
Abstract
Idiopathic nephrotic syndrome is the most common glomerulopathy in childhood characterised by heavy proteinuria, hypoalbuminemia and edema. Most of the patients have mild and transient edema but those with difficult to treat nephrotic syndrome can develop severe edema which may have serious consequences such as immobility, cellulitis and peritonitis. Understanding of the pathophysiology of edema is still evolving with recent research elucidating newer mechanism of sodium retention through plasmin mediated epithelial sodium channel activation in collecting duct. Patients with mild edema do not require specific diuretic therapy as it improves with steroid induced diuresis. In this review, the authors describe the current perspective in management of moderate to severe edema in childhood nephrotic syndrome including various parameters to assess intravascular volume status which is important for planning overall treatment strategy. Then they briefly discuss about various classes of diuretics, aquaretics and evidence based use of furosemide albumin combination therapy for treatment of edema. Management strategy for a small proportion of patients, who are unresponsive to furosemide therapy, includes diuretic synergism, intravenous furosemide albumin combination therapy and continuous intravenous furosemide infusion.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Edema is the cardinal manifestation of nephrotic syndrome, with severity varying from mild facial puffiness to severe edema; some patients may accumulate additional fluid approximating 30% of body weight. Patients with steroid sensitive nephrotic syndrome usually have mild edema that resolves following therapy with corticosteroids. Difficult to manage severe edema mostly occurs in patients with steroid resistant nephrotic syndrome and is associated with discomfort, restricted physical activity and an increased risk of infections. While patients with severe edema might require therapy with a combination of oral and intravenous (IV) diuretics, those with associated hypovolemia are best managed using IV albumin infusion. This article reviews current understanding about pathophysiology of edema formation in nephrotic syndrome, and a rational approach to its management.
Pathophysiology of Edema
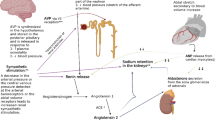
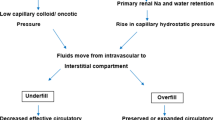
Massive proteinuria resulting in hypoalbuminemia and excessive sodium retention are chief contributing factors for edema formation in nephrotic syndrome. Two competing theories have been proposed to explain the pathophysiology of edema, the ‘underfill’ and the ‘overfill’ hypotheses [1,2,3,4]. The underfill hypothesis states that nephrotic range proteinuria leads to hypoalbuminemia, which results in decrease in plasma oncotic pressure and altered balance of Starling forces with consequent leak of fluid from the intravascular space to interstitium, resulting in edema. Depletion of intravascular volume, due to shifting of fluid into the interstitium, results in compensatory neurohormonal processes including activation of renin-angiotensin-aldosterone system (RAAS) and increased secretion of vasopressin. Sodium and water retention occur as a consequence of the compensatory response. Many observations contradict the underfill hypothesis as only pathophysiological mechanism responsible for edema formation. Patients with congenital analbuminemia do not develop edema [5], and all patients with nephrotic syndrome and severe edema do not show adequate response to albumin infusion therapy alone [5, 6]. Many patients show brisk diuresis following administration of corticosteroids well before normalization of serum albumin [7, 8]. Similarly, blockade of RAAS activation with angiotensin converting enzyme inhibitors does not result in reduction of edema.
According to the overfill hypothesis, sodium retention is the primary mechanism in nephrotic syndrome that leads to an expanded intravascular volume and extravasation of fluid into the interstitium and formation of edema [5, 9,10,11]. The mechanism for primary sodium retention is unclear, although the distal tubules and collecting ducts are considered to contribute to this phenomenon [12, 13]. The epithelial sodium channel (ENaC) is activated by plasmin that is generated by the urokinase-type plasminogen activator by cleavage of plasminogen, present in urine of patients with nephrotic syndrome [14,15,16]. Chen et al. recently showed that the urinary plasminogen-plasmin to creatinine ratio was an independent risk factor for occurrence of edema in adult patients with nephrotic syndrome [17]. Primary sodium retention in nephrotic syndrome may thus be mediated by plasmin dependent ENaC activation. While sodium retention by the kidney is important for pathogenesis of edema, other mechanisms including altered capillary permeability might also contribute [4, 18, 19].
Assessment of Intravascular Volume
Edema is the chief symptom of nephrotic syndrome at the onset of illness. Researchers report that patients with nephrotic syndrome show heterogeneity in clinical and biochemical features, including intravascular volume status (hypovolemia, euvolemia and hypervolemia) [20, 21]. It is important to distinguish between the pathophysiological processes of edema formation, since this would determine the strategy for management of edema [4, 22]. A combination of clinical and biochemical features is used to assess the intravascular volume status. Patients with volume contracted state present with abdominal pain, nausea, vomiting, dizziness, lethargy, tachycardia, pallor, cold peripheries, delayed capillary refill, low blood pressure, postural hypotension, which may progress to hypovolemic shock [23]. Hypovolemic shock is defined by blood pressure less than 5th centile in presence of other evidence of hypovolemia. Patients in volume expanded state usually show refractory edema, hypertension, bloating and dyspnea.
Two reliable biochemical markers of intravascular volume are fractional excretion of sodium (FENa), and potassium index i.e., ratio of urinary potassium to sodium plus potassium [UK+/(UK+ + UNa+)] [22, 24]. In patients with nephrotic syndrome and edema, the observed value of FENa is less than 1%; it is less than 0.2–0.5% in volume contracted state. Therefore FENa value of <0.2–0.5% can reliably identify children with hypovolemia [22, 24, 25]. Similarly, potassium index >0.6 is a marker of increased aldosterone activity in the distal nephron and suggests intravascular volume contraction [23,24,25]. In a recent study by Keenswijk et al., this ratio showed a good correlation with reduced renal plasma flow and was a satisfactory indicator of hypovolemia [24]. The above parameters do not reliably estimate the status of intravascular volume when the patient is receiving therapy with diuretics.
Other parameters for identifying hypovolemia are rise in hematocrit (>8–10%), high blood urea to creatinine ratio, and hyponatremia. Some studies have examined the role of ultrasonography, for assessing intravascular volume, using inferior vena cava indices. Very small number of patients have been evaluated, and the results do not support the use of ultrasonography as a reliable bedside tool for fluid volume assessment [26, 27]. Bioelectrical impedance analysis has also been used for assessment of fluid volume in childhood nephrotic syndrome [28, 29]. This analysis is considered to be better tool than ultrasonography for assessment of total body volume; larger studies are required before recommending it as a useful tool.
Diuretic Therapy
Diuretics interrupt sodium and water transport along specific segments of nephron, resulting in augmentation of urine output. Clinicians should be aware about specific properties of various classes of diuretics to optimize their use. These agents are classified according to their site of action, chemical structure, efficacy, and sparing of urinary potassium excretion. Loop and osmotic diuretics are highly potent resulting in almost ~25% glomerular filtrate being excreted as urine. Thiazides are moderately efficacious, while ENaC blockers and aldosterone antagonists are less effective. With better understanding of molecular mechanism of action, a classification schema based on mechanism of action of diuretics is possible [30].
Na+-K+-2Cl− Cotransport Inhibitors
This class of drugs acts by blocking sodium reabsorption through Na+-K+-2Cl− cotransporter (NKCC2) in the thick ascending limb of Henle. NKCC2 blockers are organic anions which bind to chloride binding site of this transporter at the luminal side. They are highly bound to albumin (~90%) in the blood, which limits their filtration through glomerulus. Hence to act from luminal surface, these agents are secreted in proximal tubules through organic anion transporters present on basolateral (OAT1) and apical surfaces (OAT3) of proximal tubular epithelial cells. Once the drug reaches the thick ascending limb, they increase sodium and chloride excretion (upto 25–30% of total filtered sodium load) in the urine by blocking salt transport through NKCC2. Increase in delivery of sodium in the distal segment results in increased secretion of potassium and hydrogen ions in cortical collecting ducts, and hypokalemic metabolic alkalosis. Interruption of sodium reabsorption by NKCC2 inhibitors also impairs the concentrating ability, resulting in massive diuresis.
Furosemide is the most commonly used agent, which also has weak carbonic anhydrase inhibiting ability. Furosemide shows erratic oral bioavailability and short half life. To initiate natriuresis, a minimal plasma concentration of furosemide (threshold) is essential. Threshold is not a fixed value and shows inter-patient variability. Once the threshold is crossed, increasing furosemide dose results in rapid rise in natriuresis that reaches a plateau after a high dose (ceiling dose); the agent thus shows a steep dose response curve. Diuresis following furosemide administration lasts for 6–8 h then fades away; during post diuretic phase there is compensatory increase in sodium retention from distal segment of nephron [31]. Based on the above principles, an adequate initial dose of furosemide should be given to cross its natriuretic threshold in plasma. The medication may be given in two divided doses, to avoid post diuretic sodium retention. Torasemide is pharmacologically superior to furosemide in terms of longer half life, and better and predictable oral bioavailability. However this agent is not approved for use in children (Table 1).
Chief adverse effects associated with use of NKCC2 inhibitors include volume depletion, hypokalemia and metabolic alkalosis; hyponatremia, hypomagnesemia and hypercalciuria are less common. Ototoxicity due to loop diuretics is reversible in most cases. This side effect is chiefly related to peak plasma concentration, hence the infusion rate of IV furosemide should not exceed 4 mg/min. Long term furosemide use can cause hyperuricemia, hypertriglyceridemia and rarely glucose intolerance. These agents can result in hypersensitivity reactions (skin rash, photosensitivity); the agents should be avoided in patients with hypersensitivity to sulfonamides. While concomitant use of amphotericin B increases the risk of nephrotoxicity and electrolyte disturbance, use of aminoglycoside and cisplatin is associated with ototoxicity. Nonsteroidal anti-inflammatory drugs (NSAIDs) attenuate the diuretic efficacy of furosemide; the anticoagulant activity of warfarin is increased by simultaneous use of furosemide.
Na+-Cl− Cotransport Inhibitors
Inhibitors of Na+-Cl− cotransporter (NCC) were derived from benzothiadiazine, hence medications in this group are known as thiazide diuretics. Some agents have different structure, but similar mechanism of action and are called thiazide-like (i.e., metolazone). Similar to NKCC2 inhibitors, drugs in this class are organic anions and highly albumin bound that restricts their filtration from glomeruli. NCC inhibitors are secreted into the proximal tubules through the organic anion transporter, and pass through loop of Henle to the distal tubule, which is their primary site of action. In distal tubules they inhibit NCC resulting in increased urine sodium excretion. NCC inhibitors are moderate efficacy drugs, and increase excretion of 5–6% of filtered sodium load through urine. Similar to NKCC2 inhibitors, these diuretics also increase sodium delivery in the collecting duct resulting in hypokalemia and metabolic alkalosis, as above. NCC inhibitors decrease urinary calcium excretion, through increased calcium reabsorption via the basolateral Na+-Ca2+ exchanger. It is proposed that NCC blockade leads to drop of intracellular sodium concentration, triggering sodium influx into cell in exchange of calcium through Na+-Ca2+ exchanger.
Two commonly used agents are hydrochlorothiazide and metolazone. Both drugs have satisfactory oral bioavailability (60–70%) and require once daily dosing (Table 1). Adverse effects are similar to loop diuretics, except for higher risk of hyponatremia and hypomagnesemia. Metabolic complications (hyperuricemia, hypertriglyceridemia, glucose intolerance) are more common with NCC inhibitors than NKCC2 inhibitors. Erectile dysfunction may be seen in adolescents using thiazides for management of hypertension.
Epithelial Sodium Channel Inhibitors
These are less effective diuretics, and used for their potassium sparing effect. Amiloride and triamterene are used in clinical practice. ENaC blockers are organic cations and their secretion in proximal tubules is mediated by organic cation transporter (OCT2) on the basolateral surface and the multidrug and toxin extruder (MATE1) on apical surface. Inhibition of ENaC results in increased sodium excretion (~2% of filtered load) in urine and loss of negative transepithelial potential difference, which is responsible for its potassium sparing effect. Both ENaC blockers are poorly absorbed from the gut. Hyperkalemia is the chief adverse effect; triamterene is associated with interstitial nephritis, nephrolithiasis and megaloblastic anemia. ENaC blockers should be used with caution in patients receiving cotrimoxazole, angiotensin converting enzyme inhibitors and NSAIDs.
Mineralocorticoid Receptor Antagonists
Mineralocorticoid receptors are located on basolateral surface of the tubular epithelial cells in late distal tubule and collecting duct. When aldosterone binds to these receptors, it regulates gene expression and increased production of ENaC, Na+-K+ ATPase and its migration to epithelial surface. This leads to reabsorption of sodium coupled with augmented secretion of potassium and hydrogen. Overall aldosterone antagonists are weak diuretics; their efficacy however depends on the endogenous level of aldosterone. Spironolactone and eplerenone are used in practice. Oral bioavailability of spironolactone and eplerenone is good (~65–70%); t1/2 of the latter is almost three times the former (Table 1). While their chief side effect is hyperkalemia, patients with cirrhosis may show metabolic acidosis; lethargy, drowsiness and ataxia may occur. Other adverse effects, chiefly related to affinity for steroid receptors in other organs, include gynecomastia, impotence, hirsutism, menstrual irregularity, gastritis and peptic ulcer.
Carbonic Anhydrase (CA) Inhibitors
Inhibition of CA results in reduced sodium, bicarbonate and water reabsorption in proximal tubules; however the losses are usually well compensated in the loop of Henle and distal nephron. Since CA in the collecting duct has an important role in acid (H+) secretion, its inhibition results in systemic acidosis. Acetzolamide has ~100% oral bioavailability and is excreted unchanged (Table 1). Systemic acidosis is the chief adverse effect; since CA inhibitors are sulfonamide derivatives they may results in bone marrow depression and hypersensitivity reactions. High dose CA inhibitors may rarely cause drowsiness and paresthesias.
Osmotic Diuretics
Osmotic diuretics are pharmacologically inert; they are freely filtered by the glomeruli and undergo limited reabsorption in tubules. Mannitol, a sugar alcohol, is currently used for therapeutic purposes. Administration of IV mannitol results in movement of water from intracellular to intravascular space, increased renal blood flow and abolition of medullary hypertonicity. As a consequence, tubules ability to concentrate urine is lost resulting in diuresis. Presence of osmotic agents within tubular lumen inhibits water reabsorption from proximal tubule and descending limb of Henle, which decreases sodium concentration in fluid reaching thin ascending limb, limiting passive reabsorption of sodium as well.
Mannitol is administered through the IV route, and its half-life is short, requiring frequent dosing or continuous IV infusion. Mannitol causes volume expansion and can result in pulmonary edema in patients with compromised cardiac functions. Hyponatremia may occur as mannitol shifts water from the intracellular to intravascular compartment. Rarely patients develop hypernatremia and dehydration due to excessive water loss through urine.
Vasopressin Receptor Antagonists (Aquaretics)
This class of drugs increases urine volume by augmenting free water excretion. Vasopressin acts through type 2 receptors (V2R) located on basolateral surface of the epithelial cells in the collecting duct. Binding of vasopressin to V2R leads to increased density of aquaporin channels (AQP2 and AQP4), resulting in water reabsorption. Tolvaptan is an oral vasopressin receptor antagonist (V2RA) with high affinity that increases urine volume. Since the medication leads to aquaresis, rise in blood sodium concentration is seen following their administration [32]. Aquaresis begins 2–4 h following oral administration of tolvaptan, and peak effect is observed at 4–8 h. Tolvaptan is chiefly excreted by non-renal route and metabolized by CYP3A. Thirst, dry mouth, pollakiuria and constipation are common side effects; few cases of liver injury have been reported following prolonged use. Clinicians should avoid rapid correction of hyponatremia, which might lead to osmotic demyelination syndrome. Evidence regarding use of V2RA in children is limited, and the drugs are not recommended for routine use [33, 34].
Combination Therapy of Furosemide and Albumin
Some patients of nephrotic syndrome with severe edema fail to respond to high dose oral or IV diuretics, and require albumin infusions. Albumin infusion is used to correct hypoalbuminemia, since it is the chief contributing factor for edema. Several mechanisms are proposed to explain the improved efficacy of combined therapy, the most accepted being that IV albumin enhances the secretion of furosemide in proximal tubules, with increased delivery at its primary site of action.
While initial studies reported that albumin infusion enhances diuresis in patients with diuretic refractory edema [35, 36], others have contradicted these findings [37]. The authors performed a meta-analysis of existing literature, in patients with nephrotic syndrome, to assess the efficacy of combined therapy with furosemide and albumin, compared to furosemide therapy alone [36,37,38,39,40,41]. Six studies, involving 69 patients were included in this meta-analysis. Of these studies, only one trial included children, and most had small sample size and high risk of bias. The meta-analysis showed that combination therapy was more effective in augmenting diuresis than furosemide therapy alone (Fig. 1). However in a recent Cochrane review authors failed to draw any conclusion, regarding use of human albumin infusion, due lack of studies as they excluded cross-over randomized trials [42].

Forest plot for meta-analysis assessing urine volume between combination of furosemide and albumin vs. furosemide therapy alone. FU Furosemide
Clinically, two groups of patients with nephrotic syndrome benefit from albumin infusions: (i) patients who present with intravascular hypovolemia, and (ii) those with severe diuretic refractory edema. Patients with hypovolemia, but not in shock, should be managed with IV infusion of human albumin (20%, 0.5–1 g/kg over 3–4 h), followed by careful administration of IV furosemide (0.5–1 mg/kg). Patients with refractory edema require IV infusion of albumin (1 g/kg) over 3–4 h, followed by higher dose of furosemide (2 mg/kg). Furosemide is administered as bolus dose during or at end of albumin infusion. Albumin is given as 20% undiluted solution, but may be diluted to 5% with normal saline in order to administer a higher infusate volume. Rate of albumin (20%) infusion should not exceed 2–4 ml/min, in order to avoid risk of fluid overload and pulmonary edema. The effect of albumin infusion is transient, and during relapse most patients would excrete the amount infused over next 24–48 h. Patients with severe hypoalbuminemia, specifically those with steroid resistant illness, require repeated courses of albumin infusion. While albumin infusion is effective in inducing diuresis and reduction of edema, it may be associated with worsening of hypertension, pulmonary edema and congestive heart failure [43]. It is recommended that adequate urine output be ensured before initiating albumin infusion; infusions should also be avoided in patients with respiratory distress. Rarely albumin infusion may cause an anaphylactic reaction [44].
Management of Edema
While almost one-half of the patients during relapse present in volume contracted state, the other half either have intravascular volume expansion or euvolemia. Patients with hypovolemic shock are managed with fluid resuscitation with either normal saline or 5% albumin to expand intravascular volume. Initially rapid infusion of normal saline (10–15 ml/kg) over 20–30 min is preferred in these patients. Five percent albumin infusion (10–15 ml/kg) over 30–60 min may be used in patients who fail to respond despite normal saline bolus [7, 45]. Patients with features suggestive of hypovolemia without evidence of shock should be managed with albumin infusion (0.5–1 g/kg over 3–4 h) with or without diuretics as described in Fig. 2.

Management of edema in patients with nephrotic syndrome
*Hypovolemia is indicated by urinary potassium index (> 0.6), fractional excretion of sodium (< 0.2%), hematocrit (rise of >10% from baseline) in presence of clinical evidence of hypovolemia.
#Avoid using diuretics in setting of diarrhea, vomiting and thrombosis.
Patients without hypovolemia are managed based on severity of edema. Patients with mild edema (weight gain <7%) are often managed conservatively and do not require diuretic therapy as administration of steroids induces brisk diuresis. These patients should be advised to avoid excessive sodium containing food (potato chips, papad, pickles, nuts and cornflakes) and minimal fluid restriction. Patients with moderate to severe edema (weight gain more than 8–15%) often can be managed with diuretics alone; furosemide is initial diuretic of choice due to its high efficacy and time tested safety profile. All patients on furosemide therapy should be advised restriction of sodium intake (<2 mEq/kg/d) as failure to adhere to this practice might result in poor response to therapy. Patients with nephrotic syndrome and moderate to severe edema should be initially managed with 2 mg/kg/d of oral furosemide. It is important to ensure an adequate dose of oral furosemide that crosses the natriuretic threshold. Practically this is assessed by asking if there is increased diuresis within 2–4 h of furosemide administration. Initial target of diuretic therapy is to achieve weight reduction of 1–2% of body weight per day. If the initial dose fails to augment urine volume, the dose might need to be increased upto 6 mg/kg/d. Lack of response to maximum oral dose is an indication for switching over to the IV route. Although furosemide is usually administered in divided doses, the total dose should not be divided until the initial dosage is adequate enough to induce diuresis.
Combination therapy, with a NCC inhibitor (metolazone is preferred) and furosemide, should be used for patients who fail to achieve adequate reduction in edema with furosemide alone. Patients receiving high dose oral or IV furosemide often require administration of spironolactone as a potassium sparing agent. Since urine from patients with nephrotic syndrome is proposed to activate ENaC, therapy with ENaC blockers seems appropriate for management of edema in these patients. However there is limited evidence that amiloride augments diuresis, and therefore, cannot be recommended for clinical practice.
Patients with diuretic resistant edema might benefit from coadministration of albumin and furosemide as mentioned above. An algorithm suggesting the practical approach for management of edema is shown in Fig. 1. Patients with moderate to severe edema on diuretics therapy require strict monitoring of body weight, urine output, evidence of hypovolemia and serum electrolytes 1–2 times daily.
Diuretic Resistance
Furosemide is often the initial diuretic of choice for weight reduction in patients with moderate to severe edema. A proportion of patients fail to achieve clinically desired reduction in weight despite maximum dose of diuretic; these patients are considered as diuretic resistant. Diuretic resistance is described in patients with heart failure, nephrotic syndrome and cirrhosis. Poor adherence to salt restriction and medications, gut edema, low serum albumin, intravascular hypovolemia, accumulation of endogenous acids, reduced glomerular filtration rate (GFR) and compensatory increase in sodium reabsorption from distal nephron, are responsible for furosemide resistance in children. Management of diuretic resistance comprises of improving compliance to salt restriction and medication intake, and reverting the likely etiology for non response [31, 46] (Table 2). Patients with severe diuretic resistant edema should also be advised to restrict fluid intake to two-thirds of maintenance. Most of these patients with diuretic resistant edema can be managed with albumin furosemide combination therapy. A small proportion of patients with severe resistant edema, who show diuretic response to IV bolus dose of furosemide which is not sustained, may benefit with continuous furosemide infusion. Following initial furosemide bolus of 1–2 mg/kg, infusion is started at 0.1 mg/kg/h and can be increased upto 1 mg/kg/h [41]. Patients on continuous furosemide infusion are at high risk of electrolyte imbalance and hence require frequent monitoring.
References
Epstein AA. Concerning the causation of edema in chronic parenchymatous nephritis: method for its alleviation. Am J Med. 1952;13:556–61.
Dorhout EJ, Roos JC, Boer P, Yoe OH, Simatupang TA. Observations on edema formation in the nephrotic syndrome in adults with minimal lesions. Am J Med. 1979;67:378–84.
Siddall EC, Radhakrishnan J. The pathophysiology of edema formation in the nephrotic syndrome. Kidney Int. 2012;82:635–42.
Ellis D. Pathophysiology, evaluation, and management of edema in childhood nephrotic syndrome. Front Pediatr. 2016;3:111.
Brown EA, Markandu ND, Sagnella GA, Jones BE, MacGregor GA. Lack of effect of captopril on the sodium retention of the nephrotic syndrome. Nephron. 1984;37:43–8.
Usberti M, Federico S, Meccariello S, et al. Role of plasma vasopressin in the impairment of water excretion in nephrotic syndrome. Kidney Int. 1984;25:422–9.
Bagga A. Revised guidelines for management of steroid-sensitive nephrotic syndrome. Indian J Nephrol. 2008;18:31–9.
Pasini A, Benetti E, Conti G, et al. The Italian Society for Pediatric Nephrology (SINePe) consensus document on the management of nephrotic syndrome in children: part I - diagnosis and treatment of the first episode and the first relapse. Ital J Pediatr. 2017;43:41.
Brown EA, Markandu ND, Sagnella GA, Squires M, Jones BE, MacGregor GA. Evidence that some mechanism other than the renin system causes sodium retention in nephrotic syndrome. Lancet. 1982;2:1237–40.
Usberti M, Gazzotti RM, Poiesi C, D’Avanzo L, Ghielmi S. Considerations on the sodium retention in nephrotic syndrome. Am J Nephrol. 1995;15:38–47.
Meltzer JI, Keim HJ, Laragh JH, Sealey JE, Jan KM, Chien S. Nephrotic syndrome: vasoconstriction and hypervolemic types indicated by renin-sodium profiling. Ann Intern Med. 1979;91:688–96.
Bohnert BN, Menacher M, Janessa A, et al. Aprotinin prevents proteolytic epithelial sodium channel (ENaC) activation and volume retention in nephrotic syndrome. Kidney Int. 2018;93:159–72.
Stæhr M, Buhl KB, Andersen RF, et al. Aberrant glomerular filtration of urokinase-type plasminogen activator in nephrotic syndrome leads to amiloride-sensitive plasminogen activation in urine. Am J Physiol Renal Physiol. 2015;309:F235–41.
Passero CJ, Hughey RP, Kleyman TR. New role for plasmin in sodium homeostasis. Curr Opin Nephrol Hypertens. 2010;19:13–9.
Hughey RP, Mueller GM, Bruns JB, et al. Maturation of the epithelial Na+ channel involves proteolytic processing of the alpha- and gamma-subunits. J Biol Chem. 2003;278:37073–82.
Svenningsen P, Bistrup C, Friis UG, et al. Plasmin in nephrotic urine activates the epithelial sodium channel. J Am Soc Nephrol. 2009;20:299–310.
Chen J-L, Wang L, Yao X-M, et al. Association of urinary plasminogen-plasmin with edema and epithelial sodium channel activation in patients with nephrotic syndrome. Am J Nephrol. 2019;50:92–104.
Rostoker G, Behar A, Lagrue G. Vascular hyperpermeability in nephrotic edema. Nephron. 2000;85:194–200.
Lagrue G, Branellec A, Blanc C, et al. A vascular permeability factor in lymphocyte culture supernants from patients with nephrotic syndrome. II. Pharmacological and physicochemical properties. Biomedicine. 1975;23:73–5.
Geers AB, Koomans HA, Roos JC, Boer P, Dorhout Mees EJ. Functional relationships in the nephrotic syndrome. Kidney Int. 1984;26:324–30.
Vande Walle J, Donckerwolcke R, Boer P, van Isselt HW, Koomans HA, Joles JA. Blood volume, colloid osmotic pressure and F-cell ratio in children with the nephrotic syndrome. Kidney Int. 1996;49:1471–7.
Kapur G, Valentini RP, Imam AA, Mattoo TK. Treatment of severe edema in children with nephrotic syndrome with diuretics alone-a prospective study. Clin J Am Soc Nephrol. 2009;4:907–13.
Vande Walle JG, Donckerwolcke RA, van Isselt JW, Derkx FH, Joles JA, Koomans HA. Volume regulation in children with early relapse of minimal-change nephrosis with or without hypovolaemic symptoms. Lancet. 1995;346:148–52.
Keenswijk W, Ilias MI, Raes A, Donckerwolcke R, Walle JV. Urinary potassium to urinary potassium plus sodium ratio can accurately identify hypovolemia in nephrotic syndrome: a provisional study. Eur J Pediatr. 2018;177:79–84.
Donckerwolcke RA, France A, Raes A, Vande WJ. Distal nephron sodium-potassium exchange in children with nephrotic syndrome. Clin Nephrol. 2003;59:259–66.
Dönmez O, Mir S, Ozyürek R, Cura A, Kabasakal C. Inferior vena cava indices determine volume load in minimal lesion nephrotic syndrome. Pediatr Nephrol. 2001;16:251–5.
Kosiak W, Swieton D, Piskunowicz M. Sonographic inferior vena cava/aorta diameter index, a new approach to the body fluid status assessment in children and young adults in emergency ultrasound--preliminary study. Am J Emerg Med. 2008;26:320–5.
Nalcacioglu H, Ozkaya O, Baysal K, et al. The role of bioelectrical impedance analysis, NT-ProBNP and inferior vena cava sonography in the assessment of body fluid volume in children with nephrotic syndrome. Nefrologia. 2018;38:48–56.
Özdemir K, Mir MS, Dinçel N, et al. Bioimpedance for assessing volume status in children with nephrotic syndrome. Turk J Med Sci. 2015;45:339–44.
Goodman LS, Brunton LL, Chabner B, Knollmann BC. Goodman & Gilman's Pharmacological Basis of Therapeutics, 14th ed. New York: McGraw-Hill; 2017.
Ellison DH. Clinical pharmacology in diuretic use. Clin J Am Soc Nephrol. 2019;14:1248–57.
Ali F, Guglin M, Vaitkevicius P, Ghali JK. Therapeutic potential of vasopressin receptor antagonists. Drugs. 2007;67:847–58.
Shimizu M, Ishikawa S, Yachi Y, et al. Tolvaptan therapy for massive edema in a patient with nephrotic syndrome. Pediatr Nephrol. 2014;29:915–7.
Meena J, Sinha A, Hari P, Bagga A. Therapy with the combination of tolvaptan and furosemide for refractory edema in nephrotic syndrome. Indian J Nephrol. 2020;30:53–5.
Eadington DW, Plant WD, Winney RJ. Albumin in the nephrotic syndrome. BMJ. 1995;310:1333.
Fliser D, Zurbrüggen I, Mutschler E, et al. Coadministration of albumin and furosemide in patients with the nephrotic syndrome. Kidney Int. 1999;55:629–34.
Akcicek F, Yalniz T, Basci A, Ok E, Mees EJ. Diuretic effect of frusemide in patients with nephrotic syndrome: is it potentiated by intravenous albumin? BMJ. 1995;310:162–3.
Na KY, Han JS, Kim YS, et al. Does albumin preinfusion potentiate diuretic action of furosemide in patients with nephrotic syndrome? J Korean Med Sci. 2001;16:448–54.
Ghafari A, Mehdizadeh A, Alavi-Darazam I, Rahimi E, Kargar C, Sepehrvand N. Co-administration of albumin-furosemide in patients with the nephrotic syndrome. Saudi J Kidney Dis Transplant. 2011;22:471–5.
Phakdeekitcharoen B, Boonyawat K. The added-up albumin enhances the diuretic effect of furosemide in patients with hypoalbuminemic chronic kidney disease: a randomized controlled study. BMC Nephrol. 2012;13:92.
Dharmaraj R, Hari P, Bagga A. Randomized cross-over trial comparing albumin and frusemide infusions in nephrotic syndrome. Pediatr Nephrol. 2009;24:775–82.
Ho JJ, Adnan AS, Kueh YC, Ambak NJ, Van Rostenberghe H, Jummaat F. Human albumin infusion for treating oedema in people with nephrotic syndrome. Cochrane Database Syst Rev. 2019;7:CD009692.
Haws RM, Baum M. Efficacy of albumin and diuretic therapy in children with nephrotic syndrome. Pediatrics. 1993;91:1142–6.
Liumbruno GM, Bennardello F, Lattanzio A, Piccoli P, Rossettias G; Italian Society of Transfusion Medicine and Immunohaematology (SIMTI). Recommendations for the use of albumin and immunoglobulins. Blood Transfus. 2009;7:216–34.
McCaffrey J, Lennon R, Webb NJA. The non-immunosuppressive management of childhood nephrotic syndrome. Pediatr Nephrol. 2016;31:1383–402.
Hoorn EJ, Ellison DH. Diuretic resistance. Am J Kidney Dis. 2017;69:136–42.
Author information
Authors and Affiliations
Contributions
JM performed a review of literature and prepared the initial draft of the manuscript. AB critically reviewed the first draft. Both authors reviewed and edited the final draft of the manuscript. JM will act as guarantor for this paper.
Corresponding author
Ethics declarations
Conflict of Interest
None.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Meena, J., Bagga, A. Current Perspectives in Management of Edema in Nephrotic Syndrome. Indian J Pediatr 87, 633–640 (2020). https://doi.org/10.1007/s12098-020-03252-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12098-020-03252-9