
Abstract
The pathogenetic basis of idiopathic nephrotic syndrome, a common childhood glomerulopathy, is being explored. While initial evidence supported an imbalance of T helper responses, recent studies suggest alterations in both innate and adaptive immune responses, including evidence for impaired T regulatory function. The central role of the podocyte in causing proteinuria is confirmed by the observation of mutations in key podocyte proteins in steroid resistant nephrotic syndrome and experimental evidence of altered podocyte signaling and cytoskeletal organization. The outcome and management of idiopathic nephrotic syndrome in children is determined by the response to corticosteroids and the frequency of relapses. While patients with steroid sensitive nephrotic syndrome have a favorable long term outcome, almost half of them relapse frequently and are at risk of adverse effects of corticosteroids. Although various non-corticosteroid immunosuppressive agents are used to prolong disease remission, careful monitoring is required for the potential adverse effects. Calcineurin inhibitors have emerged as the choice of therapy in patients with steroid resistant nephrotic syndrome. However, the management of this form of the disease is particularly challenging because of the variable response to immunosuppression, therapy-related significant adverse effects and high rates of disease progression to end stage renal disease. Patients with both corticosteroid sensitive and resistant forms of the disease are at risk of complications of disease, and require close monitoring and repeated counseling.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nephrotic syndrome is characterized by heavy proteinuria, hypoalbuminemia (serum albumin <2.5 g/dl), hyperlipidemia (serum cholesterol >200 mg/dl) and edema [1]. The large majority (>90 %) is primary (idiopathic); a secondary cause, e.g., amyloidosis, systemic lupus, Henoch Schonlein purpura, IgA nephropathy, and infection with human immunodeficiency virus, hepatitis B and C is seen rarely. This article describes the current view on the pathogenesis of the condition, and principles of management of steroid sensitive and steroid resistant nephrotic syndrome. The authors summarize the importance of supportive care for these patients and the long-term outcome of the disease.
Pathogenesis
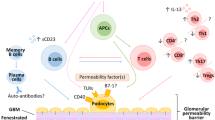
The pathogenesis of nephrotic syndrome, in context of observed immunological dysfunction, and its relationship with proteins of the slit diaphragm and the podocyte cytoskeleton, is unclear. A unifying hypothesis that combines the immunological perturbations with abnormal function at the level of the slit diaphragm is lacking. Fig. 1 summarizes the current view on pathogenesis.

Pathogenesis of idiopathic nephrotic syndrome. Schematic view of podocyte foot process demonstrates its components of the slit diaphragm complex, formed by nephrin, Neph1 and podocin (Po), and the actin cytoskeleton, which also receives inputs from the basolateral domain of the foot process, containing the α3β1 integrins and α and β dystroglycans. The dominant paradigm in nephrotic syndrome is an imbalance between T helper 1 (Th1) and T helper 2 (Th2) cytokines, and that a soluble mediator, presumably IL-13, increases glomerular permeability. Other molecules suggested as mediators include soluble urokinase plasminogen activator receptor (suPAR), vascular endothelial growth factor (VEGF) and angiopoietin-like-4 (ANGPTL4). Signaling through nuclear factor kappa B (NF-κB) and toll like receptors (TLRs) mediated pathways may polarize adaptive immune responses towards Th2 cells, or directly increase CD80 expression in podocytes. An imbalance of Th17 and T regulatory responses allows persistent CD80 activation on podocytes, and/or helper responses. Finally, therapeutic agents have direct effects on podocytes; glucocorticoids (G) on gene expression, calcineurin inhibitors (C) on stabilization of synaptopodin (S) or inhibition of TRPC6 channel, and binding of rituximab (R) to the SMPDL-3b protein
Genetics
Some patients with steroid resistant nephrotic syndrome show mutations in genes encoding key podocyte proteins that constitute the slit diaphragm or the podocyte cytoskeleton (NPHS1, NPHS2, CD2AP, TRCP6 and ACTN4); others are expressed in the glomerular basement membrane (LAMB2) or mitochondria (COQ2), or encode transcription factors necessary for normal development (WT1, LMX1B) (Table 1) [2]. Genetic mutations are present in 10–20 % patients with sporadic steroid resistance, and in a higher proportion of patients with familial nephrotic syndrome [3].
Immunological Dysfunction
Nephrotic syndrome is considered a T cell disorder, where these cells release cytokine(s) that act on the glomeruli to induce an increase in permeability to plasma proteins. Evidence for the hypothesis is provided by: absence of immune deposits in the glomeruli; occurrence of remission following measles infection (that suppresses T cells); association with Hodgkin lymphoma; and response to treatment with agents that inhibit T cell function (corticosteroids, cyclosporine) [4]. It has been speculated that immune dysfunction might result in production of an, as yet uncharacterized, circulating factor that affects the slit diaphragm, resulting in selective proteinuria. A number of proteins are proposed, including interleukin 13, soluble urokinase plasminogen activator receptor, soluble CD80 and vascular endothelial growth factor; their role in pathogenesis needs confirmation and better characterization [5].
Recent evidence suggests an activation of innate immune responses in response to triggering of toll like receptors (TLRs) by microbial products, even directly on podocytes (Fig. 1) [6]. Proteinuria in experimental models is linked to an increase in CD80 expression within podocytes. An imbalance of Th17 and T regulatory responses allows persistent CD80 activation on podocytes, and/or helper responses [7]. Cross talk between B cells and T helper cells is suggested by the response to rituximab, an anti-B cell agent.
Pathology
In over 80 % patients, the histology shows insignificant glomerular abnormalities on light microscopy, termed minimal change disease (Fig. 2a) [8]. Immunofluorescence examination does not show glomerular deposits of immunoglobulins or complement. Electron microscopy reveals effacement of podocytes foot processes with disruption and disorganization of actin filaments. Mesangial proliferation, characterized by the presence of >3 cells per mesangial lobule, is found in a small proportion.

a Light microscopy (hematoxylin and eosin staining) in a 4-y-old girl with initial steroid resistance shows no abnormality, typical of minimal change disease. b Light microscopy (hematoxylin and eosin staining) in a 12-y-old boy with initial steroid resistant nephrotic syndrome shows segmental sclerosis and enlarged glomerulus. Findings suggest focal segmental glomerulosclerosis
The underlying pathology in patients with steroid resistant nephrotic syndrome, and in 5–10 % cases with sensitive nephrotic syndrome, is focal segmental glomerulosclerosis (FSGS) (Fig. 2b) [9]. Based on location of sclerosis, various subtypes are: (i) tip lesions, (ii) cellular variant, (iii) perihilar lesions, and (iv) FSGS not otherwise specified alterations. Collapsing FSGS, a subtype which is associated with human immunodeficiency virus or parvovirus infections, shows rapid progression to end stage renal disease within 2–3 y. Increased incidence of FSGS has been reported recently, the reasons for which are unclear [10].
Histology in patients with steroid resistant nephrotic syndrome shows minimal change disease and FSGS in 30–40 % patients each, and mesangioproliferative glomerulonephritis in a small group. Approximately 15 % patients with steroid resistance show membranoproliferative glomerulonephritis, membranous nephropathy, IgA nephropathy or amyloidosis. Several syndromic forms of nephrotic syndrome are associated with diffuse mesangial sclerosis.
Steroid Sensitive Nephrotic Syndrome
The majority of patients with idiopathic nephrotic syndrome are steroid responsive [1]. The prognosis in these cases is favorable, as compared to patients who do not respond to corticosteroids (steroid resistance). The disease course is variable; while almost 40 % have no relapses or a single relapse, more than 55 % show multiple relapses that occur either infrequently or frequently (Table 2).
Evaluation at Onset
Evaluation of a patient with suspected nephrotic syndrome includes history and physical examination, with attention to a possible secondary etiology, prior therapies and evidence of infections [11]. ‘Nephrotic range’ proteinuria is defined as 3+/4+ (300–2000 mg/dl) urine protein by dipstick on first morning urine for 3 consecutive days, spot protein/creatinine ratio >2 mg/mg, or urine protein excretion >40 mg/m2 per hour on a timed-sample. A precise quantitative assessment of proteinuria is not necessary for the diagnosis. Relevant investigations are listed in Table 3.
Indications for Renal Biopsy
Most children with idiopathic nephrotic syndrome do not require a renal biopsy. Renal biopsy is required at the onset of nephrotic syndrome in situations where a cause other than minimal change nephrotic syndrome is suspected, such as: (i) age at onset <1 y or >16 y, (ii) gross hematuria, persistent microscopic hematuria or low serum C3; (iii) renal failure, not attributable to hypovolemia; (iv) suspected secondary causes; and (v) sustained severe hypertension. A renal biopsy is considered later if (i) diagnosis of steroid resistance is made, or (ii) therapy with calcineurin inhibitors is planned.
The specimen should be examined by light and immunofluorescence microscopy. Electron microscopy helps confirm the diagnosis of minimal change disease (MCD), and early membranous nephropathy, membranoproliferative glomerulonephritis and Alport syndrome.
Management of the Initial Episode
The initial (first) episode of nephrotic syndrome should be treated adequately, both in terms of dose and duration of corticosteroids, since this is an important determinant of long term course [12].
Only prednisolone and prednisone are of proven benefit in the treatment of proteinuria [13]. Other agents such as deflazacort, methylprednisolone, dexamethasone, betamethasone, triamcinolone or hydrocortisone should not be used. Prednisolone is administered after meals to reduce its gastrointestinal side effects; antacids are not required unless there is gastrointestinal intolerance. Prednisolone is given at a dose of 2 mg/kg per day (maximum 60 mg) in single or divided doses for 6 wk, followed by 1.5 mg/kg (maximum 40 mg) as a single morning dose on alternate days for the next 6 wk. Therapy with corticosteroids is then stopped.
Prolongation of initial steroid therapy for 12 wk or longer is associated with significantly reduced risk for subsequent relapses [1]. While it is proposed that therapy with corticosteroids should not be stopped abruptly, but further tapered over the next 8–12 wk, the benefits of prolonged therapy should be balanced by the risk of steroid adverse effects.
Therapy for Relapse
Relapses are often triggered by minor infections. Symptomatic therapy of infectious illness might result in remission of low grade (1+/2+) proteinuria. However, persistence of 3+/4+ proteinuria with infections requires treatment for relapse. Prednisolone is given at a dose of 2 mg/kg/d (single or divided doses) until urine protein is negative or trace for three consecutive days (remission), and subsequently as a single morning dose of 1.5 mg/kg on alternate days for 4 wk, and then discontinued. Treatment for a relapse thus usually lasts for 5 to 6 wk, and there is no evidence that prolonged therapy of a relapse determines the long-term outcome of the illness.
If the patient is not in remission despite 2 wk treatment with daily prednisolone, the treatment is extended for 2 more weeks. Patients showing no remission despite 4 wks’ treatment are labelled as late steroid resistance.
Infrequent Relapsers
Patients suffering from three or fewer relapses a year should receive 5–6 wk treatment for each disease relapse as described above [8].
Frequent Relapses and Steroid Dependence
Patients with frequent relapses or steroid dependence require prolonged treatment in order to maintain disease remission. The strategies used to maintain remission in such patients are as follows [8].
Long Term, Alternate Day Steroids
Following treatment of a relapse, the dose of prednisolone is gradually tapered to maintain the patient in remission; usually a dose of 0.3–0.7 mg/kg is given on alternate days for 9–18 mo. This strategy is effective in maintaining remission in many patients.
Relapses are often precipitated following minor infections. Evidence from multiple prospective studies suggest that increasing the frequency of administration of prednisolone from alternate day to daily during minor infectious illnesses is effective in preventing infection precipitated relapses [14].
Steroid Sparing Agents
The additional use of an alternative agent should be considered in patients with: (i) prednisolone threshold (for maintaining remission) higher than 0.5–0.7 mg/kg on alternate days, or (ii) features of corticosteroid toxicity (growth failure, hypertension and cataract). The agents used, usually in successive order, are listed below and in Fig. 3.

Alternative agents in steroid sensitive nephrotic syndrome. The agents are usually used in a successive order from top to bottom. Agents marked with an asterisk (*) are preferred in patients with significant steroid toxicity (cataracts, severe stunting or obesity) or if disease relapses are associated with life threatening complications (thrombosis, severe infections)
Levamisole
This medication is effective in reducing relapse rates and steroid requirement in some patients with frequent relapses and steroid dependence. The medication is given at a dosage of 2–2.5 mg/kg on alternate days for 12–24 mo. The dose of prednisolone is decreased every 2–4 wk to 0.25–0.5 mg/kg on alternate days. While therapy with prednisolone may be discontinued in some cases, many patients continue to require a small dose, given on alternate days. Adverse effects are uncommon, but comprise flu-like symptoms, neutropenia, hepatotoxicity, convulsions and skin rash. Leukocyte counts are monitored every 2–3 mo.
Cyclophosphamide
This agent is preferred in patients with: (a) significant steroid toxicity, (b) severe relapses with episodes of hypovolemia, life threatening infections or thrombosis, and (c) poor compliance or difficult follow up. Therapy with cyclophosphamide is effective in ensuring sustained steroid free remission in a significant proportion of patients. Children older than 7–8 y, those with frequent relapses and lower steroid threshold do better than those who are younger and have steroid dependence or high steroid threshold.
The dose of cyclophosphamide is 2–2.5 mg/kg/d for 8–12 wk. Its cumulative dose should not exceed 168 mg/kg, and repeat courses are avoided. Leukocyte counts should be monitored every 2 wk and the medication discontinued if below 3000–4000/mm3. Fluid intake is increased and the child encouraged to void frequently. The dose of prednisolone is 1–1.5 mg/kg on alternate days during cyclophosphamide therapy; subsequently steroids are tapered and discontinued over 4–6 wk.
The chief adverse effects of treatment include leukopenia, alopecia, nausea, vomiting and hemorrhagic cystitis; long-term toxicity includes the risk of gonadal toxicity and malignancies. Another alkylating agent, chlorambucil, though effective has significant additional toxicities and a low margin of safety, and is not recommended.
Mycophenolate Mofetil
Despite limited randomized controlled studies, the use of mycophenolate mofetil has increased in patients with relapsing nephrotic syndrome. Prolonged therapy results in reduced relapse rates and significant steroid sparing [15]. The dose of the medication is 600–1000 mg/m2/d or 20–25 mg/kg/d in two divided doses for 12–36 mo. Tapering doses of prednisolone are administered for 6–12 mo.
Therapy is generally safe and associated with few side effects. Leukocyte counts are monitored every 1–2 mo and treatment withheld if below 4000/mm3. Other adverse effects include abdominal pain and diarrhea.
Cyclosporine and Tacrolimus
Therapy with either of these agents is indicated in patients with frequent relapses or steroid dependence that fail to benefit with levamisole, cyclophosphamide and/or mycophenolate mofetil. Treatment may be associated with significant adverse effects and cautious use under the supervision of an expert is necessary.
Cyclosporine A (4–5 mg/kg/d) or tacrolimus (0.1–0.2 mg/kg/d) are administered, in two divided doses, for 12–24 mo aiming for respective trough levels of 80–120 ng/ml and 3–7 ng/ml. Both these agents have strong steroid sparing potential, with steroid discontinuation achieved in a majority of patients following taper over 6–9 mo.
Adverse effects are common and include acute and chronic nephrotoxicity. Renal functions are monitored every 3 mo; rise of creatinine more than 25 % is of concern. Patients receiving cyclosporine show more cosmetic side effects (hirsutism, gum hyperplasia), hypertension and hypercholesterolemia. Treatment with tacrolimus is associated with risk of hyperglycemia, elevated transaminases, diarrhea, tremors, headache, and seizures. In view of a significant long-term risk of nephrotoxicity, a renal biopsy is done after 2–3 y of continuous therapy.
Rituximab
This is a monoclonal anti-CD20 antibody, used with moderate success in patients with steroid dependent nephrotic syndrome, with remission lasting 6–18 mo [16]. Rituximab is recommended in patients with marked steroid dependence who fail to respond satisfactorily to other therapies, or in patients with toxicity secondary to other drugs. Rituximab should be administered under close supervision at a specialized center.
Steroid Resistant Nephrotic Syndrome
The management of patients with steroid resistant nephrotic syndrome (SRNS) is difficult, with patients showing a variable response to immunosuppression, adverse effects of prolonged therapy and risk of progressive renal damage.
A patient is considered to have SRNS if there is lack of remission despite treatment with prednisolone, at a dose of 2 mg/kg/d (60 mg/m2/d) for 4 wk (Table 2). This definition is based on knowledge that 95 % patients with steroid sensitive nephrotic syndrome achieve remission within 4 wk of steroid therapy, and that longer therapies are associated with high incidence of medication related adverse effects. Care is taken to exclude systemic infections (e.g., peritonitis, cellulitis, respiratory tract infections), which might result in persistent proteinuria. Initial resistance is defined by the lack of remission at the first episode of nephrotic syndrome, and late resistance is considered in patients who are steroid sensitive initially, but show steroid resistance during a subsequent relapse [17].
Evaluation
Baseline assessment of renal function, blood levels of albumin and cholesterol, and quantification of urinary protein loss (spot urine protein to creatinine ratio in young children; 24-h protein excretion in older children) guides future evaluation of response to therapy (Table 3). Patients should be evaluated for hepatitis B and C virus infection, if renal histology shows membranous nephropathy or membranoproliferative glomerulonephritis, or in those having deranged liver functions.
Children diagnosed with SRNS (initial or late) should undergo renal biopsy before instituting specific treatment. While patients with minimal change disease show a satisfactory response to therapy, the presence of FSGS with chronic tubulointerstitial changes is associated with less satisfactory outcomes. The diagnosis of entities such as membranoproliferative glomerulonephritis and membranous nephropathy is important, since their management differs from idiopathic minimal change and FSGS.
About 10–20 % patients with familial and sporadic SRNS might carry homozygous or compound heterozygous mutations in genes encoding podocyte proteins, including podocin (NPHS2), nephrin (NPHS1) and Wilms tumor (WT1) genes [18]. These patients are usually unresponsive to immunosuppressive medications, progresses rapidly to end stage renal disease, and unlike non-genetic FSGS (which recurs after transplantation in 30 %), does not recur. Patients with syndromic forms have onset of disease in early childhood; e.g., Denys Drash syndrome (diffuse mesangial sclerosis, Wilm’s tumor), Frasier syndrome (FSGS, male pseudohermaphroditism, gonadoblastoma) and Pierson syndrome (diffuse mesangial sclerosis, microcoria, neurological abnormalities). The frequency of mutations in these genes in children in east and south Asia is lower.
Where facilities exist, mutational analysis should be offered to patients with: (i) congenital nephrotic syndrome (onset below 3 mo of age), (ii) family history of SRNS, (iii) sporadic initial steroid resistance that does not respond to therapy with cyclophosphamide or calcineurin inhibitors, and (iv) girls with steroid resistant FSGS.
Management
Patients with idiopathic SRNS secondary to minimal change disease, FSGS or mesangioproliferative glomerulonephritis are treated similarly. Patients with minimal change disease demonstrate higher rates of remission and better prognosis.
The chief factor predicting renal outcome is the response of proteinuria to therapy rather than the renal histology. The aim of therapy in patients is thus to induce and maintain remission of proteinuria, while avoiding medication related adverse effects. Most regimens use a combination of an immunosuppressive agent with prednisolone (given on alternate days) and an angiotensin converting enzyme inhibitor (Table 4) [19, 20].
Calcineurin Inhibitors (Cyclosporine or Tacrolimus)
Treatment with calcineurin inhibitors (CNI) is considered ‘first line’ for patients with steroid resistance. Cyclosporine and tacrolimus are effective in inducing complete or partial remission in 60–80 % patients [21]. However, a recent randomized multicentric trial in the United States reported disappointing results with either cyclosporine or mycophenolate mofetil combined with alternate day steroids in steroid resistant FSGS [22]. Since adverse effects are common, immunosuppressive therapy should be stopped if there is no response after 6 mo.
Cyclophosphamide
Oral cyclophosphamide, administered alone or with oral steroids, has limited efficacy in inducing remission. Cyclophosphamide has also been used, with modest success, either in the intravenous form or in combination with intravenous steroids. Intravenous cyclophosphamide, when administered monthly for six doses along with tapering doses of alternate day prednisolone, induces remission in 40–50 % patients with SRNS.
Pulse Corticosteroids with Oral Cyclophosphamide
Pulses of IV methylprednisolone or dexamethasone have been used in combination with oral cyclophosphamide with moderate efficacy. The risk of steroid toxicity is high, with a significant proportion of patients developing systemic infections, hypertension and electrolyte abnormalities (Table 4 ).
Other Agents
Rituximab has been reported to be successful in inducing disease remission in 25–30 % patients. Its safety and efficacy needs to be established in larger series. Other therapies that have shown promise, in anecdotal reports, include the combination of cyclosporine and mycophenolate mofetil, and plasmapheresis.
Adjunctive Therapies
Prednisolone
Prednisolone is a component of all regimens used in therapy of SRNS. It is administered on alternate days at 1 mg/kg d for 1–3 mo, following which the dose may be tapered. Prednisolone may be discontinued if the child is in sustained complete remission for 6–12 mo.
Angiotensin Converting Enzyme Inhibitors, Angiotensin Receptor Blockers
Therapy with angiotensin converting enzyme (ACE) inhibitors (e.g., enalapril 0.3–0.6 mg/kg/d, ramipril 6 mg/m2/d) is associated with decrease in proteinuria and control of hypertension. Adverse effects include dry cough, hyperkalemia and decline in renal function. The dose of the ACE inhibitor is decreased or therapy discontinued if hyperkalemia develops or the estimated GFR falls below 30 ml/min/1.73 m2. Angiotensin receptor blockers (e.g., losartan, valsartan) may be used in case of persistent dry cough with ACE inhibitors, or as add-on therapy to achieve better antiproteinuric effect. There are limited studies on the efficacy of this combination in children.
Monitoring Response to Therapy
Patients should be monitored every month until response to therapy is demonstrated, and then every 2–3 mo. Complete remission is defined as presence of trace or negative proteinuria (by dipstick) or spot urine protein to creatinine ratio (Up/Uc) <0.2 mg/mg. Patients are considered to be in partial remission if they show 1-2+ proteinuria (or Up/Uc between 0.2–2), blood albumin >2.5 g/dl and no edema. Non-response is defined as 3-4+ proteinuria (Up/Uc >2), blood albumin <2.5 g/dL or edema.
The aim of treatment is the achievement of complete remission, but occurrence of partial remission is also satisfactory. Patients who respond to treatment do so within 3–6 mo; those that fail therapy with one regimen may show response to different agents.
Drug Monitoring
Most agents used in the therapy of SRNS require monitoring for adverse effects (Table 4 ). Monitoring for drug levels is recommended when using either cyclosporine or tacrolimus, because individual variations in bioavailability may result in occurrence of either sub-therapeutic or toxic levels. A 12-h trough level should be estimated about 2-wk after introduction of therapy, after any dose change, and if suspecting drug toxicity or poor compliance. Trough levels in the range of 80–120 ng/ml for cyclosporine and 4–7 ng/ml for tacrolimus are acceptable.
Examination of renal histology is advised in patients receiving prolonged therapy (2–3 y) with calcineurin inhibitors. Histological features of nephrotoxicity include nodular hyalinosis or striped interstitial fibrosis and tubular atrophy. Prolonged duration of therapy (exceeding 2–3 y) and persistent heavy proteinuria (beyond 30 d) are risk factors for nephrotoxicity. The decision to continue calcineurin inhibitors therapy should be reviewed in presence of these changes.
Duration of Therapy
Consensus is lacking on the optimal duration of treatment with calcineurin inhibitors. Therapy is continued for 2–3 y in patients that show complete or partial remission, followed by the options: (i) taper dose of the medication to the lowest effective dose, and continue for another 1–2 y; (ii) exclude medication-related nephrotoxicity on renal histology and then continue therapy; (iii) switch treatment to a less toxic agent, e.g., mycophenolate mofetil, rituximab.
Recurrence of FSGS after Renal Transplantation
FSGS recurs in 30–50 % of children following renal transplantation, leading to graft loss in half of these patients. Risk factors for recurrence include (i) non-genetic forms of FSGS, (ii) progression to ESRD within 3-y of onset of disease, (iii) mesangial proliferation on the original biopsies and (iv) nephrectomy of native kidneys prior to transplant. Pre-transplant plasmapheresis is an important preventive strategy. While there is no consensus on optimal therapy of patients with recurrent FSGS, options include: (i) intensive plasmapheresis; (ii) intravenous immunoglobulin (500 mg/kg/dose once a wk after pheresis); (iii) rituximab (375 mg/m2/wk for 4 wk); and (iv) oral cyclophosphamide (2–2.5 mg/kg/d for 3 mo) instead of MMF.
Supportive Care and Management of Complications
Patients with relapsing steroid sensitive nephrotic syndrome and those with steroid resistant disease require attention for complications resulting from disease or its therapy.
Edema
Presence of significant edema or anasarca is associated with discomfort and an increased risk of infections. Since daily administration of corticosteroids results in diuresis within 2–4 d, specific therapy for edema is not required in most patients. Those with significant edema and weight gain require treatment with diuretics.
Management
Figure 4. outlines shows the stepwise treatment of edema in children with nephrotic syndrome [23]. Edema that does not respond to maximal doses of oral furosemide requires co-administration of a thiazide diuretic (e.g., hydrochlorthiazide, metolazone). If furosemide is used for prolonged duration (>7 d) or in high doses (3–6 mg/kg/d), additional treatment with spironolactone (2–4 mg/kg/d) prevents the occurrence of hypokalemia. Patients with refractory edema should be hospitalized and administered IV furosemide either as bolus injections (1–3 mg/kg/dose, infused over 15–20 min) or as continuous infusions (0.1–1 mg/kg/h), under careful monitoring.

Management of edema in patients with nephrotic syndrome. *Hypovolemia is suggested by the presence of tachycardia, feeble pulses, cold extremities or hypotension. Additional features include an elevated hematocrit, disproportionately high blood urea, low fractional excretion of sodium (<0.5 %) and urinary K+/ [K+ + Na+] >0.6. **Management of hypovolemia consists of rapid infusion of normal saline at a dose of 15–20 mL/kg over 20–30 min; repeated as required. Infusion of 5 % albumin (10–15 mL/kg) or 20 % albumin (0.5–1 g/kg) are useful in subjects who do not respond to two boluses of saline
Infusions of albumin (20 % albumin, 0.5–1 g/kg, over 2–4 h), with an IV bolus of furosemide, administered at the end of the infusion, are useful in patients with severe hypoalbuminemia (albumin <1.5 g/dl). The effect of these infusions is transient, necessitating repeat administration in patients with severe edema. Infusion of IV albumin in patients with oliguria can worsen hypertension and may result in pulmonary edema and congestive cardiac failure. Adequacy of urine output must be ensured prior to albumin infusion, and its administration should be avoided in individuals with respiratory distress of any cause.
Abdominal paracentesis might be necessary in patients with refractory severe ascites and respiratory distress, but should be done carefully under aseptic precautions. A daily record of weight and urine output, frequent monitoring of vital signs and daily estimation of electrolytes (hypokalemia, hyponatremia and metabolic alkalosis) are essential. Parents should be instructed to discontinue diuretics if symptoms of hypovolemia (abdominal pain, dizziness) appear, or the child has diarrhea, vomiting or poor oral intake.
Hypovolemia
Hypovolemia may occur during a severe disease relapse or following administration of diuretics, particularly in children with poor oral intake, diarrhea and vomiting. Features include abdominal pain, lethargy, dizziness and leg cramps, tachycardia, hypotension, delayed capillary refill, low volume pulses, and cool clammy distal extremities. An elevated ratio of blood urea to creatinine and rising hematocrit suggests the presence of hypovolemia. The level of urine sodium and its fractional excretion are reduced to <20 mEq/L and 0.2–0.4 % respectively. A high urinary potassium index, urine K+/ (urine K+ + urine Na+), exceeding 0.6 suggests the presence of hypovolemia.
Therapy with diuretics should be discontinued. When signs of hypovolemia are absent, an increase in oral fluid intake alone may suffice. With features of hypovolemia, patients require admission and rapid infusion of normal saline (10–20 mL/kg) over 20–30 min. Patients who do not respond to two boluses of saline should receive infusion of 5 % albumin (10–15 mL/kg) or 20 % albumin (0.5–1 g/kg).
Infections
Common infections include peritonitis and cellulitis, which should be treated using appropriate antibiotics. Since varicella may be life-threatening in patients with nephrotic syndrome receiving corticosteroids or other immunosuppressive drugs, these patients should be monitored for complications, during the illness and for 7–10 d afterwards. All patients with varicella must receive oral acyclovir for 7 d; severe illness requires admission and administration of IV acyclovir. Administration of varicella zoster immunoglobulin (in a single dose within 96 h of exposure), or intravenous immunoglobulin (400 mg/kg, single dose) prevents or lessens the severity of the disease in susceptible individuals exposed to varicella.
Hypertension
Hypertension may be noted at the onset of disease or develop secondarily as a result of high dose steroid therapy. Persistent elevation in blood pressure, refractory to control of edema and decline of corticosteroid dose, merits treatment. An angiotensin converting enzyme inhibitor, enalapril (0.3–0.6 mg/kg/d in 2 divided doses) or ramipril is the medication of choice. The dose is increased to permissible limits in patients with persistent hypertension, while monitoring renal function and levels of potassium. Some patients require additional therapy with calcium channel blockers (e.g., amlodipine) or adrenergic blockers. The target blood pressure is between the 75–90th percentile for age, gender and height.
Dyslipidemia
Hyperlipidemia associated with disease relapses does not merit treatment. However, patients with steroid resistant nephrotic syndrome with persistent proteinuria have continued dyslipidemia that is undesirable, and requires treatment with statins.
Therapy with HMG CoA reductase inhibitors (e.g., atorvastatin 10–20 mg daily in children >5 y) is recommended in presence of biochemical abnormalities that persist for 3–6 mo: total cholesterol >200 mg/dl, LDL cholesterol >130 mg/dl or triglycerides >200 mg/dl.
Thrombosis
Children with nephrotic syndrome are predisposed to venous thromboembolism during relapses, due to multiple reasons including loss of antithrombin III, protein S, and high levels of lipoprotein A, factor V, factor VIII and fibrinogen levels. In addition, low intravascular volume (particularly with aggressive diuretic use, or diarrheal dehydration), immobilization, indwelling vascular catheters and puncture of deep vessels also predispose to thrombosis. Thrombosis should be suspected in patients with hematuria or flank pain (renal vein thrombosis); venous congestion, pain, reduced mobility of limbs (deep vein thrombosis); or seizures, vomiting, altered sensorium and neurological deficits (saggital sinus or cortical venous thrombosis). Deep vein thrombosis can lead to pulmonary embolism. Diagnosis requires confirmation with ultrasonography, Doppler studies and cranial MRI if required. Therapy includes the use of heparin (IV) or low-molecular-weight heparin (subcutaneously) initially, followed by oral anticoagulants for 4–6 mo.
Vaccination
The administration of live vaccines (oral polio, varicella) should be deferred until the child is off immunosuppressive medications for at least 4 wk. If essential, these vaccines may be given to patients receiving alternate day prednisolone at a dose <0.5 mg/kg.
The administration of pneumococcal vaccine is desirable. Children below 2 y of age should receive the pneumococcal conjugated vaccine, 0.5 ml intramuscularly, in the schedule advised by the Indian Academy of Pediatrics (Age <6 mo: 3 doses 4–8 wk apart and booster at 15–18 mo; Age 6–12 mo: 2 doses 4–8 wk apart and booster at 15–18 mo; Age between 12–23 mo: 2 doses 8 wk apart). Above 2 y of age, one dose of the polysaccharide vaccine (PPV23) is administered, following one dose of the conjugate vaccine; the gap between the injections should be at least 2 mo. Children who continue to have relapses of nephrotic syndrome may receive one repeat dose of PPV23, 5 y after the primary vaccination.
Two doses of the varicella vaccine should be administered 4 wk apart while the child is in remission and off immunosuppressive medications. Injectable polio vaccine should be administered to children with nephrotic syndrome and their siblings. If the child has received primary immunization with oral polio vaccine (at 6, 10, 14 wk), two doses of the parenteral vaccine are given at 2 mo interval followed by a third dose 6 mo after the first dose, and a booster at 5 y.
Nutrition
During remission, children should eat a balanced, nutritious diet without restrictions. If disease relapses are associated with edema, salt restriction is advised, by curbing the intake of snacks and foods with high salt content. Undue restriction that makes food unpalatable is not required.
Patients with persistent or recurrent proteinuria should increase their daily intake of proteins to 2–2.5 g/kg. Increase in physical activity can help achieve desirable body weight in those with Cushingoid features or obesity. Patients receiving prolonged therapy with prednisolone (>3 mo) should receive supplements of calcium carbonate (250–500 mg) and vitamin D (125–250 IU).
Stress Dose of Steroids
Patients who have received steroids at high doses for more than 2 wk in the past year are at risk of suppression of the hypothalamo-pituitary- adrenal axis. These children require steroid supplements during surgery, anesthesia or serious infections. Corticosteroids are supplemented, as parenteral hydrocortisone at a dose of 2–4 mg/kg/d, followed by oral prednisolone at 0.3–0.6 mg/kg/d. This is given for the duration of stress and then tapered rapidly.
The long term management of nephrotic syndrome requires frequent interactions with patients’ families to ensure their cooperation in disease management. Parents are explained the natural history of the disease and its outcome, and the expected adverse effects of repeated courses of high dose steroid therapy and other medications. While steroid responsiveness remains the most important prognostic feature, patients with both steroid sensitive and steroid resistant disease require frequent monitoring of clinical course and biochemical response, and timely management of disease- and therapy-related complications in order to enable satisfactory long term outcomes.
References
Bagga A, Mantan M. Nephrotic syndrome in children. Indian J Med Res. 2005;122:13–28.
Löwik MM, Groenen PJ, Levtchenko EN, Monnens LA, van den Heuvel LP. Molecular genetic analysis of podocyte genes in focal segmental glomerulosclerosis-A review. Eur J Pediatr. 2009;168:1291–304.
Gigante M, Piemontese M, Gesualdo L, Iolascon A, Aucella F. Molecular and genetic basis of inherited nephrotic syndrome. Int J Nephrol. 2011; doi:10.4061/2011/792195
Araya CE, Wasserfall CH, Brusko TM, et al. A case of unfulfilled expectations. Cytokines in idiopathic minimal lesion nephrotic syndrome. Pediatr Nephrol. 2006;21:603–10.
Parikh SM. Circulating mediators of focal segmental glomerulosclerosis: soluble urokinase plasminogen activator receptor in context. Am J Kidney Dis. 2011; doi:10.1053/j.ajkd.2011.09.011
Zhang SY, Audard V, Fan Q, Pawlak A, Lang P, Sahali D. Immunopathogenesis of Idiopathic Nephrotic Syndrome. Contrib Nephrol. 2011;169:94–106.
Taylor PA, Lees CJ, Fournier S, Allison JP, Sharpe AH, Blazar BR. B7 expression on T cells down-regulates immune responses through CTLA-4 ligation via T-T interactions. J Immunol. 2004;172:34–9.
Barnett HL, Edelmann CM, Greifer I. The primary nephrotic syndrome in children. Identification of patients with minimal change nephrotic syndrome from initial response to prednisone. A report of the International Study of Kidney Disease in Children. J Pediatr. 1981;98:561–4.
Gbadegesin R, Lavin P, Foreman J, Winn M. Pathogenesis and therapy of focal segmental glomerulosclerosis: An update. Pediatr Nephrol. 2011;26:1001–15.
Dragovic D, Rosenstock JL, Wahl SJ, Panagopoulos G, DeVita MV, Michelis MF. Increasing incidence of focal segmental glomerulosclerosis and an examination of demographic patterns. Clin Nephrol. 2005;63:1–7.
Indian Pediatric Nephrology Group, Indian Academy of Pediatrics; Bagga A, Ali U, Benerjee S, et al. Management of steroid sensitive nephrotic syndrome: Revised guidelines. Indian Pediatr. 2008;45:203–14.
Hodson EM, Willis NS, Craig JC. Corticosteroid therapy for nephrotic syndrome in children. Cochrane Database Syst Rev. 2007; 4:CD001533.
Gipson DS, Massengill SF, Yao L, et al. Management of childhood onset nephrotic syndrome. Pediatrics. 2009;124:747–57.
Gulati A, Sinha A, Hari P, Bagga A. Daily corticosteroids reduce infection associated relapses in frequently relapsing nephrotic syndrome: a randomized controlled trial. Clin J Am Soc Nephrol. 2011;6:63–9.
Moudgil A, Bagga A, Jordan SC. Mycophenolate mofetil therapy in frequently relapsing steroid-dependent and steroid-resistant nephrotic syndrome of childhood: current status and future directions. Pediatr Nephrol. 2005;20:1376–81.
Gulati A, Sinha A, Jordan S, et al. Efficacy and safety of treatment with rituximab for difficult steroid resistant and dependent nephrotic syndrome. Clin J Am Soc Nephrol. 2010;5:2207–12.
Gulati A, Bagga A, Gulati S, Mehta KP, Vijaykumar M, on behalf of the Indian Society of Pediatric Nephrology. Management of children with steroid resistant nephrotic syndrome. Indian Pediatr. 2009;46:35–47.
Benoit G, Machuca E, Antignac C. Hereditary nephrotic syndrome: a systematic approach for genetic testing and a review of associated podocyte gene mutations. Pediatr Nephrol. 2010;25:1621–32.
Hodson EM, Willis NS, Craig JC. Interventions for idiopathic steroid-resistant nephrotic syndrome in children. Cochrane Database Syst Rev. 2010; 11: CD003594.
Colquitt JL, Kirby J, Green C, Cooper K, Trompeter RS. The clinical and cost effectiveness of treatments for children with idiopathic steroid-resistant nephrotic syndrome: a systematic review. Health Technol Assess. 2007;11:1–93.
Choudhry S, Bagga A, Hari P, Sharma S, Kalaivani M, Dinda A. Efficacy and safety of tacrolimus versus cyclosporine in children with steroid-resistant nephrotic syndrome: a randomized controlled trial. Am J Kidney Dis. 2009;53:760–9.
Gipson DS, Trachtman H, Kaskel FJ, et al. Clinical trial of focal segmental glomerulosclerosis in children and young adults. Kidney Int. 2011;80:868–78.
Vasudevan A, Mantan M, Bagga A. Management of edema in nephrotic syndrome. Indian Pediatr. 2004;41:787–95.
Conflict of Interest
None.
Role of Funding Source
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sinha, A., Bagga, A. Nephrotic Syndrome. Indian J Pediatr 79, 1045–1055 (2012). https://doi.org/10.1007/s12098-012-0776-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12098-012-0776-y