incidence adjusted for age, sarcoidosis is more common in patients younger than 40 years of age, with a peak at 20–29 years [3].
Signs/Symptoms
Sarcoidosis presents a variable and complicated clinical picture. In attempts to properly categorize this multifaceted disease, three distinct clinical scenarios have been recognized: asymptomatic, nonspecific constitutional symptoms, and organ specific symptoms. On initial diagnosis, 1/3rd of patients are asymptomatic and another 1/3rd experience constitutional symptoms such as fever, weight loss, and fatigue [3]. Clinical suspicion is commonly increased by the initial presence of persistent dry cough, eye, or skin manifestations, leading to a diagnostic work up [4]. Constitutional symptoms can be present, including fever and weight loss [5].
In addition, organ involvement can be further classified by disease course, into acute or chronic onset. The acute organ involvement is more common in Caucasians, presenting with bilateral hilar lymphadenopathy, erythema nodosum, and ankle arthritis (Lofgren’s syndrome). Patients with this acute course have a good prognosis with over 90 % having resolution of the disease anywhere from 12 to 36 months [3, 5].
Diagnosis
The diagnosis of sarcoidosis begins with clinical suspicion based on history, then is furthered with imaging, and finally confirmed with tissue biopsy. The process commonly begins with a chest X-ray, followed by transbronchial lung biopsy to obtain a biopsy specimen. Alongside this histological diagnosis, other granulomatous diseases must be ruled out with pulmonary function tests, blood counts, chemistries, PPD, acid-fast bacilli testing, cANCA, and others [3]. Table 3 shows several other types of granulomatous diseases, certainly not an exhaustive collection however. Biopsies of peripheral lymphadenopathy or cutaneous lesions are additional means of obtaining adequate tissue. These may be within the realm of otolaryngologists, who can contribute to the diagnosis by obtaining the pertinent samples [2].
Advances in technology have expanded the role for the use of imaging such as MRI and PET scans to identify sarcoidosis. In addition, a high CD4/CD8 T-cell ratio (above 3.5) obtained from bronchoalveolar lavage represents another piece of diagnostic evidence [4]. Serum levels of ACE help to stage and estimate the disease progress. A minor mention must be given to the Kveim-Siltzbach reaction, an earlier tool used in the diagnosis of sarcoidosis. Suspected patients are injected intradermally with a mixture of sarcoid positive lymph or spleen tissue and, within 2–4 weeks, a clear local nodular granulomatous reaction should appear [4]. When initially posited, it was found that 80 % of patients demonstrated a positive reaction to the test [4].
Although imaging can be helpful in diagnosis, it is important to keep in mind that the disease has been known to mimic multiple disorders, ranging from inflammatory reactions to cancer. A recent study shows a second case of cervical neck sarcoidosis being mistaken for a parathyroid adenoma. A ‘hot’ nodule was identified on Tc-99 m sestamibi scan, a common tool to locate possible parathyroid adenomas prior to surgical resection [8].
Ultrasound has become a common imaging tool to visualize the parotid gland. However, Teymoortash, et al. [9].found that ultrasound findings had poor diagnostic specificity for sarcoidosis. While there are pathognomonic findings on certain imaging and histologic studies, a reliable modality for parotid assessment is yet to be determined.
Finally, a preliminary study done on 18f-FDG PET/CT scan showed promise as an additional clinical modality to aid in diagnosis. While guided biopsy is still the overwhelming gold standard for the diagnosis of sarcoidosis, in view of its various presentations, more diagnostic methods are desirable [10].
Head and Neck Manifestations
Extrathoracic manifestations in isolation are fairly rare and can involve nearly every organ. 10–15 % of patients with sarcoidosis demonstrate head and neck manifestations, with the possibility to involve nearly every subsite: sinonasal, oropharynx, hypopharynx, skin, eye, salivary glands, neck, larynx, bone, and even intracranial infiltration [4]. As substantial literature is not available for some of these subsites such as hypopharynx, we have provided an overview of the findings with incorporation of recent literature in the discussion of the more well reported regions.
Sinonasal
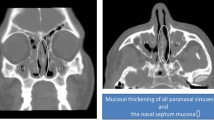
Sinonasal sarcoidosis is difficult to diagnose since the symptoms of nasal obstruction and rhinitis are nonspecific. It is estimated that nasal involvement is found in 1–4 % of patients with both rhinitis and chronic obstruction [4]. One study reported on a patient who had 6 months of chronic nasal congestion, subsequently found to be due to sarcoid, which went undiagnosed due to the nonspecific nature of findings [11].
While symptoms may be fairly stratified, sarcoidosis can manifest within the entire depth of the system from mucosal surfaces to bony structures. A recent study mentioned the first case of isolated extrapulmonary sarcoidosis within the sinonasal region with associated intracranial extensions—an ethmoid sinus mass that eroded through the cribiform plate [12]. Lastly,
Bianchini et al. [13] presented a case of nasal subcutaneous sarcoidosis, an extremely rare finding in comparison to the commonly found subcutaneous nodules found in sarcoidosis.
Treatment is debated. Gulati et al. conducted a study on specific treatment regimens for patients with sinonasal involvement, entailing endoscopic sinus surgery in combination with local intranasal or systemic corticosteroids. Sinus surgery along with medical management may yield significant short and long-term success [14].
Laryngeal
Isolated laryngeal sarcoidosis is found in only 0.5 % of patients [15]. Plaschke, et al. published a recent study outlining a successful treatment option, supraglottoplasty using CO2-laser excision of tissue, a technique that removes involved mucosa and allows suturing to promote primary healing of mucosal surfaces. This results in reduced symptoms and recurrence of swelling. This mucosal sparing surgery, even when demonstrated on only six patients, shows potential as an additional management tool [15]. Figures 2, 3 show laryngeal sarcoidosis at low and high magnification.

20 × magnification of laryngeal specimen showing granuloma in a patient with sarcoidosis (Original image provided courtesy of Dr. Codrin Iacob, Department of Pathology at New York Eye and Ear Infirmary-Mount Sinai Health System)

40 × magnification of the same laryngeal specimen seen previously in Fig. 7 (Original image provided courtesy of Dr. Codrin Iacob, Department of Pathology at New York Eye and Ear Infirmary-Mount Sinai Health System)
Cervical
Jalisi et al. report the first patient with a sarcoid lesion involving the common carotid artery circumferentially. This extrathoracic manifestation masked itself as a neoplastic process [16]. As noted previously, adenopathy is a fairly common head and neck manifestation [1, 2].
Ocular
Bodaghi et al. [18] in 2012 studied a cohort of 1,800 patients and found 21 % of patients to have ocular manifestations of sarcoidosis on initial presentation. Of these ocular manifestations, the most common was anterior uveitis (76.4 %), followed by intermediate (17.3 %) and posterior uveitis (4.7 %) [18]. These patients were also found to have optic nerve involvement and lacrimal gland swelling. Within this large patient cohort, ocular manifestations were the most common site for clinical symptoms, second only to those of the thorax (n = 662, or 36.7 %) [17]. Frequently anterior uveitis is the only ocular finding within patients, occurring in up to 85 % of patients with ocular sarcoidosis. It presents commonly as chronic, bilateral granulomatous reactions with the presence of mutton-fat keratic precipitates [18].
In order to give the readers an appreciation of imaging findings, Figs. 4, 5 show a magnetic resonance image of the brain with enhancement of the optic chiasm in a patient with neurosarcoidosis. Figure 6 shows lacrimal gland specimens with granulomata in low magnification.

Post contrast coronal T1 weighted image showing bilateral temporal leptomeningeal enhancement, enhancement of the optic chiasm, and bilateral cavernous sinus enhancement. (Original image provided courtesy of Dr. Azita Khorsandi, Department of Radiology at New York Eye and Ear Infirmary-Mount Sinai Medical Center)

Post contrast sagittal T1 weighted image demonstrating suprasellar cistern enhancement along the optic chiasm and radiation. Frontal leptomeningeal enhancement is also noted (Original image provided courtesy of Dr. Azita Khorsandi, Department of Radiology at New York Eye and Ear Infirmary-Mount Sinai Medical Center)

20 × image of a lacrimal gland specimen showing mucinous and serious acini, along with granuloma with chronic inflammation (Original image provided courtesy of Dr. Codrin Iacob, Department of Pathology at New York Eye and Ear Infirmary-Mount Sinai Health System)
Salivary Gland
There is sparse literature regarding salivary gland involvement in sarcoidosis. Reports estimate that 6 % of patients can have parotid gland manifestations ranging from xerostomia to facial nerve palsy. The most common presentation however is a painless enlargement of the gland. This can at times be associated with Heerfordt’s syndrome (Table 4), uveoparotid fever, a sarcoidosis syndrome characterized by mild fever, painless parotid gland enlargement, cranial nerve involvement, and uveitis [19]. There have been additional reports of chronic bilateral parotid swelling, which represented a hard elastic subcutaneous tumor demonstrating sarcoidosis [20].
Submandibular gland involvement also presents as a painless swelling of the gland that can at times accompany parotid enlargement. Reports of unilateral and bilateral findings have been equally reported. On review of the literature it was reported that the majority of submandibular cases were in women, demonstrating a strong female preponderance [21]. There have been rare reports of sublingual involvement, such as an excised ranula on being diagnosed as sarcoidosis on histopathologic specimen [22].
Cutaneous
Cutaneous eruptions caused by sarcoidosis are classified into either specific (demonstrating noncaseating granulomas on biopsy) or non-specific (demonstrating general reactive process without granuloma formation) [23]. Cutaneous sarcoidosis lesions are found at times to be strong prognostic indicators. Erythema nodosum is the typical non-specific lesion, which is commonly associated with Lofgren’s syndrome (Table 5). These manifestations have been found to be associated with a good prognosis and spontaneous resolution. Maculopapular lesions and subcutaneous nodules are associated with systemic disease remission, while plaques and lupus pernio are distinct hallmarks of chronic disease. Cutaneous sarcoidosis lesions have also been referred to as the great imitator due to the need for advanced clinical suspicion for diagnosis. Manifestations are generally multiple brown erythematous lesions [24].
Otologic
Literature regarding ear involvement in sarcoidosis is scant, usually in the form of rare reports of manifestations within specific compartments. Hearing impairment can be secondary to granulomatous meningitis involving the posterior fossa–causing cranial nerve and brainstem dysfunction. It is suggested that this is the most common mechanism for vestibulo-acoustic manifestations of sarcoidosis [25]. Sarcoidosis can also rarely affect the helix, external auditory canal, middle ear, and temporal bone. The most common manifestation as reported by Ozdogan et al. is infiltration into the skin and cartilage of the pinna. There also has been reports of conductive hearing loss, but this is likely secondary to otitis media caused by sinonasal involvement [26].
Neurosarcoidosis
Neurological presentations of sarcoidosis are found in 5–26 % of patients. One of the first CNS involvements to be described was Heerfordt’s syndrome, a constellation of uveitis, parotiditis, facial palsy, and fever–deemed diagnostic for sarcoidosis. However, the leptomeninges at the base of the brain are the most commonly affected region [27]. Additionally, it can involve or infiltrate the brain parenchyma (with surrounding astrocyte gliosis) and persistent inflammation can obstruct the CSF pathways to cause hydrocephalus. Figure 7 shows leptomeningeal enhancement on MRI in a patient with sarcoidosis. The most common clinical symptoms are cranial nerve palsies, more specifically the facial nerve and optic nerve [27]. Endocrine abnormalities caused by alterations in the hypothalamic-pituitary axis are also within the range of sarcoidosis manifestations. Regardless of CNS manifestations it was found that only 50 % of patients with neurosarcoidosis are diagnosed pre-mortem [27].

Post contrast coronal T1 weighted image demonstrating cerebellar and frontal nodular leptomeningeal enhancement (Original image provided courtesy of Dr. Azita Khorsandi, Department of Radiology at New York Eye and Ear Infirmary-Mount Sinai Medical Center)
Similar to other extrathoracic manifestations, neurosarcoidosis requires confirmatory tissue biopsy. In some patients a biopsy can be difficult, such as those with neurological disease and those with involvement at sites with limited accessibility. As a result, the literature cites the use of multiple criteria, involving the constellation of clinical suggestive symptoms in addition to ruling out other causes, in order to diagnose neurosarcoidosis [27]. MRI and CSF analysis, while not specific, have been found to aid in diagnosis, demonstrating leptomeningeal enhancement and CSF lymphocytosis. Due to the lack of firm recommendations, systemic corticosteroids remain as the mainstay treatment [27].
There have been recent cases in the literature of novel manifestations of neurosarcoidosis. Amin reported a case involving the trigeminal nerve, in which a patient had bilateral cavernous sinus masses and trigeminal neuropathy. Imaging suggested a meningioma; however, histopathology revealed non-caseating granulomas [28]. Another patient, presenting with generalized tonic–clonic-seizures, had progressively enlarging cystic lesions expanding into the right temporal lobe on MRI and abnormal enhancing lesions on CT scan. Urgent biopsy revealed sarcoidosis with neurological infiltration [29]. These two patients illustrate that neurosarcoidosis, similar to typical sarcoidosis, can present with complex findings that can be further clouded by imaging.
Pituitary Involvement
Hypothalamic-pituitary manifestations of sarcoidosis are very rare, comprising only 1 % of lesions found within the sella turcica [30]. Although rare, hypothalamic-pituitary involvement can be one of the first manifestations, with associated endocrine disorders as presenting symptoms. Diabetes insipidus and hyperprolactinemia are the most common presenting disorders [30]. A study by Langrand et al., reports a comprehensive retrospective review of 24 patients with hypothalamic-pituitary sarcoidosis. 22 of these patients had anterior pituitary dysfunction, 21 of them had clinical symptoms of a deficiency, and 12 presented with diabetes insipidus [30]. Additionally, 14 patients demonstrated enhancing regions indicative of pituitary gland involvement on MRI. Patients with sarcoidosis were compared demonstrating that clinically sarcoidosis patients have a higher frequency of neurological findings, 58 versus 12 % than patients without involvement of the hypothalamic-pituitary axis [30]. Furthermore, patients with hypothalamic-pituitary involvement demonstrated increased sinonasal manifestations, 25 versus 2 % in non-hypothalamic-pituitary sarcoidosis patients. Finally when these patients were treated with a standardized corticosteroid regimen, it was found that the incidence of remission was inferior, 8 versus 39 %. Ultimately, treatment with corticosteroids has been the only established treatment for sarcoidosis. In this retrospective study, MRI abnormalities improved or disappeared in 7/24 cases after treatment with prednisone or other dose equivalent corticosteroid. Unfortunately, nearly all of the endocrine defects were irreversible regardless of treatment [30].
Treatment
There has been considerable debate regarding the treatment of sarcoidosis, but the current accepted therapy has been corticosteroids. A pilot study comparing observation, pulse corticosteroids, stable dose corticosteroids, or TNF-α antagonist adalimumab therapy, was conducted in 31 patients, 21 having isolated head and neck manifestations [31]. It was suggested that pulse therapy was the most effective treatment option, preventing the associated complications of continuous systemic steroids. This study provided a major recommendation for management of head and neck sarcoidosis, an important step without precedent in literature [31].
TNF-α antagonist therapy is a novel treatment for systemic inflammatory diseases such as sarcoidosis. While there have been multiple studies outlining the positive effects of the therapy, a recent study outlines a potential complication. A patient treated with adalimumab for psoriatic arthritis went on to develop sarcoidosis of the hypopharynx [32]. This has been the first reported case of sarcoidosis of the hypopharynx, and demonstrated a complex clinical scenario in that discontinuation of the drug resulted in resolution of the sarcoidosis. This is an unforeseen complication of TNF-α therapy resulting in an extremely rare manifestation of sarcoidosis [32].
Recently, a new study recommended the use of minimally invasive surgery with intralesional corticosteroid injection along with laser reduction to be used as an effective alternative therapy. [33]. Combining two previously supported therapies, intralesional steroids and surgical debulking, showed some promise. This study also represents the largest single-institution series on laryngeal sarcoidosis cases treated with this combination of therapies [33].
Surgical intervention may be indicated in patients that do not respond to corticosteroids, or when sarcoidosis threatens vital structures. Like any surgical procedures, resection of sarcoidosis lesions is weighted based on risk and benefits to the patients. This risk benefit analysis is duly noted when examining the treatment options for patients with pituitary sarcoidosis. These lesions serve as a prime example of how such an assessment can drastically alter the treatment regimen, surgical indications, and surgical approach to resection of a lesion.
There are commonly two types of approaches to pituitary lesions, the trans-cranial approach and a less invasive ‘extracranial’ technique. The first transnasal resection was done in 1907, and became more common when Cushing used it for general sellar lesions. Guiot and Hardy, who added the adjunctive use of fluoroscopy and the surgical microscope, refined the technique. With the advent of the endoscope, the endoscopic transphenoidal approach to pituitary lesions was described [34]. Before the benefits of endoscopic surgery became widely known, transnasal approaches were favored solely for better direct visualization of the pituitary. It was a Viennese otolaryngologist, Oskar Hirsch, who described the classical endonasal approach that was then used for over 50 years, avoiding the high morbidity and mortality that was associated with trans-cranial surgery [35].
In current day, the endoscopic transnasal approach has become an accepted technique to excise 96 % of pituitary adenomas. The rod-lens endoscope provides for superior direct and multi-angle visualization of the area–compared to the operative microscope. An endoscopic endonasal approach provides the most direct access to pertinent structures without need for retraction of the brain or manipulation of optic nerve tracts. Endoscopic endonasal approaches adequately access not only the sella turcica, but also the entire anterior skull base. In addition, the two team approach in which both neurosurgery and otolaryngology teams coordinate the management, presents direct advantages to the patient as the specialties provide complementary care [36].
However, the endoscopic approach is not a panacea and is associated with its own array of complications such as diabetes insipidus, cerebrospinal fluid leak, and meningitis [35] [36]. There are a few limitations to the surgical technique that should be mentioned. The first is the difficulty in controlling major bleeding that can occur due to the space limitations of using an endoscope. Reconstructions can become complicated when manipulating endoscopically, however adequate bony exposure can still yield good results [36]. Despite these, the benefits of the technique, coupled with favorable surgical outcomes and patient satisfaction, have allowed for the wide acceptance of the procedure. The surgical technique and treatment of associated complications is not within the scope of this manuscript; as such, the reader is encouraged to turn to the literature for a full discussion of endoscopic skull base surgery.
Conclusion
Sarcoidosis is a multifaceted disorder with the potential to invade any organ. Because of its unclear etiology and non-specific presentation, sarcoidosis poses a significant diagnostic hurdle for the otolaryngologist. Extrathoracic manifestations are rare and can affect multiple subsystems ranging from sinonasal deeply invasive cranial lesions. Although the mainstay of treatment for all of these manifestations is systemic corticosteroids, physicians should be up to date on current evidence demonstrating combination of corticosteroid with minimally invasive surgery, and other alternative treatment options.
References
Dash GI, Kimmelman CP. Head and neck manifestations of sarcoidosis. The Laryngoscope. 1988;98(1):50–3.
Shah UK, White JA, Gooey JE, Hybels RL. Otolaryngologic manifestations of sarcoidosis: presentation and diagnosis. The Laryngoscope. 1997;107(1):67–75.
Schwartzbauer HR, Tami TA. Ear, nose, and throat manifestations of sarcoidosis. Otolaryngol clin N Am. 2003;36(4):673–84.
Mrowka-Kata K, Kata D, Lange D, Namyslowski G, Czecior E, Banert K. Sarcoidosis and its otolaryngological implications. Eur Arch Otorhinolaryngol. 2010;267(10):1507–14.
Dastoori M, Fedele S, Leao JC, Porter SR. Sarcoidosis: a clinically orientated review. J Oral Pathol Med. 2013;42(4):281–9.
Chen ES, Moller DR. Etiology of sarcoidosis. Clin Chest Med. 2008;29(3):365–77.
Semenzato G. ACCESS: a case control etiologic study of sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2005;22(2):83–6.
Calo PG, Pisano G, Tatti A, Loi G, Furcas S, Nicolosi A. Cervical lymph node sarcoidosis mimicking a parathyroid adenoma: a clinical case. Clin med insights Case Rep. 2013;6:159–63.
Teymoortash A, Werner JA. Parotid gland involvement in sarcoidosis: sonographic features. J clin ultrasound JCU. 2009;37(9):507–10.
Braun JJ, Kessler R, Constantinesco A, Imperiale A. 18F-FDG PET/CT in sarcoidosis management: review and report of 20 cases. Eur j nuclear med mol imaging. 2008;35(8):1537–43.
Mallis A, Mastronikolis NS, Koumoundourou D, Stathas T, Papadas TA. Sinonasal sarcoidosis. A case report. Eur rev med pharmacol sci. 2010;14(12):1097–9.
Dessouky OY. Isolated sinonasal sarcoidosis with intracranial extension: case report. Acta Otorhinolaryngol Ital. 2008;28(6):306–8.
Bianchini C, Tosin E, Ciorba A, Pelucchi S, Pastore A. Subcutaneous sarcoidosis: a rare involvement of the paralateral nasal region. Acta Otorhinolaryngol Ital. 2011;31(2):118–20.
Gulati S, Krossnes B, Olofsson J, Danielsen A. Sinonasal involvement in sarcoidosis: a report of seven cases and review of literature. Eur Arch Otorhinolaryngol. 2012;269(3):891–6.
Plaschke CC, Owen HH, Rasmussen N. Clinically isolated laryngeal sarcoidosis. Eur Arch Otorhinolaryngol. 2011;268(4):575–80.
Jalisi S, Winter LK, Elackattu A, Sakai O. Sarcoidosis masquerading as carotid body tumor. Arch Otolaryngol Head Neck Surg. 2010;136(11):1132–5.
Heiligenhaus A, Wefelmeyer D, Wefelmeyer E, Rosel M, Schrenk M. The eye as a common site for the early clinical manifestation of sarcoidosis. Ophthalmic Res. 2011;46(1):9–12.
Bodaghi B, Touitou V, Fardeau C, Chapelon C, LeHoang P. Ocular sarcoidosis. Presse medicale (Paris, France: 1983). 2012;41(6 Pt 2):e349–54.
Vairaktaris E, Vassiliou S, Yapijakis C, Papakosta V, Kavantzas N, Martis C, et al. Salivary gland manifestations of sarcoidosis: report of three cases. J Oral Maxillofac Surg. 2005;63(7):1016–21.
Ohtsuka S, Yanadori A, Tabata H, Yamakage A, Yamazaki S. Sarcoidosis with giant parotomegaly. Cutis. 2001;68(3):199–200.
Vourexakis Z, Dulguerov P, Bouayed S, Burkhardt K, Landis BN. Sarcoidosis of the submandibular gland: a systematic review. Am J Otolaryngol. 2010;31(6):424–8.
Narang R, Dixon RA Jr. Sarcoidosis and ranula of a sublingual gland. Oral surg oral med oral pathol. 1975;39(3):376–81.
Haimovic A, Sanchez M, Judson MA, Prystowsky S. Sarcoidosis: a comprehensive review and update for the dermatologist: part I. Cutaneous disease. J Am Acad Dermatol. 2012;66(5):699.e1–18.
Mana J, Marcoval J. Skin manifestations of sarcoidosis. Presse medicale (Paris, France: 1983). 2012;41(6 Pt 2):e355–74.
Hybels RL, Rice DH. Neuro-otologic manifestations of sarcoidosis. The Laryngoscope. 1976;86(12):1873–8.
Ozdogan A, Acioglu E, Karaman E, Oz B, Musellim B. A difficult case: sarcoidosis of the middle ear. Am J Otolaryngol. 2009;30(4):281–4.
Vargas DL, Stern BJ. Neurosarcoidosis: diagnosis and management. Semin respir crit care med. 2010;31(4):419–27.
Amin A, Balderacchi JL. Trigeminal neurosarcoidosis: case report and literature review. Ear Nose Throat J. 2010;89(7):320–2.
Al hajri FA, Muqim AT, Muttikkal TJ. Neurosarcoidosis with intraparenchymal cystic lesions. A case report. Neuroradiol J. 2009;21(6):810–6.
Langrand C, Bihan H, Raverot G, et al. Hypothalamo-pituitary sarcoidosis: a multicenter study of 24 patients. QJM. 2012;105(10):981–95.
Knopf A, Lahmer T, Chaker A, et al. Head and neck sarcoidosis, from wait and see to tumor necrosis factor alpha therapy: a pilot study. Head Neck. 2013;35(5):715–9.
Christoforidou A, Goudakos J, Bobos M, Lefkaditis E, Vital V, Markou K. Sarcoidosis-like granulomatosis of the hypopharynx as a complication of anti-TNF therapy. Am J Otolaryngol. 2013;34(3):268–72.
Butler CR, Nouraei SA, Mace AD, Khalil S, Sandhu SK, Sandhu GS. Endoscopic airway management of laryngeal sarcoidosis. Arch Otolaryngol Head Neck Surg. 2010;136(3):251–5.
Santos AR, Fonseca neto RM, Veiga JC, et al. Endoscopic endonasal transsphenoidal approach for pituitary adenomas: technical aspects and report of casuistic. Arq Neuropsiquiatr. 2010;68(4):608–12.
Gendeh BS. Doi M, Selladurai BM, Khalid BA, Jegan T, Misiran K The role of endoscopic endonasal approach to pituitary tumours: HUKM experience. Med J Malaysia. 2006;61(3):343–8.
Oostra A, van Furth W, Georgalas C. Extended endoscopic endonasal skull base surgery: from the sella to the anterior and posterior cranial fossa. ANZ J Surg. 2012;82(3):122–30.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Badhey, A.K., Kadakia, S., Carrau, R.L. et al. Sarcoidosis of the Head and Neck. Head and Neck Pathol 9, 260–268 (2015). https://doi.org/10.1007/s12105-014-0568-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12105-014-0568-y