
Abstract
This study investigated the role of human papillomavirus (HPV) in Schneiderian papillomas (SPs) to determine whether HPV is associated with the pathogenesis of particular histologic subtypes and whether p16INK4a can be used as a surrogate marker for HPV detection. Twenty-seven papilloma specimens (19 inverted [IPs], 6 exophytic [EPs], 1 oncocytic [OP] and 1 mixed) were collected from 23 patients. Purified SP DNA extracts were tested for HPV by PCR using GP5 +/GP6 + primers; HPV genotyping was performed by dot blot hybridization. PCR positive specimens were screened for HPV by biotinyl-tyramide-based chromogenic in situ hybridization (CISH). Immunohistochemsistry (IHC) for the HPV L1 capsid protein and for p16INK4a was performed on all specimens. HPV was detected by PCR in 16/27 (59.3%) SPs; 9/19 (47.4%) IPs; 6/6 (100%) EPs [p = 0.051], and 1/1 (100%) mixed SP. HPV was not detected in the single OP. High risk genotypes were detected in 4/9 IPs (44.4%) and 0/6 EPs (0%) [p = 0.10]. Seven of 16 PCR positive SPs were also CISH positive for HPV: 5/6 EPs (83.3%) and 1/9 IP (11.1%) [p = 0.01]. IHC for the L1 capsid protein was positive in 2 SPs (1 EP and 1 mixed). p16INK4a staining was seen in 14/16 (87.5%) PCR positive SPs and in 10/11 (90.9%) PCR negative SPs (p = 1.00). In summary, this study demonstrates a strong association between HPV and EPs, however, its role in IPs remains less well-defined. Further, p16INK4a is not a useful surrogate marker for HPV detection across the various SPs.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Sinonasal or Schneiderian papillomas (SPs) are benign neoplasms that originate from the Schneiderian mucosa; an ectodermally derived ciliated respiratory mucosa of the nasal cavity and paranasal sinuses [1]. Although known by a variety of names SPs, have been classically divided into three histologic subtypes: (1) fungiform or exophytic (EP) (2) inverted (IP) and (3) cylindrical cell or oncocytic (OP) papillomas [2, 3]. Etiological investigations of SPs began in the 1980s, with a viral etiology, specifically human papillomavirus (HPV), being of great interest [4]. To date HPV has been detected in EPs and IPs with HPV6 and 11 being the most consistently detected genotypes [1, 4–6]. The rates of HPV detection are highly variable with EPs demonstrating a higher HPV detection rate compared to IPs [6–10]. HPV has not been detected in OPs [6–9].
Clinically, SPs can be difficult to manage as they have a tendency to recur if not completely excised [3, 11]. Beck et al. demonstrated that both the extent of surgical excision and presence of HPV were important in predicting recurrence [12]. Importantly, SPs have also been associated with malignancy [3, 11, 13–15], with IPs and OPs being best documented [13–15]. However, there have been only case reports of EPs and carcinomas [16, 17]. It is this association with malignancy and a high rate of recurrence that drives the interest in understanding the etiology of SPs. If a causative role for HPV in SPs, particularly the IP subtype, is established this would have significant implications in surveillance and treatment; in many ways perhaps paralleling the advances seen in cervical cancer screening and prevention.
The p16INK4a protein, a cyclin dependent kinase inhibitor, is upregulated in cells infected with high-risk HPV genotypes [18]. Hence, p16INK4a has been shown to be a surrogate marker for HPV detection in head and neck cancers particularly tonsillar carcinomas [19]. It is also used as a biomarker in aiding the diagnosis of cervical neoplasia [20]. There have been many studies geared towards detecting HPV in SPs by using both PCR and in situ hybridization, however, the literature on p16INK4a and SPs is relatively sparse [21, 22]. The objectives of this study were three-fold: What proportion of SPs in a Vermont population are HPV positive? Does HPV correspond with particular SP histologic subtypes? Can p16 INK4a be used as a surrogate marker for HPV positive SPs?
Materials and Methods
Case Selection
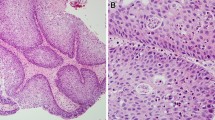
With Institutional Review Board approval, 27 formalin-fixed, paraffin-embedded (FFPE) SP specimens (1991–2008) from 23 patients were collected from Fletcher Allen Health Care archives. The 23 patients consisted of 7 females and 16 males with ages ranging from 16 to 94 years (mean = 58.9; median = 60; SD = 16.6). SPs were categorized into their standard histologic subtypes resulting in 19 IPs, 6 EPs and 1 OP (Fig. 1). In addition 1 SP was described as mixed as it displayed features of both an IP and an EP. Mild dysplasia was noted in two EPs. Multifocal moderate dysplasia and focal severe dysplasia were seen in 2 IPs.

Schneiderian papilloma histologic subtypes: a exophytic papilloma, b oncocytic papilloma and c inverted papilloma
DNA Extraction
Up to ten 5 μm sections were cut from FFPE tissue blocks into micro-centrifuge tubes. Tissues were dewaxed with xylene followed by ethanol rinses. DNA was extracted using a combined proteinase K digestion and mini-column purification procedure (DNeasy tissue kit, Qiagen, Valencia, CA). DNA concentration was estimated using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Wilmington, DE).
Detection of HPV by PCR
Purified SP DNA extracts were tested for HPV using GP5+/GP6+ PCR primers. Two PCR strategies were applied, a ‘touchdown’ and ‘slow ramping’, as previously described [23]. This widely used assay detects at least thirty-seven different alpha-genus HPV types, which are the types most commonly associated with anogenital lesions.
Identification of Genotype by Dot Blot Hybridization
HPV positive specimens were identified by the detection of a 150 base-pair amplicon after electrophoresis in a 2.0% ethidium bromide stained agarose gel. HPV genotype was identified by dot blot hybridization of PCR products with genotype-specific oligonucleotide probes [24].
Detection of HPV by Chromogenic In Situ Hybridization
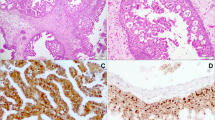
PCR positive specimens were screened for HPV by biotinyl-tyramide-based chromogenic in situ hybridization (CISH) as previously described [24, 25]. Briefly, 5 μm section FFPE specimens were dewaxed through xylene and ethanol washes and air dried. The slides were then incubated in 10 mM sodium citrate pH 6.0 at 98°C for 40 min followed by a 20 min cool down period. Tissues were then digested with 100 μg/pepsin (Sigma P7012, Sigma–Aldrich, St. Louis, MO) in 0.2 M HCL for 5–12 min (incubation time optimized for each tumor specimen) and were then treated with 0.3% hydrogen peroxide in methanol for 20 min. Next, tissue sections were overlaid with biotinylated Wide Spectrum HPV DNA Probe Cocktail [that detects HPV types 6, 11, 16, 18, 31, 33, 35, 45, 51, and 52 (Dako North America Inc., Carpinteria, CA)] sealed under a cover slip with rubber cement. Slides were placed in a Vysis HYBrite hybridization system (Abbott Molecular, Des Plaines, IL) and heated to 95°C for 5 min to denature specimen and probe DNA and then allowed to hybridize overnight at 37°C. The next day, coverslips were removed under 2 × SSC and then washed 3 × 10 min with 0.1 × SSC at 50°C followed by rinses with 2 × SSC at room temperature. Tissues were then incubated serially with primary streptavidin horseradish peroxidase (HRP) conjugate, biotinyl-tyramide and secondary streptavidin HRP conjugate using a GenPoint™, Catalyzed Signal Amplification System according to supplier instructions (Dako North America Inc., Carpinteria, CA). CISH signals (Fig. 2) were recorded as diffuse (indicative of episomal HPV) or as punctate (indicative of HPV integrated into the cell genome). Using this method, it is possible to detect integrated HPV16 in FFPE preparations of SiHa cells, which are known to contain approximately one copy of the HPV16 genome in chromosome 13q [25]. The detection threshold for episomal HPV is unknown because of difficulties in establishing a model tissue [25, 26].

Chromogenic in situ hybridization (CISH) for HPV. Inset: diffuse or episomal signal (rectangle) and punctate or integrated signal (circles)
Immunohistochemistry for HPV L1 Capsid Protein
Immunohistochemsistry (IHC) was performed for the HPV major (L1) capsid protein (clone K1H8, Dako North America, Inc., Carpinteria, CA). Following tissue section dewaxing and high temperature antigen retrieval with sodium citrate (pH 6.0), the antibody (1:50 dilution) was applied to tissue sections for 30 min and after buffer washes, was detected using Envison™ reagents (Dako North America, Inc., Carpinteria, CA) and 3,3′-diaminobenzidine tetrahydrochloride (DAB) staining.
Immunohistochemistry for p16INK4a
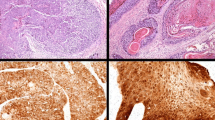
IHC for p16INK4a was performed on all specimens using the CINtec® p16INK4a Histology kit (mtm laboratories, Inc (USA), Westborough, MA) according to supplier instructions and with appropriate positive and negative controls. A positive reaction was defined by strong cytoplasmic and nuclear staining. Four patterns of positive staining were observed and described as follows: pattern a—full-thickness, roman column-like, pattern b—up to the lower ½ of the thickness of the epithelium, pattern c—greater than the lower ½ to full-thickness of the epithelium and pattern d—isolated or sporadic cells distributed throughout the epithelium (Fig. 3).

p16INK4a immunohistochemistry positive staining patterns. a—pattern a: full-thickness, roman column-like; b—pattern b: up to the lower ½ of the thickness of the epithelium; c—pattern c: greater than the lower ½ to full-thickness of the epithelium; d—pattern d: isolated or sporadic cells distributed throughout the epithelium
Statistical Analysis
p values were obtained using Fisher’s exact T test.
Results
HPV Genotypes Detected by PCR
HPV was detected in 16 of 27 (59.3%) SPs by PCR. HPV positive SPs consisted of 9/19 (47.4%) IPs, 6/6 (100%) EPs [p = 0.18], and 1/1 (100%) mixed SP (Fig. 4). HPV was not detected in the single OP in the study. In the PCR positive SPs, both low risk (HPV6 and HPV11) and high-risk (HPV16 and HPV18) genotypes were detected by dot blot hybridization. There were 3 SPs (18.8%) positive for HPV16, 1 with both HPV16 and HPV18 (6.3%), 5 with HPV6 (31.3%) and 7 with HPV11 (43.8%) (Fig. 5). HPV16/18 was detected in 4 of 9 IPs (44.4%) and 0 of 6 EPs (0%) [p = 0.10].

Correlation of Schneiderian papilloma histologic subtypes with HPV detection by PCR

Correlation of HPV genotypes with schneiderian papilloma histologic subtypes
HPV Detected by Chromogenic In Situ Hybridization
Of the PCR positive SPs, 7/16 (43.8%) were also positive for HPV by CISH. CISH positive SPs were composed of 5/6 EPs (83.3%) and 1/9 IPs (11.1%) [p = 0.01] as well as 1 mixed SP. Punctuate and diffuse signals were seen in 6 of the 7 SPs (1 mixed and 5 EPs) (Fig. 6). Focal diffuse CISH positivity was seen in 1 IP. All CISH positive SPs were either HPV6 or HPV11.

Distribution of HPV genotypes and Schneiderian papilloma histologic subtypes in CISH positive (+) and CISH negative (−) Schneiderian papillomas
L1 Capsid and p16INK4a Protein Detection by Immunohistochemistry
IHC for the L1 capsid protein (Fig. 7) was positive in two specimens (1 mixed and 1 EP). Both of these specimens were CISH positive and infected with low risk HPV subtypes (1 HPV6 and 1 HPV11). IHC for p16INK4a (Table 1) was positive in 14/16 (87.5%) PCR positive SPs and in 10/11 (90.9%) PCR negative SPs [p = 1.000]. The distribution of the four patterns observed with respect to PCR positivity, CISH positivity, HPV genotypes and histologic subtype is shown in Table 1. A combination of staining patterns was the most common staining pattern seen in 12 of 28 SPs (42.9%). A combination staining pattern is defined as SPs not having a single A, B, C or D pattern but rather demonstrating more than one of the previously defined staining patterns for example an SP may show patterns A, B and C. No SP demonstrated only pattern B or pattern C.

Immunohistochemistry for the HPV L1 capsid protein. Positive staining defined by strong nuclear positivity
Schneiderian Papillomas with Dysplasia
Of the 2 EPs displaying mild dysplasia, both were positive for HPV by PCR and ISH. HPV was not detected in the IP with multifocal moderate dysplasia, however, HPV was detected by PCR only in the IP with focal severe dysplasia. Low risk HPV was detected in the 3 of 4 SPs (75%) displaying dysplasia (2 HPV6 and 1 HPV11).
Discussion
In this study, the prevalence of HPV in SPs was 59.3%. HPV was detected in higher frequency in EPs (100%) as compared to IPs (47.4%), which is consistent with current literature [6–10]. The presence of HPV DNA in 100% of EPs by PCR suggests that HPV may be the major causative factor in these SPs. The lower detection rate of HPV in IPs suggests that the pathogenesis may be multifactorial; alternatively there may be an HPV pathway for a subset of IPs versus a non-HPV pathway as has been found in vulvar squamous cell carcinoma [27].
In an attempt to explain the disparate detection rates between IPs and EPs, Lawson et al. suggested a “hit and run” mechanism [10]. The latter was first described by Skinner (1976) which proposes that a virus may initiate cell transformation, and then disappear without leaving viral “footprints” [28]. This hypothesis was extended to IPs suggesting that the non-keratinizing nature of IPs lends itself to reduced viral replication and with epithelial shedding the virus is lost. In contrast, the hyperkeratotic epithelium of EPs is permissive for replication accounting for the higher detection rates of HPV in EPs [10]. A contradiction to Lawson’s hypothesis is that usually only a small number of EPs exhibit surface keratination [3, 29]. Nevertheless, the “hit and run” mechanism remains the most plausible explanation for the lower detection rate of HPV in EPs.
HPV was not detected in the single OP, similar to other studies [6–9]. The majority of SPs (75%) were infected with low-risk HPV6/11 genotypes. Interestingly, 3 of 4 SPs (2 EPs and 2 IPs) with dysplasia were positive for low-risk HPV genotypes. However, the four SPs with high-risk HPV 16/18 genotypes were IPs without dysplasia. There was no statistical difference between high-risk HPV genotype detection in IPs (44.4%) as compared to EPs (0%) [p = 0.10]. Given the association of IPs with malignancy, it seems logical to assume that high-risk HPV genotypes would be found in greater frequency in IPs as compared to EPs.
The integration of viral specific oncogenes E6 and E7 is characteristic of high-risk HPV infections in high-grade pre-invasive and invasive cervical neoplasia and may be critical for the conversion to a malignant phenotype [18]. E7 degrades RB and releases p16INK4a from negative feedback control, resulting in p16INK4a upregulation. This relationship between p16INK4a overexpression and HPV infection has made p16INK4a a useful surrogate marker for high-risk HPV infection in tonsillar carcinoma and cervical neoplasia [19, 20]. Although most SPs are benign with low-risk HPV genotypes most commonly detected [5], the relationship with p16INK4a has not been fully explored. Two previous studies investigated p16INK4a and SPs [21, 22]: the first examined p16INK4a and p53 in the dysplasia-carcinoma pathway in SPs and the second investigated whether p16 could be used to differentiate EPs from squamous papillomas [21, 22]. Our interpretation of positive p16INK4a staining was based on strong nuclear and cytoplasmic staining and demonstrated that there was no statistical difference between p16INK4a staining in PCR positive (87.5%) versus PCR negative (90.9%) SPs [p = 1.000]. These findings suggest that p16INK4a cannot be used as a surrogate marker for HPV in SPs.
HPV is increasingly recognized as important in the etiology of many head and neck tumors and has patient management implications [30]. Thus, there is a need for a standardized HPV assay or assays for the definitive detection of clinically significant HPV. A distinction has been made between ‘driver’ HPV that is causal for a tumor, and ‘passenger’ HPV that is an incidental bystander infection unrelated to tumorigenesis [31]. PCR assays allow the sensitive identification of HPV in a tumor; however, the very sensitivity of PCR may confound the discrimination of driver from passenger HPV. The visualization of HPV directly in cells and tissues by CISH assays is consistent with the detection of driver HPV; however, the possibility of CISH sub-threshold driver HPV cannot be entirely excluded as CISH data may depend on tissue fixation, assay protease treatments, HPV probe character, CISH detection strategy, and assay application in a given laboratory. p16INK4a IHC is the simplest test to apply and has been reported not only as an efficient HPV surrogate marker for tonsillar carcinomas but as a test with good clinical outcome correlates [30]; HPV testing by CISH was also reported as a suitable approach [30]. Our study does not support the application of p16INK4a IHC to papillomas; a combination of HPV screening and typing by PCR and CISH may be the best approach for HPV testing of SPs at the present time.
In summary, this study demonstrates a strong association between HPV and EPs, whilst its role in IPs is less well-defined. Alternatively, HPV could be a single factor contributing to the multifactorial pathogenesis of IPs. Finally, p16INK4a is not a useful surrogate marker for HPV detection in SPs.
References
Perez-Ordonez B. Special tumours of the head and neck. Curr Diagn Pathol. 2003;9:366–83.
Barnes L, Eveson JW, Reichart P, Sidransky D, editors. Pathology and genetics of head and neck tumours. World health organization classification of tumours. Lyon, France: IARC Press; 2005.
Hyams VJ. Papillomas of the nasal cavity and paranasal sinuses. A clinicopathological study of 315 cases. Ann Otol Rhinol Laryngol. 1971;80:192–206.
Syrjanen S, Happonen RP, Virolainen E, et al. Detection of human papillomavirus (HPV) structural antigens and DNA types in inverted papillomas and squamous cell carcinomas of the nasal cavities and paranasal sinuses. Acta Otolaryngol. 1987;104:334–41.
Syrjanen KJ. HPV infections in benign and malignant sinonasal lesions. J Clin Pathol. 2003;56:174–81.
Gaffey MJ, Frierson HF, Weiss LM, et al. Human papillomavirus and Epstein-Barr virus in sinonasal Schneiderian papillomas. An in situ hybridization and polymerase chain reaction study. Am J Clin Pathol. 1996;106:475–82.
Judd R, Zaki SR, Coffield LM, et al. Sinonasal papillomas and HPV: HPV 11 detected in fungiform Schneiderian papillomas by in situ hybridization and PCR. Hum Pathol. 1991;22:550–6.
Weiner JS, Sherris D, Kasperbauer J, et al. Relationship of HPV to Schneiderian papillomas. Laryngoscope. 1999;109:21–6.
Buchwald C, Franzmann MB, Jacobsen GK, et al. Human papillomavirus (HPV) in sinonasal papillomas: a study of 78 cases using in situ hybridization and polymerase chain reaction. Laryngoscope. 1995;105:66–71.
Lawson W, Schlecht NF, Brandwein-Gensler M. The role of the human papillomavirus in the pathogenesis of Schneiderian inverted papillomas: an analytic overview of the evidence. Head Neck Pathol. 2008;2:49–59.
Lawson W, Ho BT, Shaari CM, et al. Inverted papilloma: a report of 112 cases. Laryngoscope. 1995;105:282–8.
Beck JC, McClatchey KD, Lesperance MM, et al. Presence of human papillomavirus predicts recurrence of inverted papilloma. Otolaryngol Head Neck Surg. 1995;113:49–55.
Ward BE, Fechner RE, Mills SE. Carcinoma arising in oncocytic Schneiderian papilloma. Am J Surg Pathol. 1990;14:364–9.
Kaufman MR, Brandwein MS, Lawson W. Sinonasal papillomas: clinicopathologic review of 40 patients with inverted and oncocytic schneiderian papillomas. Laryngoscope. 2002;112:1372–7.
Christensen WN, Smith RR. Schneiderian papillomas: a clinicopathologic study of 67 cases. Hum Pathol. 1986;17:393–400.
Buchwald C, Franzmann MB, Jacobsen GK, et al. Carcinomas occurring in papillomas of the nasal septum associated with human papilloma virus (HPV). Rhinology. 1997;35:74–8.
Norris HJ. Papillary lesions of the nasal cavity and paranasal sinuses. Part I. Exophytic (squamous) papillomas. A study of 28 cases. Laryngoscope. 1962;72:1784–97.
Zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2:342–50.
Klussmann JP, Gultekin E, Weissenborn SJ, et al. Expression of p16 protein identifies a distinct entity of tonsillar carcinomas associated with human papillomavirus. Am J Pathol. 2003;162:747–53.
Kalof AN, Cooper K. p16INK4a immunoexpression: surrogate marker of high-risk HPV and high-grade cervical intraepithelial neoplasia. Adv Anat Pathol. 2006;13:190–4.
Allende DS, Hoschar A, Batra P, et al. Dysplasia and carcinomas arising in Schneiderian papillomas, 20 years experience. Mod Pathol. 2008;21:231A.
Allende DS, Hoschar A, Hunt JL. Exophytic Schneiderian papillomas and their differential diagnosis: Can p16 help in the diagnosis of these two entities? Mod Pathol. 2008;21:231A.
Evans MF, Adamson CS, Simmons-Arnold L, et al. Touchdown general primer (GP5 +/GP6 +) PCR and optimized sample DNA concentration support the sensitive detection of human papillomavirus. BMC Clin Pathol. 2005;5:10–1.
Evans MF, Mount SL, Beatty BG, et al. Biotinyl-tyramide-based in situ hybridization signal patterns distinguish human papillomavirus type and grade of cervical intraepithelial neoplasia. Mod Pathol. 2002;15:1339–47.
Evans MF, Aliesky HA, Cooper K. Optimization of biotinyl-tyramide-based in situ hybridization for sensitive background-free applications on formalin-fixed, paraffin-embedded tissue specimens. BMC Clin Pathol. 2003;3(1):2.
Evans MF, Cooper K. Human papillomavirus integration: detection by in situ hybridization and potential clinical application. J Pathol. 2004;202:1–4.
Yang B, Hart WR. Vulvar intraepithelial neoplasia of the simplex (differentiated) type: a clinicopathologic study including analysis of HPV and p53 expression. Am J Surg Pathol. 2000;24:429–41.
Skinner GR. Transformation of primary hamster embryo fibroblasts by type 2 simplex virus: evidence for a “hit and run” mechanism. Br J Exp Pathol. 1976;57:361–76.
Barnes L. Schneiderian papillomas and nonsalivary glandular neoplasms of the head and neck. Mod Pathol. 2002;15:279–97.
Kuo KT, Hsiao CH, Lin CH, Kuo LT, Huang SH, Lin MC. The biomarkers of human papillomavirus infection in tonsillar squamous cell carcinoma-molecular basis and predicting favorable outcome. Mod Pathol. 2008;21:376–86.
Matsukura T, Sugase M. Pitfalls in the epidemiologic classification of human papillomavirus types associated with cervical cancer using polymerase chain reaction: driver and passenger. Int J Gynecol Cancer. 2008;18:1042–50.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shah, A.A., Evans, M.F., Adamson, C.SC. et al. HPV DNA is Associated with a Subset of Schneiderian Papillomas but Does not Correlate with p16INK4a Immunoreactivity. Head and Neck Pathol 4, 106–112 (2010). https://doi.org/10.1007/s12105-010-0176-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12105-010-0176-4