
Abstract
Objectives
To report the distribution pattern of various categories of primary immunodeficiency disorders (PIDs) in children from North India, frequency of warning signs and critical parameters for evaluation.
Methods
In this retrospective study, 528 children below 18 y of age after clinical assessment and presentation suggestive of PID were further screened by immunophenotyping for immune cell markers by flow cytometry.
Results
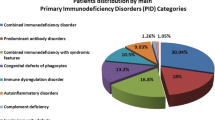
A total of 120 (23%) children were diagnosed with PID with median age at diagnosis being 2.5 y in males and 3.5 y in females and an average delay in diagnosis from onset of symptoms being approximately 5 y. Chronic lower respiratory tract infections, gastrointestinal symptoms like persistent diarrhea and failure to thrive were amongst the most common warning signs in these patients. PIDs were classified according to the International Union of Immunological Societies’ (IUIS) criteria. The diagnosis of index study subjects included combined humoral and cellular immunodeficiency (29%), phagocytic defects (29%), followed by predominantly antibody deficiency (18%), innate immunity and dysregulation (17%) and other well-defined syndromes (7%). A family history of PID (23%), consanguineous marriage (8%) and previous sibling death (23%) were observed as major clinical predictors/clues for underlying PID. All children received prophylactic antibiotics and/or antifungals in addition to specific therapy for underlying immune deficiency.
Conclusions
The field of PIDs in India remains largely unexplored and we are faced with various challenges in the diagnosis of PIDs due to lack of awareness as well as absence of equipped immunological laboratory support. The authors propose a methodical step-wise laboratory diagnostic approach that can facilitate early diagnosis and timely intervention of these mis/underdiagnosed disorders.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Primary immunodeficiency disorders (PIDs) comprise of 300 rare chronic disorders which primarily affect the components of immune system and in some cases, non-immunological abnormalities. The International Union of Immunological Societies (IUIS) Expert Committee categorized PIDs into 9 groups based on the genetic and molecular diagnosis of patients [1]. The initial cases of PIDs reported from India almost 50 y ago were based on clinical manifestations [2, 3]. In the last two decades, a transition in the diagnoses of PIDs has been based on flow cytometric analysis, in selective cases by mutational analysis and is evolving towards deep sequencing approaches [4,5,6,7,8,9,10].
Of the 10 warning signs of PIDs, family history is the strongest predictor and parental consanguinity along with previous sibling mortality are strongly associated [11]. The distribution pattern of PIDs in India varied significantly from those reported by western studies [12]. There is an urgent need for development of more advanced facilities for diagnosis and management of PID in India and also the need for establishing population, hospital based registries [13, 14] and awareness programs [11].
The authors report here, the distribution pattern of various categories of PIDs in children from North India, frequency of warning signs and critical parameters for evaluation.
Material and Methods
Children less than 18 y of age with suspected PID [15, 16], seeking either inpatient or outpatient care with well-established clinical and diagnostic features since January 2014 through December 2016, in authors’ tertiary care hospital were included. PID was classified according to the IUIS criteria [1]. The diagnosis was established based on characteristic clinical manifestations and corroborated by immunological markers using flow cytometry analyses. All children with PID were assessed for any new infection, development of any new sign or symptom and monitored for growth during each follow-up visit every 12–16 wk. The Institute Ethics Committee approved the study protocol (Reference No. IEC-55/03.03.2017).
Laboratory investigations included complete blood count, peripheral blood smear, measurement of serum immunoglobulin (IgG, IgA, IgM, and IgE) in all cases and isohemagglutinins, peripheral blood lymphocyte subsets including T cell subsets (CD3, CD4 and CD8), B cells (CD19, CD20) and natural killer cells (CD56/16) by flow cytometry and CH50 complement assay by ELISA. Specific immunological tests performed included CD56, perforin and CD107a, Double negative TCRαβ subset, Dihydrorhodamine (DHR), CD18/CD11b & CD11c, intracellular STAT-3/IL-17 and Btk protein expression in monocytes by flow cytometry assay, as needed.
Results
Of the 528 patients evaluated for suspected PID, 120 (total 23% of which 71% were boys) cases were identified with PID (Fig. 1a). Eighty six percent of the cases had a varied age at presentation of PID after 3 mo till 17 y of age. A substantial number of patients (14%) however, presented with severe clinical manifestations before 3 mo of age. The mean duration in delay of diagnosis from onset of symptoms was 5 y and varied with the underlying PID; antibody predominant deficiencies [X-linked agammaglobulinemia (XLA), Common variable immunodeficiency (CVID)] were diagnosed on an average after 5 y of disease onset, whereas the gap between onset of clinical symptoms and diagnosis was within 3 mo in SCID. Eighty percent of PID cases had a family history of consanguinity and in 23% cases previous sibling deaths were noted.

(a) Flow chart showing cases screened for PID (b) Frequency of warning signs for inclusion of PID cases (c) Diagnostic algorithm for screening of patients suspected of primary immunodeficiency. Ab Antibody; CBC Complete blood count; CID Combined immunodeficiency; DHR Dihydrorhodamine; HIV Human immunodeficiency virus; Igs Immunoglobulins; IUIS International union of immunological societies; LAD Leukocyte adhesion deficiency; NK Natural killer; PBS Peripheral blood smear; PID Primary immunodeficiency disorder
Half of the patients presented primarily with repeated lower respiratory tract infections i.e., pneumonia requiring multiple antibiotics (Fig. 1b). Other symptoms noted were gastrointestinal, like persistent diarrhea and failure to thrive that was also seen in CVID cases. Recurrent deep skin abscess was noted in 15% of the cases. One case of Hyper IgM syndrome presented with disseminated cryptococcosis.
The screening and diagnosis of PID was achieved by analyzing the absence or reduced expression of various immunological markers by flow cytometry based assays. The prevalence of PID positive cases were divided into 5 broad categories. The combined T and B cell deficiency and phagocytic defects occupied majority of the cases followed by predominantly antibody deficiency and diseases of immune-dysregulation. Well-defined deficiencies were rare in the present patient cohort (Table 1).
At authors’ center, 29% of patients were diagnosed with combined T and B cell immunodeficiency. Phagocytic cell defect, chronic granulomatous disease (X-linked and carriers) was diagnosed by Dihydrorhodamine (DHR) flow cytometry assay in 30 children with PID. Reduced (<5%) or absent neutrophil oxidative index (NOI) was considered as a X-linked CGD against a healthy control that showed more than 85–90% NOI. Carrier females usually showed mosaic pattern with 2 subsets of neutrophils, one with normal NOI and the other population with reduced NOI. Ten cases (six males) of LAD I were diagnosed based on CD18, CD11b and CD11c deficiency on surface of leucocytes.
In this cohort, the diagnosis of XLA was based on markedly reduced or absent B cells (<1%), markedly reduced serum immunoglobulin levels and lack of expression of Brutons tyrosine kinase (Btk) protein in the monocytes. Ten male patients (two siblings from two families) who presented to authors’ hospital with recurrent severe infections with onset of symptoms before 1 y of age were diagnosed with XLA. These patients showed profound reduction in serum immunoglobulin levels and absent or reduced Btk protein expression in the monocytes by flow cytometry.
The authors also identified three male pediatric cases of CVID (diagnosed after 15 y) who had onset of clinical symptoms at 3–4 y of age. All patients had reduced serum immunoglobulins with normal B cell frequency. The baseline immunological workup carried out in authors’ laboratory showed markedly reduced serum immunoglobulin levels (IgG, IgA and IgM), normal T, B and NK cell frequency with reversal of CD4:CD8 ratio (1:1) and profoundly decreased (<1%) class switched memory B cells (CD19 + CD27 + IgD-) by flow cytometry.
Twenty PID positive cases were of primary Hemophagocytic lymphohistiocytosis (HLH) [18 Familial hemophagocytic lymphohistiocytosis type 2 (FHL2) and 2 Griscelli syndrome]. FHL2 patients had perforin deficiency and normal CD107a degranulation in NK and T cells whereas Griscelli patients expressed reduced CD107a degranulation marker but normal perforin levels. Four cases of autoimmune lymphoproliferative syndrome were detected to have increased levels of CD3 double negative (TCRαβ+CD4-CD8-) T cells (constituting >2.5% of total lymphocytes).
Other Well defined immunodeficiency syndromes or disorders were rare comprising 7% of the total PID cases. These included ataxia telangiectasia (1 case), Wiskott Aldrich syndrome (2 cases) and Hyper IgE syndrome (7 cases). Of the 7 cases of Hyper IgE (5 were males), authors performed intracellular STAT-3 and phospho-STAT3 staining in the CD3+ T cells and assessed positivity by flow cytometry. Phospho-STAT3 expression was reduced in all 7 cases with Hyper IgE syndrome.
Discussion
Primary immunodeficiencies still remain underdiagnosed in the less affluent regions of the world lacking state-of-the-art facilities for its diagnosis and leave clinicians in dilemma for its treatment. Till date, scarcity of epidemiological data on PID in India is primarily attributed to early mortality because of undiagnosed disease in absence of reliable screening assays. In the past decade, India has witnessed significant advancement in the field of PID diagnosis [17]. Here, authors present a retrospective analysis of children after clinical and immunological assessment for PID.
According to the predominance of the five PID categories in the present study (summarized in Table 1), Category I of IUIS classification of PIDs include Combined B and T cell immunodeficiency, also called as Severe combined immune deficiency (SCID), which is a heterogeneous group of PID conditions. Accurate incidence of SCID in India is unknown but is postulated to be similar to the rest of the world [5]. One of the subtype of SCID with distinctive clinical features is Hyper IgM syndrome (HIGM) caused by defects of immunoglobulin class switch recombination (CSR) and somatic hyper mutation (SHM) [18]. The commonest X-linked type of Hyper IgM syndrome has defective expression of CD40 ligand (CD40L, CD154) on T cells upon activation which was noted in three of index cases [19].
The next PID category with comparable frequency to combined immune deficiency was Category V in IUIS PID classification, which includes congenital defects of phagocytes number, function or both. Chronic granulomatous disease (CGD) and leukocyte adhesion deficiency (LAD) syndromes are the two broad sub-categories in phagocytic defects [20]. The frequency, clinical presentation and treatment of these patients were as per the protocols (Table 1). Jindal et al. reported a comparative study between 153 vs. 122 cases of PID from major institutes of Chandigarh and Mumbai, respectively. The spectrum of PIDs reported at these centres varied because of the regional differences in the population of North and West India. However, in the present study patients from all over India were enrolled hence the regional bias was precluded. The present data reflects a more uniform distribution of cases and combined and phagocytic defects were found as commonest PIDs followed by antibody deficiencies and diseases of immune dysregulation as compared to previous studies from India, which reflected skewed distribution in patterns and number of referrals to other centers [17].
Antibody deficiencies, are enlisted under category III of IUIS PID classification and include X-linked agammaglobulinemia, which is caused by mutations involving gene mapped on chromosome Xq21.3-Xq22, coding for Btk protein [21, 22]. The incidence of XLA is reported to be around 1/200,000 male births [23]. Individuals affected by this disorder present with severe, recurrent bacterial infections involving various organ systems after the protective maternal antibodies start declining in the infant circulation. On the other end of the spectrum is CVID, most common symptomatic humoral primary immunodeficiency disorder that presents in late childhood or in adults [24,25,26]. It is a condition marked by defect in maturation and differentiation of naïve B cells to memory B and plasma cells. Patients are prone to chronic sinusitis, pneumonias, gastrointestinal complications and autoimmune features. Initial diagnosis of CVID was established based on European Society for Immunodeficiencies (ESID) 2014 diagnostic criteria in the present cases [27]. The frequency, clinical presentation and management of these cases is as summarized in Table 1.
Diseases of immune dysregulation comprising of 17% of the cases were clubbed in category IV of IUIS classification, which included hemophagocytic lymphohistiocytosis (HLH), autoimmune lymphoproliferative syndrome (ALPS), immune dysregulation with colitis and type I interferonopathies [1]. Hemophagocytic lymphohistiocytosis, is a hyperinflammatory syndrome induced by uncontrolled activation and proliferation of histiocytes/ macrophages and T cells with defect in cytotoxic pathways [28]. The frequency of disorders of immune dysregulation ranked as the fourth category, similar to global frequency reported by Modell et al. [12].
Rare syndromes included Hyper IgE syndrome also known as Job’s syndrome, is a rare primary immunodeficiency characterized by a distinct clinical syndrome and molecular pathophysiology. Patients present with skin eczema, staphylococcal skin and lung abscesses, fractures, dental abnormalities and a high serum IgE levels (>2000 IU/ml). Wiskott–Aldrich syndrome (WAS) is an X-linked primary immunodeficiency disorder that is characterized by the classic triad of severe immunodeficiency, eczema and a bleeding diathesis due to microthrombocytopenia that affects approximately one to four cases per 1,00,000 live male births [29]. As documented by Modell et al. in their data of global overview of primary immunodeficiencies, antibody mediated deficiencies showed the highest frequency followed by other well defined immunodeficiency syndromes and combined T and B cell defects [12].
The complement system is a part of the innate immune system that facilitates clearance of microbes and damaged cells from an organism. The authors performed complement assay in forty cases over the period of this study. However, no patient was identified with complement deficiency.
In the present study, consanguineous marriage and family history of previous sibling deaths emerged as strong indicators of PID. The authors defined a diagnostic algorithm for screening of PIDs in pediatric population visiting their center in North India (Fig. 1c), as early diagnosis may allow timely intervention and reduce morbidity and mortality associated with these diseases. Hematopoietic stem cell transplant (HSCT) remains the only curative option and if intervened early after disease onset, can offer long term survival benefits. Unfortunately, lack of early diagnosis, awareness and late referral for HSCT are likely contributory factors to the hitherto poor outcome in these children. At authors’ centre too, most cases were diagnosed relatively late after disease onset and hence succumbed to the disease. Several patients are on regular follow-up in pediatric OPD and few underwent successful HSCT.
The present study depicts the wide spectrum of commonly encountered PIDs from an apex tertiary care referral hospital equipped with laboratory workup of children with suspected primary immune deficiencies. This report highlights the importance of suspecting PIDs in children often managed under the broad rubric of infectious disorders. Although the limitation of the present study is that it is cross-sectional, therefore the long-term outcome of these cases are not available. The diagnosis of most PIDs in this study is based mainly on clinical, laboratory parameters including flow cytometry studies, however the genetic confirmation of the underlying disease is lacking. The exhaustive IUIS classification system of Primary immune deficiencies enunciate the plethora of gene defects that can manifest in patients as well as reiterates the importance of having facilities of mutational analysis by whole exome/whole genome sequencing and advanced genomic techniques like Next Generation Sequencing for the early diagnosis of these conditions. The authors admit that the lack of genetic tests and Enzyme assays during this study limited the spectrum of diagnosis. Nonetheless, scientific awareness about PID disorders among clinicians in different specialties accompanied by a methodical step-wise laboratory diagnostic approach can facilitate early diagnosis and timely intervention. A routine immune screening for PIDs and increased awareness among clinicians and researchers alike is strongly recommended.
References
Bousfiha A, Jeddane L, Al-Herz W, et al. The 2015 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol. 2015;35:727–38.
Chandra RK, Ghai OP. Primary immunodeficiency states in Indian children. Indian J Med Res. 1976;64:68–75.
Datta U, Kumar L, Mehta S, Walia BN, Sharma BK, Sehgal S. Primary immunodeficiency defects seen in PGI--one year study. J Assoc Physicians India. 1984;32:701–4.
Verma S, Sharma PK, Sivanandan S, et al. Spectrum of primary immune deficiency at a tertiary care hospital. Indian J Pediatr. 2008;75:143–8.
Gupta S, Madkaikar M, Singh S, Sehgal S. Primary immunodeficiencies in India: a perspective. Ann N Y Acad Sci. 2012;1250:73–9.
Al-Saud BK, Al-Sum Z, Alassiri H, et al. Clinical, immunological, and molecular characterization of hyper-IgM syndrome due to CD40 deficiency in eleven patients. J Clin Immunol. 2013;33:1325–35.
Chopra YR, Yadav SP. Status of primary immunodeficiency disorders in India. Indian Pediatr. 2013;50:974.
Madkaikar M, Mishra A, Ghosh K. Diagnostic approach to primary immunodeficiency disorders. Indian Pediatr. 2013;50:579–86.
Mishra A, Gupta M, Dalvi A, Ghosh K, Madkaikar M. Rapid flow cytometric prenatal diagnosis of primary immunodeficiency (PID) disorders. J Clin Immunol. 2014;34:316–22.
Fleisher TA, Madkaikar M, Rosenzweig SD. Application of flow cytometry in the evaluation of primary immunodeficiencies. Indian J Pediatr. 2016;83:444–9.
Reda SM, El-Ghoneimy DH, Afifi HM. Clinical predictors of primary immunodeficiency diseases in children. Allergy Asthma Immunol Res. 2013;5:88–95.
Modell V, Knaus M, Modell F, Roifman C, Orange J, Notarangelo LD. Global overview of primary immunodeficiencies: a report from Jeffrey Modell centers worldwide focused on diagnosis, treatment, and discovery. Immunol Res. 2014;60:132–44.
Keerthikumar S, Raju R, Kandasamy K, et al. RAPID: resource of Asian primary immunodeficiency diseases. Nucleic Acids Res. 2009;37:D863–7.
Chinnabhandar V, Yadav SP, Kaul D, Verma IC, Sachdeva A. Primary immunodeficiency disorders in the developing world: data from a hospital-based registry in India. Pediatr Hematol Oncol. 2014;31:207–11.
Subbarayan A, Colarusso G, Hughes SM, et al. Clinical features that identify children with primary immunodeficiency diseases. Pediatrics. 2011;127:810–6.
Slatter MA, Gennery AR. Clinical immunology review series: an approach to the patient with recurrent infections in childhood. Clin Exp Immunol. 2008;152:389–96.
Jindal AK, Pilania RK, Rawat A, Singh S. Primary immunodeficiency disorders in India—a situational review. Front Immunol. 2017;8:714.
Notarangelo LD, Duse M, Ugazio AG. Immunodeficiency with hyper-IgM (HIM). Immunodefic Rev. 1992;3:101–21.
Korthäuer U, Graf D, Mages HW, et al. Defective expression of T-cell CD40 ligand causes X-linked immunodeficiency with hyper-IgM. Nature. 1993;361:539–41.
Harris ES, Weyrich AS, Zimmerman GA. Lessons from rare maladies: leukocyte adhesion deficiency syndromes. Curr Opin Hematol. 2013;20:16–25.
Kristufek D, Aspalter RM, Eibi MM, Wolf HM. Characterisation of novel Brutons tyrosine kinase gene mutations in central European patients with agammaglobulinemia. Mol Immunol. 2007;44:1639–43.
Valiaho J, Smith CI, Vihinen M. BTKbase: the mutation database for X-linked agammaglobulinemia. Hum Mutat. 2006;27:1209–17.
Chun JK, Lee TJ, Song JW, Linton JA, Kim DS. Analysis of clinical presentations of Bruton disease: a review of 20 years of accumulated data from pediatric patients at severance hospital. Yonsei Med J. 2008;49:28–36.
Jolles S. The variable in common variable immunodeficiency: a disease of complex phenotypes. J Allergy Clin Immunol Pract. 2013;1:545–56.
Notarangelo LD. Primary immunodeficiencies. J Allergy Clin Immunol. 2010;125:S182–94.
Mohammadinejad P, Aghamohammadi A, Abolhassani H, et al. Pediatric patients with common variable immunodeficiency: long-term follow-up. J Investig Allergol Clin Immunol. 2012;22:208–14.
Ameratunga R, Brewerton M, Slade C, et al. Comparison of diagnostic criteria for common variable immunodeficiency disorder. Front Immunol. 2014;5:415.
Usmani GN, Woda BA, Newburger PE. Advances in understanding the pathogenesis of HLH. Br J Haematol. 2013;161:609–22.
Buchbinder D, Nugent DJ, Fillipovich AH. Wiskott–Aldrich syndrome: diagnosis, current management, and emerging treatments. Appl Clin Genet. 2014;7:55–66.
Acknowledgements
The authors thank the study subjects, for providing samples and supporting this work; the participating clinical staff, for their dedication to this research; Ms. Poonam, Ms. Jyoti, Mr. Pankaj, Mr. Anoop (Department of Transplant Immunology and Immunotherapeutics, All India Institute of Medical Sciences), for their help in processing of samples.
Author information
Authors and Affiliations
Contributions
DG and DT: Concepts, design, definition of intellectual content, literature search, experimental studies, data analysis, manuscript preparation, editing, and review; SKK: Concepts, design, definition of intellectual content, experimental studies, data analysis, manuscript preparation and review; RL: Concepts, definition of intellectual content, clinical studies, manuscript editing, and review; NB: Design, literature search, experimental studies, data acquisition and analysis and manuscript preparation and review; SKM: Concepts, definition of intellectual content, clinical and experimental studies, data analysis, manuscript preparation and review; PK and RK: Definition of intellectual content, clinical and experimental studies, data analysis, manuscript preparation; SC: Definition of intellectual content, clinical studies, manuscript preparation, editing, and review; DKM: Concepts, design, definition of intellectual content, manuscript preparation, editing and review. DKM is the guarantor for this paper.
Corresponding author
Ethics declarations
Conflict of Interest
None.
Source of Funding
Institutional (AIIMS, New Delhi, India).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gupta, D., Thakral, D., Kumar, P. et al. Primary Immunodeficiency Disorders Among North Indian Children. Indian J Pediatr 86, 885–891 (2019). https://doi.org/10.1007/s12098-019-02971-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12098-019-02971-y