
Abstract
This article overviews the pathogenesis and management of idiopathic nephrotic syndrome, a common childhood glomerulopathy. While initial evidence supported an imbalance of T helper responses in patients with nephrotic syndrome, recent studies suggest alterations in both innate and adaptive immune responses, including evidence for impaired T regulatory function. The central role of the podocyte in causing proteinuria is confirmed by the observation of mutations in key podocyte proteins in steroid-resistant nephrotic syndrome and experimental evidence of altered podocyte signaling and cytoskeletal organization, which might be corrected by medications. The outcome and management of idiopathic nephrotic syndrome in children are determined by the response to corticosteroids and the frequency of relapses. While patients with steroid-sensitive nephrotic syndrome have a favorable long-term outcome, almost half of them relapse frequently and are at risk of adverse effects of corticosteroids. Various non-corticosteroid immunosuppressive agents are used to prolong disease remission, but require careful monitoring for potential adverse effects. While calcineurin inhibitors are the choice of therapy for patients with steroid-resistant nephrotic syndrome, long-term management of disease is challenging due to variable response to immunosuppression, therapy-related adverse effects, and high rates of disease progression to end-stage renal disease. Information from recent randomized clinical trials has helped to clarify and improve the standard of care for childhood nephrotic syndrome and underscore the need for well-designed collaborative studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nephrotic syndrome is a common cause of chronic kidney disease, characterized by heavy proteinuria, hypoalbuminemia (serum albumin <2.5 g/dl), hyperlipidemia, and edema [1•]. The large majority (>90 %) is primary (idiopathic); a secondary cause, e.g., amyloidosis, systemic lupus, Henoch Schonlein purpura is seen rarely. Most (>80 %) children with idiopathic nephrotic syndrome show remission following therapy with oral corticosteroids. The prognosis in these cases is favorable, in contrast to patients who do not respond to such treatment, termed ‘steroid resistant.’
This article overviews our current understanding of the pathogenesis of idiopathic nephrotic syndrome, including the pivotal role of podocytes and the immune system. Specific management and outcome of patients with steroid sensitive and resistant nephrotic syndrome are discussed. Details on congenital nephrotic syndrome and on dietary management, immunization, and management of complications are not discussed.
Pathology
Histological studies by the International Society for Kidney Disease in Children (ISKDC) show that over 80 % patients have insignificant glomerular abnormalities on light microscopy (minimal change disease) [2]. While immunofluorescence examination is negative, electron microscopy reveals effacement of podocytes with disruption and disorganization of actin filaments. About 40–70 % patients with steroid-resistant nephrotic syndrome and 5–10 % cases with sensitive nephrotic syndrome have focal segmental glomerulosclerosis (FSGS), with sclerosis involving a segment of the glomerular tuft. While reports from referral centers suggest an increase in the diagnosis of FSGS in children, incidence studies in defined populations are necessary to determine temporal trends [3].
Based on the location of sclerosis, the Columbia Classification distinguishes FSGS into five morphologic variants: tip lesions, cellular variant, perihilar lesions, collapsing FSGS, and FSGS not otherwise specified [4]. While retrospective studies show differences in baseline features and outcomes between these variants [5•], the FSGS clinical trial was the first prospective study with protocol-defined therapies to confirm adverse and favorable renal outcomes in collapsing and tip variants, respectively [6]. Collapsing glomerulopathy, characterized by segmental and/or global collapse of the glomerular capillary tufts and podocyte hypertrophy, is an aggressive form of FSGS that was classically described in HIV-associated nephropathy but is now recognized to have multiple disease associations [7].
About 10–15 % of patients with steroid resistance, especially those above 12–14-year old, may also present with membranoproliferative glomerulonephritis, membranous nephropathy, or IgA nephropathy.
Pathogenesis
The glomerular filtration barrier consists of capillary endothelial fenestrations, glomerular basement membrane (GBM), and interdigitating podocyte foot processes. Recent studies suggest that the podocytes are critical in maintaining the selective filtering function.
Genetics
Pathogenic defects in genes encoding slit diaphragm or cytoskeletal proteins are detected in 80–100 % cases with congenital nephrotic syndrome (onset <3 months age), 50–60 % of infantile-onset steroid resistance, 65–70 % of familial steroid resistance, and 25 % of sporadic steroid-resistant nephrotic syndrome, particularly with use of high-throughput next-generation sequencing [8, 9, 10••]. Mutations in large number of genes are recognized, including those encoding structural elements of the slit diaphragm or the podocyte cytoskeleton (NPHS1, NPHS2, CD2AP, TRCP6, and ACTN4), proteins deposited in the GBM (LAMB2), mitochondrial genes (COQ2), or transcription factors (WT1, LMX1B) (Table 1), with presentation ranging from congenital nephrotic syndrome to disease onset in adulthood.
Homozygous or compound heterozygous mutations in NPHS1 and NPHS2 continue to be the chief causes of non-syndromic early onset nephrotic syndrome [8]. Other genes implicated in childhood onset steroid resistance are those encoding Wilms’ tumor 1 (WT1), laminin beta 2 (LAMB2), phospholipase C epsilon (PLCE1), myosin (MYO1E) [11], cubilin (CUBN) [12], and rhoGDIα (ARHGDIA) [13]. Heterozygous mutations in alpha-actinin 4 (ACTN4), transient-receptor potential cation channel subfamily C member 6 (TRCP6), and inverted formin 2 (INF2) present with later onset of illness and FSGS; INF2 mutations are suspected in patients with concomitant Charcot Marie Tooth disease [14]. The risk of developing FSGS with high-risk haplotypes in MYH9 or APOL1 genes requires to be ascertained across ethnicities [15, 16]. While most patients with inherited forms of steroid resistance do not respond to immunosuppressive agents, partial response to calcineurin inhibitors has been reported [17, 18]. Disease due to genetic defects does not relapse after transplantation, but recurrence is reported in patients with truncating deletions of NPHS1 and occasionally, NPHS2 mutations [19].
Immune Dysfunction
There is considerable evidence of immune dysfunction in patients with steroid-sensitive nephrotic syndrome. It is proposed that altered cell-mediated immunity and T helper type 2 polarization cause the release of an uncharacterized circulating factor that increases glomerular permeability. Evidence favoring Th2 bias includes association with atopy, and elevated plasma levels of IgE, decreased induction of the Th1-specific transcription factor T-bet, and induction of c-maf, a Th2-specific transcription factor [20].
Recent studies suggest that disease activity is associated with an imbalance between T helper 17 cells, upregulated in many autoimmune conditions, and regulatory T cells [21–23]. Deficiency or dysfunction of T regulatory cells [24, 25] may allow activation of effector T cells to secrete factors that mediate glomerular permeability [25] or increase oxidant production by polymorphonuclear leukocytes [26]. Conversely, stimulation of T regulatory cells, demonstrated to cause remission in an experimental model of nephrotic syndrome [27], follows measles [28] as well as B cell depletion with rituximab [26, 29], both of which are reported to induce prolonged remission or eradicate the disease.
The rapidity with which relapses follow infections is suggested to indicate involvement of innate immunity. It is proposed that pathogen-triggered activation of toll-like receptors induces nuclear factor kappa β (NF-κB) signaling or alters its regulatory feedback leading to activation of innate immune responses and/or T cell polarization toward the Th2 phenotype through the proto oncogene c-maf [20]. The beneficial influence of saquinavir, a protease inhibitor that inhibits NF-κB activation in selected patients with steroid resistance, supports the hypothesis [30].
CD80 is normally expressed on antigen presenting cells and engages either CD28 on effector T cells or cytotoxic T-lymphocyte-associated (CTLA)-4 on regulatory T cells [31]. In vitro models of nephrotic range proteinuria show increased podocyte expression of CD80 [31]. A ‘two-hit’ hypothesis proposes that podocyte CD80 expression, initiated through engagement of toll-like receptors 3 or 4 by a virus or cytokine, remains upregulated due to inadequate censoring by regulatory T cells or podocytes [32, 33]. Studies report increased urinary excretion of CD80 in patients with minimal change disease and not in FSGS [34•, 35, 36••]. Finally, increased podocyte CD80 expression and proteinuria in patients with recurrent FSGS or minimal change disease were attenuated by therapy with CTLA-4 agonists, abatacept, and belatacept [37, 38].
Circulating Factors
The soluble mediator hypothesis, supported by the recurrence of nephrotic proteinuria immediately post-transplant in 20–40 % of patients with idiopathic FSGS [39], and the induction of proteinuria and podocyte foot process effacement in rats or increase in vascular permeability in guinea pigs [40] by supernatants from T cells of patients with nephrotic syndrome, is the most widely believed paradigm for disease pathogenesis in FSGS. Proposed circulating factors include interleukin (IL)-13 [41], cardiotrophin-like cytokine-1 [40], tumor necrosis factor α [4], hemopexin [40], and the c-maf-inducing protein, c-mip [42].
The debate on the identity of the elusive circulating factor was stimulated by findings of Wei et al., who described elevated levels of soluble urokinase plasminogen activating receptor (suPAR) in Caucasians with primary and recurrent FSGS, and showed that membrane bound uPAR activated podocyte β3 integrin signaling, foot process effacement, and histology resembling FSGS [43••]. Further studies demonstrated that levels of serum suPAR were elevated in two well-characterized cohorts of children and adults with primary FSGS [44], distinguished FSGS from minimal change disease [43], primary from secondary FSGS [45] and recurrent from non-recurrent disease [43], and declined with therapy with mycophenolate mofetil [46] or plasmapheresis [47]. Recent studies have contested the role of suPAR in distinguishing between FSGS, MCD and other glomerular diseases [48••, 49, 50] and suggest that blood levels relate more closely to reduced glomerular filtration and systemic inflammation than renal histology [48, 51].
Recent studies link a hyposialylated form of podocyte glycoprotein angiopoietin like 4 (ANGPTL4) to proteinuria in experimental models [52] and soluble ANGPTL4 to proteinuria and hypertriglyceridemia through feedback loops [53]. Modification of ANGPTL4 at a key lipoprotein lipase interacting site using recombinant technology [54], and administering oral N-acetyl-d-mannosamine, a naturally occurring precursor of sialic acid [55], reduced proteinuria without inducing hypertriglyceridemia, suggesting that sialylation of ANGPTL4 might be a potential anti-proteinuric intervention.
Serum and urinary levels of several microRNAs (miRNAs), short non-coding RNA molecules that regulate gene expression, are altered in nephrotic syndrome and relate to underlying histology or to degree of tubulointerstitial injury [56]. Blood levels of mir-186 [57], miR-192, and miR-205 might differentiate FSGS from minimal change disease [58]. The precise role of the miRNAs in disease pathogenesis needs to be defined.
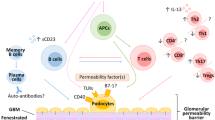
The podocyte is increasingly recognized as the target for anti-proteinuric interventions [59]. Co-incubation of podocytes with glucocorticoids enhances nephrin transport, reduces actin disruption and increases its polymerization, prevents upregulation of miRNA-30, and enhances podocyte recovery [60, 61]. Calcineurin inhibitors stabilize podocytes by changing the expression, distribution, or phosphorylation of nephrin, synaptopodin, cofilin-1, and TRPC6 [62]. Rituximab has been shown to bind to podocyte sphingomyelin phosphodiesterase acid like 3b, SMPDL-3b, preventing disruption of the actin cytoskeleton and podocyte apoptosis by plasma from patients with recurrent FSGS [63]. Figure 1 summarizes prevailing hypotheses regarding the pathogenesis of nephrotic syndrome, including immune dysfunction and permeability factors and their relationship with inherited or acquired defects in podocyte slit diaphragm proteins and the actin cytoskeleton.

Pathogenesis of idiopathic nephrotic syndrome. Schematic view of podocyte foot process demonstrates components of the slit diaphragm complex, formed by nephrin, Neph1, and podocin, and the actin cytoskeleton, which also receives inputs from the basolateral domain of the foot process, containing the α3β1 integrins and α and β dystroglycans. The dominant paradigm in nephrotic syndrome is an imbalance between T helper 1 (Th1) and T helper 2 (Th2) cytokines, and that a cytokine soluble mediator, presumably IL-13, increases glomerular permeability. Other molecules suggested as mediators include soluble urokinase plasminogen activator receptor (suPAR) that activates podocyte β3 integrin signaling to integrin-linked kinase (ILK), circulating proteases that phosphorylate vasodilator stimulated phosphoprotein (VASP), vascular endothelial growth factor, and angiopoietin-like-4 (ANGPTL4). Signaling through nuclear factor kappa B (NF-κB) and toll-like receptor (TLR) mediated pathways may polarize adaptive immune responses toward Th2 cells, or directly increase CD80 expression in podocytes. An imbalance of Th17 and T regulatory responses allows persistent CD80 activation on podocytes, and/or helper responses. MicroRNAs may influence glomerular permeability through changes in gene transcription. Finally, therapeutic agents have direct effects on podocytes; glucocorticoids (G) on gene expression, calcineurin inhibitors (C) on stabilization of synaptopodin (S) or inhibition of TRPC6 channel, binding of rituximab (R) to the SMPDL-3b protein and of abatacept (A) to CD80. Parts of the figure have been used by the authors in two publications [64, 65]. The figure is original and has not been adapted from any other manuscript
Evaluation
Majority of patients with idiopathic nephrotic syndrome have steroid-sensitive illness; the disease course varies with 35–40 % having a single episode or 1–2 relapses, and 55–60 % showing multiple relapses that occur infrequently or frequently [66, 67]. Nephrotic range proteinuria is the presence of 3–4+ protein by dipstick on first morning urine for 3 consecutive days, spot protein/creatinine ratio >2 mg/mg, or protein excretion >40 mg/m2/h. Investigations at the onset include (i) urinalysis, (ii) blood levels of urea, creatinine, albumin, cholesterol, and (iii) complete blood counts. Additional investigations based on clinical suspicion are (i) tuberculin test, chest X-ray (areas with high prevalence of tuberculosis); (ii) C3, anti-streptolysin O (gross or persistent microscopic hematuria); (iii) hepatitis B surface antigen (recent jaundice, raised transaminases); (iv) anti-nuclear antibodies; and (v) urine culture (features of urinary tract infection).
Most children with nephrotic syndrome do not require a kidney biopsy. A biopsy is required at onset if a cause other than minimal change disease is suspected, such as (i) age at onset <1 year, (ii) gross or persistent microscopic hematuria, or low C3; (iii) renal failure not attributable to hypovolemia; (iv) suspected secondary cause; and (v) sustained severe hypertension. While no upper age cutoff is identified, the likelihood of corticosteroid resistance and finding of significant glomerular lesions is higher in older children [68]. A renal biopsy is considered later (i) for steroid resistance and (ii) if therapy with calcineurin inhibitors is planned. The specimen should be examined by light, immunofluorescence and electron microscopy.
The diagnosis of steroid resistance is based on demonstration of non-response (3–4+ proteinuria, edema, or hypoalbuminemia), despite therapy with prednisone in adequate doses for 4–8 weeks [69–71]. Recent recommendations suggest awaiting remission for 6–8 weeks while tapering corticosteroids since a small proportion of patients might show remission, while on alternate-day prednisone the use of pulse steroids to confirm resistance is not recommended [70]. Patients with steroid-resistant nephrotic syndrome require (i) 24-h quantitation of proteinuria, (ii) estimation of glomerular filtration rate, and (iii) renal biopsy. Those patients with the collapsing variant of FSGS are screened for anti-HIV and anti-parvovirus B19 IgM antibodies. Testing for inherited mutations is not recommended due to variable availability and high cost of testing, low prevalence of defects, and unclear association with response to therapy [8, 70]. Screening for genetic mutations should be offered to patients with a family history of similar renal disease, and those presenting in the first 3 months of life (congenital nephrotic syndrome).
Management of Steroid-Sensitive Nephrotic Syndrome
International collaborative efforts by the ISKDC [66] and Arbeitsgemeinschaft für Pädiatrische Nephrologie (APN) [72] have had a crucial role in defining the treatment of nephrotic syndrome. Randomized controlled trials (RCTs) using corticosteroids and non-corticosteroid immunosuppressive agents, summarized in systematic reviews [73, 74], form the bases for recommendations by the Kidney Disease: Improving Global Outcomes (KDIGO) Glomerulonephritis Work Group [68, 75], and organizations such as the Indian Society of Pediatric Nephrology [76] and Canadian Society of Pediatric Nephrology [71] (Table 2). Selected randomized controlled trials on nephrotic syndrome, published in the last 4 year, are summarized in Table 3 [77–86].
Initial Episode
Although the ISKDC proposed that initial prednisone therapy comprise of 4 weeks daily and 4-weeks intermittent therapy [66], refinements were proposed over the last four decades. Ever since an APN-supported randomized trial showed reduced relapse rates on prolonging initial therapy from 8 to 12 weeks [72], experts have suggested that extending therapy to 24 weeks was even better. Results from a meta-analysis showed that, compared to 3 months, therapy for 6 months led to reduced risk of frequent relapses (relative risk, 0.55; 95 % confidence interval, CI 0.39,0.80) and reduced number of annual relapses (mean difference −0.44; 95 % CI −0.82, −0.07) [73]. The analysis suggested an inverse relationship between relapses and duration of initial therapy, such that the relative risk for relapse at 12–24 months would fall by 11 % of baseline relapse rate for every 1 month increase in duration of therapy from 2 to 7 months. Further, while the reduction in risk of relapse appeared to relate primarily to an increase in duration and not the cumulative dose of prednisolone, a meta-analysis of two studies using differing doses over similar duration of therapy suggested reduced risk of relapse with higher dose [73].
Three recently published, well-designed randomized studies contest the above view (Table 3) [77–79]. A multicenter placebo-controlled parallel group trial from Netherlands, on 150 children, showed no differences in the cumulative proportion of children with frequent relapses or any relapse when initial therapy was prolonged from 12 to 24 weeks without increasing the cumulative dose [79]. Similarly, a randomized placebo-controlled blinded trial conducted across 5 centers in north India on 181 patients randomized to receive 6 or 3 months of prednisolone differing in cumulative dose by 739 mg/m2, showed no differences in the frequency of relapses at one year (P = 0.24), hazard ratio for frequent relapses (1.01; 95 % CI 0.61, 1.67; P = 0.96), and proportions with sustained remission or steroid adverse effects [77]. Post hoc subgroup analysis showed that children younger than 3 year might benefit from 6-months therapy with reduced risk for first relapse, but not frequent relapses. An open label multicenter randomized trial from Japan, published simultaneously with the above study, examined the non-inferiority of 2-months to 6-months therapy with prednisone at higher cumulative dose (2240 vs. 3840 mg/m2) in 255 patients, 1–15-year old followed for 2 years [78]. Time to first relapse and frequent relapses were similar between the two groups and the ratio of the number of relapses per person year was 0.94 (95 % CI 0.71, 1.22; P = 0.65). The authors concluded that 2-months’ initial steroid therapy, despite less medication exposure was not inferior to 6-months treatment in affecting the rates of frequent relapses.
Results from these RCTs that enrolled almost 600 patients emphasize that prolongation of initial therapy to 6 months is not useful in modifying the course of the disease, or reducing subsequent needs for corticosteroids and steroid-sparing agents. One study showed that the benefit of extended initial therapy was limited to the period while steroids were being administered [77]. Since the intent of intensive initial therapy is to alter the disease course rather than delay the time to first relapse, lower rates of relapses during steroid tapering do not appear to be a valid outcome. Given the current data and risk of adverse effects due to steroid intake, prolongation of initial therapy beyond 12 weeks [72] is perhaps not required. Results from a British study comparing 2- and 4-months initial corticosteroid therapy [NCT00308321] are awaited.
One retrospective study suggested that prednisone underdosing, where dosing for the initial episode is prescribed according to weight rather than body surface area, increased the likelihood of frequent relapses [87]. While the results suggest that corticosteroids be dosed by body surface area rather than by weight, confirmation from prospective studies is required.
Management of Frequent Relapses
Therapy for relapses comprises of daily prednisolone until remission followed by on alternate days for 4 weeks. There is no evidence that prolonged therapy for a relapse determines the long-term outcome of the illness. Patients with two or more relapses in 6 months or >4 in 1 year are classified as frequent relapsers. Steroid dependence is defined by occurrence of at least two relapses while receiving prednisolone or within 2 weeks of its stoppage. Frequent relapses and steroid dependence are more common in patients with age at onset below 3 year, delayed time to initial remission, brief corticosteroid therapy at disease onset, and short duration of initial remission [66, 67, 88, 89].
Therapy with multiple courses of high-dose prednisolone is associated with significant toxicity, including behavioral problems, cataract, glaucoma, hypertension, avascular hip necrosis, and diabetes. Relapses may be associated with significant complications, including infections, thrombosis, and dyslipidemia. Hence, patients with frequent relapses or steroid dependence are treated with long-term alternate day steroids, or alternative steroid-sparing medications to maintain remission while limiting exposure to steroids. Since very few RCTs have compared the relative efficacy and safety of these strategies, guidelines do not specify the order or specific choices of alternative therapy [68, 74, 76] (Table 2).
Long-Term, Alternate Day Steroids
The dose of prednisolone is tapered to the lowest dose required to maintain the patient in remission without significant adverse effects. A dose of 0.3–0.7 mg/kg given on alternate days for 6–18 months is effective in reducing relapses or maintaining remission in 30–40 % patients. Since relapses are precipitated following minor infections, 3 studies examined the role of short-term (5–7 days) daily administration of corticosteroids in reducing infection-related relapses [80, 90, 91]. Although all studies found an effect of this intervention on relapse rates, one had enrolled a small number of patients [90] and another did not examine long-term benefits [91]. A prospective well-powered RCT showed that daily administration of small dose of prednisolone, during intercurrent infections, independently reduced annual relapse rates by 59 % (rate ratio, 0.41; 95 % CI 0.3, 0.6) without increase in steroid toxicity; 6 patients were required to be treated to prevent occurrence of frequent relapses in one [80]. Based on the above findings, KDIGO currently suggests that the frequency of administration of prednisolone is to be increased from alternate day to daily during episodes of fever with or without upper respiratory tract infection [68]. A British trial (PREDNOS2; ISRCTN10900733) is evaluating the effectiveness of 6-day daily prednisolone during upper respiratory infections in preventing a subsequent relapse in such patients.
Based on the hypothesis that suppression of the hypothalamo-pituitary axis is a risk factor for relapses of nephrotic syndrome, small reports suggest that administration of low dose daily prednisolone may in some patients be more effective in preventing relapses, as compared to administration of corticosteroids on alternate days [92, 93]. While evidence from randomized trials comparing daily low dose with standard alternate-day prednisolone is awaited [CTRI/2012/12/003194], KDIGO guidelines suggest that daily prednisone may be given at the lowest dose to maintain remission without major adverse effects in children with steroid-dependent disease where alternate-day prednisone therapy is not effective [68].
Findings from 2 double blind, placebo-controlled, RCT from the Indian subcontinent suggest that supplementation with zinc at 10–20 mg daily for 6–12 months was associated with reduction in relapses in patients with nephrotic syndrome [94, 95]. Subgroup analysis in one study showed 28 % reduction in relapse rates and a significantly higher likelihood of sustained remission (P = 0.02) in patients with frequent relapses [94]. However, the latter findings need confirmation.
Corticosteroid-Sparing Agents
The additional use of an alternative agent should be considered in patients with (i) prednisolone threshold (for maintaining remission) higher than 1.0 mg/kg on alternate days or (ii) features of corticosteroid toxicity (growth failure, hypertension, cataract) [68, 71, 76]. The agents used are listed below and in Fig. 2 and Table 2. Based on findings of a meta-analysis that mizoribine and azathioprine are no more effective than placebo or prednisone alone in maintaining remission [74], these agents are not recommended for management of frequent relapses or steroid dependence.
Levamisole This is an important steroid-sparing agent in many parts of the world, especially in patients with mild degree of steroid dependency. Levamisole, used in 5 randomized studies at 2–2.5 mg/kg on alternate days for 4–12 months, showed reduction in risk of relapses by 57 % [relative risk, RR 0.43; 95 % CI 0.27,0.68] [74]. The medication is well tolerated with few adverse effects including neutropenia, seizures, cutaneous vasculitis, and hepatotoxicity. Since levamisole does not reduce steroid requirement substantially, patients with high threshold or marked toxicity might not do very well, and should receive more potent agents. While KDIGO guidelines recommend levamisole as a steroid-sparing agent, two RCTs are evaluating its efficacy in comparison to placebo and mycophenolate mofetil [The Netherlands, ISRCTN23853712 and India, CTRI/2012/02/002394, respectively].
Alkylating agents Meta-analysis of 5 RCT, on 134 patients, comparing cyclophosphamide or chlorambucil and prednisone to prednisone alone shows that these agents reduce the risk of relapse at 6–12 months [RR 0.43; 95 % CI 0.31, 0.60] and 12–24 months [RR 0.20; 95 % CI 0.09, 0.46] [74]. Adverse effects are common, including leukopenia, hemorrhagic cystitis, alopecia, nausea, and vomiting, and with prolonged use, gonadal toxicity, and malignancies. While the efficacy of the two agents is similar, therapy with chlorambucil is avoided due to high toxicity, including seizures. The efficacy of IV and oral cyclophosphamide is perhaps similar; the former might be a choice if there are concerns about compliance with oral therapy. Benefits of therapy with cyclophosphamide are better in patients with frequent relapses compared to steroid dependence, and in older (>7 year) than younger patients [68, 71].
Mycophenolate mofetil (MMF) MMF inhibits inosine monophosphate dehydrogenase, limiting T and B lymphocyte proliferation and cytokine gene expression. Over the last decade, several uncontrolled retrospective and prospective studies have reported that MMF (dose 600–1000 mg/m2/day or 20–25 mg/kg/day) has steroid-sparing effects and reduces relapse rates without significant toxicity in patients with frequent relapses or steroid dependence [96, 97]. Side effects are few and include leukopenia, abdominal pain, and diarrhea. A recent multicenter, open label, crossover study in 60 children showed no difference in the proportions in sustained remission during 1-year therapy with cyclosporine (85 %) and MMF (64 %), but the time to first relapse was longer with cyclosporine therapy (Table 3) [81]. Adverse effects were comparable, but therapy with MMF was associated with higher levels of cystatin clearance, estimated GFR, and hemoglobin. The authors concluded that MMF is a useful agent particularly due to the lack of nephrotoxicity. In a small Bayesian trial (n = 24), Baudouin et al. demonstrated that use of MMF in patients with steroid-dependent nephrotic syndrome was associated with significantly reduced risk of relapse (17.6 % at 6 months; 95 % CI 5.4, 35.0) and 75 % reduction in steroid requirement [98]. Studies are examining the relative efficacy of MMF in patients with frequent relapses or steroid dependence [cyclophosphamide NCT01092962; levamisole CTRI/2012/02/002394]. KDIGO suggests the use of MMF, for 12–24 months, in patients with steroid-dependent nephrotic syndrome [68].
Calcineurin inhibitors (CNI) Observational studies show that cyclosporine A and tacrolimus maintain remission and enable steroid sparing in 60–90 % patients with steroid-dependent nephrotic syndrome who fail treatment with alkylating agents [99, 100]. The efficacy of these agents relative to placebo or to each other has not been examined. While 2 RCT found that efficacy of cyclosporine was comparable to alkylating agents [RR 0.91; 95 % CI 0.55, 1.48], patients relapsed when therapy with cyclosporine was discontinued [68, 74]. Information on tacrolimus use is limited to non-randomized studies, but the agent is preferred due to lack of cosmetic adverse effects (hirsutism, gum hyperplasia) [100]. With either agent, therapy requires to be administered for at least 12 months with monitoring of drug levels (target trough 4–7 ng/ml for tacrolimus and 80–150 ng/ml for cyclosporine) [68]. Chief adverse effects include acute and chronic nephrotoxicity (with both CNI), hypertension and hyperlipidemia (chiefly with cyclosporine), and hyperglycemia (chiefly with tacrolimus).
Rituximab Multiple case series, summarized in a recent review [64], have reported benefits of B cell depletion with rituximab in patients with steroid-dependent nephrotic syndrome who fail treatment with immunosuppressive agents. Treatment with one or more doses of rituximab is reported to result in remission lasting 3–12 months, with 25–83 % patients showing sustained remission. A large series, on 101 patients with steroid dependence, found that therapy with rituximab was associated with 81.8 % reduction in relapse rates [101•], comparable to 62–95 % in previous reports [64].
Controlled studies on the efficacy of rituximab are limited to three recent studies, one showing superior efficacy of rituximab when compared to placebo [82], one showing non-inferiority of rituximab, low doses of CNI and prednisone to CNI and prednisone alone [83] (Table 3), and a single limb study showing steroid sparing with improved growth velocity in patients with difficult steroid dependence [102•]. A retrospective case control study showed that 2 doses of rituximab were as effective as 12-months’ treatment with tacrolimus [103]. Current clinical practice guidelines-suggest that treatment with rituximab is to be considered in patients with steroid-dependent nephrotic syndrome who fail to respond to conventional agents, including CNI, and/or have serious adverse effects of therapy [68, 71]. However, reports of efficacy and safety of rituximab are likely to result in increased use of this agent in preference to CNI and/or MMF.
Management of Steroid-Resistant Nephrotic Syndrome
Therapy of patients with steroid-resistant nephrotic syndrome is difficult, with variable response to immunosuppression, adverse effects of prolonged therapy, and risk of progressive renal damage. Table 4 summarizes recent guidelines on the evaluation and management of patients with steroid resistance [69–71]. The aim of therapy is to induce and maintain remission of proteinuria, while avoiding medication-related adverse effects. Definitions of response to therapy are listed in Table 4. Patients are monitored closely until response to therapy is demonstrated, and then every 2–3 months [69, 75]. While complete remission is associated with high rates of survival free of end-stage renal disease (ESRD) [104], there are data that partial remission is associated with satisfactory outcomes, compared to those with non-response [105].
Immunosuppressive Protocols
Most regimens use a combination of an immunosuppressive agent with prednisolone given on alternate days, and an angiotensin converting enzyme inhibitor (ACE-I) or angiotensin-receptor blocker (ARB) (Table 4) [69–71, 106].
CNI (Cyclosporine, Tacrolimus)
Following evidence from RCTs demonstrating superior efficacy for cyclosporine (5 studies) and tacrolimus (2 studies) in inducing and maintaining remission in 46–100 % patients, therapy with CNI is considered ‘first line’ for patients with steroid resistance [70, 71, 106]. While a randomized multicenter trial in the United States enrolling adults and children with FSGS showed relatively low rates of remission with either cyclosporine (45.8 %) or the combination of mycophenolate mofetil with pulse dexamethasone (33 %) (P = 0.11) [832], a study from India showed high rates of complete or partial remission using tacrolimus (82.5 %) as compared to IV cyclophosphamide (45.9 %) in children with FSGS or MCD (P < 0.001) (Table 3) [85]. The reasons for variable outcomes may relate to differences in definitions of steroid resistance, histology, or prevalence of inherited defects. Cyclosporine and tacrolimus showed comparable efficacy and low rates of adverse effects in another RCT [107].
Examination of renal histology is advised in patients receiving prolonged therapy (2-3 years) with CNI; specific features of CNI nephrotoxicity include nodular arteriolar hyalinosis and striped interstitial fibrosis [69]. Risk factors for nephrotoxicity include prolonged duration of therapy (>2–3 years) and persistent heavy proteinuria (>1–3 months) [108]. The decision to continue calcineurin inhibitors therapy should be reviewed in patients showing non-response to proteinuria despite 4-6 months of therapy.
Consensus is lacking on the optimal duration of treatment with CNI. While guidelines suggest continuing therapy for 12 months or longer in patients that show complete or partial remission (2C) [69–71], in practice, the agent is continued for 2–3 years, followed by one of the following options: (i) taper to the lowest effective dose, and continue for another 1–2 years; (ii) exclude nephrotoxicity on renal histology and then continue therapy; (iii) switch treatment to a less toxic agent, e.g., MMF or rituximab. An RCT is evaluating the non-inferiority of switching therapy at 6 months to MMF, after inducing remission with tacrolimus [CTRI/2012/05/002636].
Cyclophosphamide
Meta-analyses demonstrate that the proportions of patients with remission are similar between patients treated with oral cyclophosphamide and prednisone compared to those receiving prednisone alone [RR 1.06; 95 % CI 0.61, 1.87] [106]. The efficacy of IV cyclophosphamide (administered monthly at 500 mg/m2 for 6 pulses) was not superior as compared to oral cyclophosphamide with [RR 3.13; 95 % CI 0.81,12.06] or without IV dexamethasone [RR 1.13; 95 % CI 0.65,1.96] in one study each [106]. Contrary to the results of a recent RCT in children [84], tacrolimus and IV cyclophosphamide had similar rates of remission (complete or partial) at 6 months (66.7 vs. 55.6 %; P = 0.77) and 12 months (77.3 vs. 66.7 %; P = 0.97) in 33 Chinese adults with steroid-dependent or steroid-resistant FSGS [109]. Given the overall limited efficacy in pediatric patients and risks of significant toxicity, KDIGO and Canadian guidelines suggest not using cyclophosphamide for patients with steroid resistance [70, 71]. However, the easy availability and relatively low cost of IV cyclophosphamide make it a useful option in resource-limited settings [69].
Pulse Corticosteroids with Oral Cyclophosphamide
Pulses of IV methylprednisolone or dexamethasone have been used in combination with oral cyclophosphamide with modest efficacy and high risk of steroid toxicity; patients are at risk of systemic infections, hypertension, and electrolyte abnormalities [106]. Combination of the above agents is no longer recommended for management of steroid resistance [69–71].
Mycophenolate Mofetil
MMF has been tried with limited success in patients with steroid resistance, including in a recent randomized trial [84]. Its use may be more beneficial in patients who have achieved remission with CNI, and may help mitigate CNI dependence and toxicity [69, 71, 110].
Rituximab
Despite initial interest [111], the efficacy of rituximab in inducing remission in patients with steroid and CNI-resistant nephrotic syndrome appears limited [64]. Experience on treatment with rituximab in various series, summarized in a recent review [64], shows that the agent induces complete remission in 0–27.3 % and partial remission in 21.2–37.5 % patients at 4–8 weeks. A randomized controlled trial on 31 children with steroid and CNI-resistant nephrotic syndrome failed to show benefits of additional rituximab therapy in ameliorating proteinuria at 3 and 6 months (Table 3) [86]. A review of experience at our center in 58 patients with steroid and CNI resistance confirms the limited efficacy, with complete and partial remission in 12.1 and 17.2 % patients, respectively [101]. Similar to previous findings [64], response to rituximab was better in patients with prior response to CNI and unsatisfactory in those with FSGS. Therapy with rituximab may be useful in maintaining remission in patients with steroid resistance that shows a relapsing illness and CNI dependence. Treatment with this agent was associated with 71 % reduction in relapse rates, with withdrawal of CNI in 95 % and corticosteroids in 72 % patients with CNI-dependent steroid resistance [101].
Other Agents
KDIGO suggests the use of MMF (2D), high-dose steroids (2D), or combination of CNI and MMF (2D) in patients with steroid and CNI resistance [70]. Other therapies that have shown promise, in anecdotal reports, include the combination of cyclosporine and mycophenolate mofetil [112], abatacept [37], lipid column apheresis [113], galactose [114], the protease inhibitor saquinavir [30], TNF blockade with etanercept [115], and ACTH gel [116]. These agents require evaluation in larger prospective studies.
Angiotensin-converting enzyme (ACE) inhibitor or angiotensin-receptor blockers Therapy with ACE inhibitors (enalapril 0.3-0.6 mg/kg/day, ramipril 6 mg/m2/day) is associated with decrease in proteinuria and control of hypertension. Adverse effects include dry cough, hyperkalemia, and decline in renal function; therapy is discontinued if hyperkalemia develops or estimated GFR < 30 ml/min/1.73 m2. Angiotensin-receptor blockers (e.g., losartan, valsartan) may be used in case of persistent dry cough with ACE inhibitors, or as add-on therapy for better anti-proteinuric effect. A recent RCT including 268 children demonstrated that losartan and enalapril reduce proteinuria comparably in children (reduction from baseline by 30.0 and 40.5 %, respectively) [117]. While there are limited studies on the efficacy of ACE inhibitor and ARB combination in children, meta-analyses of studies in adult patients caution against significant adverse events (hyperkalemia, hypotension, nephrotoxicity) associated with dual blockade of the renin angiotensin aldosterone system (RAAS) [118••].
Vitamin D A systematic review of 6 randomized controlled studies in 688 adult patients showed that vitamin D analogs (paricalcitol, calcitriol) reduce residual proteinuria [weighed mean difference -16 (95 % CI -13, -18) % vs. 6 (95 % CI 0, 12) %; P < 0.0001] with high odds of achieving ≥ 15 % proteinuria reduction from baseline [OR, 2.72; 95 % CI 1.82, 4.07; P < 0.001] [119•]. Therapy may be associated with risk of hypercalcemia [RR 4.78; 95 % CI 2.20, 10.37] [120] and there is no evidence to suggest that the intervention retards the decline of renal function [119•, 120].
Therapeutic options available for patients with steroid-dependent and steroid-resistant nephrotic syndrome are summarized in Fig. 2.

Immunosuppressive agents in nephrotic syndrome. The agents listed for frequent relapses or steroid dependence are usually recommended in a successive order from top to bottom. Agents marked with an asterisk (*) are preferred in patients with significant steroid toxicity (cataracts, severe stunting or obesity) or if disease relapses are associated with life-threatening complications (thrombosis, severe infections). Patients with steroid resistance should receive treatment with a calcineurin inhibitor along with an angiotensin converting enzyme inhibitor (ACEI) or angiotensin-receptor blocker and tapering doses of prednisone. Since response to other immunosuppressive strategies is less satisfactory and non-response to CNI is associated with adverse outcomes, further immunosuppression should be considered only following counseling regarding efficacy, safety, and costs of various options, including withdrawing immunosuppression
Outcomes
Most patients with steroid-sensitive nephrotic syndrome show highly satisfactory renal outcomes. While morbidity due to infections has declined with rapid diagnosis and use of vaccines, toxicities associated with repeated course of corticosteroids remain a major concern in managing patients with frequent relapses or steroid dependence. Follow up of the initial ISKDC cohort revealed that almost 80 % patients were in sustained remission at 8 years from diagnosis [66]; however, other series suggest that 27–42 % patients continue to relapse into adulthood [121, 122]. Ten year follow up of patients who received cyclosporine for frequent relapses in a randomized study showed that 17.4 and 50 % continued to suffer infrequent or frequent relapses, respectively, into adulthood [123•]. Relapses recurred in adulthood in 16.4 % patients, chiefly among patients with frequent relapses [124].
Outcomes in patients with steroid resistance are less satisfactory. Patients with minimal change disease demonstrate higher rates of remission and better prognosis than those with FSGS; however, the chief factor predicting renal outcome is the response of proteinuria to therapy rather than histology. Renal survival varies from 71.5 % [125] to 94.3 % [126] at 5 years. Resistance to calcineurin inhibitors and FSGS on histology predicts adverse outcomes. A recent study on 29 patients with late steroid resistance followed for 85 ± 47 months reported that 66 % patients were in complete or partial remission and <10 % showed end-stage renal disease (ESRD) [127].
Recurrence of FSGS After Renal Transplantation
Almost 30 % of patients with idiopathic FSGS that undergo renal transplantation develop recurrence of FSGS in the first allograft, leading to increased risk of delayed graft function and high rates of graft loss (30–50 % at 5 year) [39]. Recurrence of proteinuria usually occurs within hours to days after the transplant, and is characterized by progressive hypoalbuminemia and foot process effacement on electron microscopy. After the loss of the first allograft, the risk of recurrence of FSGS in subsequent kidney transplants is 80–100 %. Features associated with FSGS recurrence include (i) rapid progression to end-stage renal disease (within 3 years from onset), (ii) disease onset in childhood (<15 year), (iii) mesangial proliferation on renal histopathology, (iv) non-genetic forms of FSGS, (v) nephrectomy of native kidneys prior to transplant, (vi) white ethnicity, and (vii) delayed rather than initial resistance [128, 129]. Disease recurrence is attributed to circulating permeability factors, with studies suggesting a role for soluble urokinase receptor (suPAR) and increased TNF-α activity [43, 115]. A panel of antibodies against 7 glomerular antigens (CD40, PTPRO, CGB5, FAS, P2RY11, SNRPB2, and APOL2) is reported to predict post-transplant FSGS recurrence with 92 % accuracy [129].
Pre-transplant plasmapheresis is the most widely used strategy to decrease the risk of recurrence of FSGS in kidney transplant recipients [130]. A recent study suggests that peri-transplant administration of rituximab prevents recurrence by stabilizing the podocyte cytoskeleton [63]. While there is no consensus on optimal therapy of patients with recurrent FSGS, options include (i) intensive plasmapheresis [131]; (ii) rituximab (375 mg/m2/week for 2–4 weeks) [132•]; (iii) aggressive immunosuppression, including administration of high-dose cyclosporine, oral cyclophosphamide (2–2.5 mg/kg/day for 3 months) instead of MMF, and use of IV immunoglobulin (500 mg/kg/dose once a week); and (iv) ACE inhibition.
Summary
Nephrotic syndrome is an important chronic disease in childhood, characterized by steroid-sensitive illness in most patients. Research on pathogenesis has emphasized the importance of T-lymphocyte dysregulation and vascular permeability factors that might alter podocyte function and glomerular permselectivity. Mutations in genes that encode important podocyte proteins and therapeutic targets within the podocytes have been identified. However, a hypothesis unifying available evidence on pathogenesis is yet to be proposed. A significant proportion of patients have difficult disease course, characterized by frequent relapses, steroid dependence, or steroid resistance, requiring therapy with alternative immunosuppressive agents. Clinical studies support the use of levamisole, cyclophosphamide, mycophenolate mofetil, calcineurin inhibitors, and rituximab in patients with frequent relapses or steroid dependence. The management of steroid-resistant nephrotic syndrome is difficult, and patients failing to achieve remission show progressive renal damage. Calcineurin inhibitors induce remission in the majority of patients but carry risk of nephrotoxicity with prolonged administration. Prospective trials in patients with frequent relapses, steroid dependence, and resistance are the bases of current international guidelines and ongoing studies are necessary to identify effective and safe therapies.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
• El Bakkali L, Rodrigues Pereira R, Kuik DJ, Ket JC, van Wijk JA. Nephrotic syndrome in The Netherlands: a population-based cohort study and a review of the literature. Pediatr Nephrol. 2011;26:1241–6. This prospective study, conducted in the Netherlands during 2003–2006, determined the incidence of nephrotic syndrome to be 1.52:100, 000 children/year, and found it comparable to disease incidences across over the world.
Churg J, Habib R, White RH. Pathology of the nephrotic syndrome in children. A report for the International Study of Kidney Disease in Children. Lancet. 1970;760(i):1299–302.
Borges FF, Shiraichi L, da Silva MP, Nishimoto EI, Nogueira PC. Is focal segmental glomerulosclerosis increasing in patients with nephrotic syndrome? Pediatr Nephrol. 2007;22:1309–13.
D’Agati V, Fogo A, Bruijn J, Jennette J. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am J Kidney Dis. 2004;43:368–82.
• Stokes MB, D’Agati VD. Morphologic variants of focal segmental glomerulosclerosis and their significance. Adv Chronic Kidney Dis. 2014;21:400–7. This review discusses the subtypes of focal segmental glomerulosclerosis proposed in the Columbia Classification in 2004 and the utility of the classification demonstrated in various retrospective studies that show differences between subtypes in their clinical presentation and final outcomes.
D’Agati VD, Alster JM, Jennette JC, Thomas DB, Pullman J, Savino DA, Cohen AH, Gipson DS, Gassman JJ, Radeva MK, Moxey-Mims MM, Friedman AL, Kaskel FJ, Trachtman H, Alpers CE, Fogo AB, Greene TH, Nast CC. Association of histologic variants in FSGS clinical trial with presenting features and outcomes. Clin J Am Soc Nephrol. 2013;8:399–406.
Mubarak M. Collapsing focal segmental glomerulosclerosis: current concepts. World J Nephrol. 2012;1:35–42.
Brown EJ, Pollak MR, Barua M. Genetic testing for nephrotic syndrome and FSGS in the era of next-generation sequencing. Kidney Int. 2014;85:1030–8.
Santin S, Bullich G, Tazon-Vega B, et al. Clinical utility of genetic testing in children and adults with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2011;6:1139–48.
•• McCarthy HJ, Bierzynska A, Wherlock M, Ognjanovic M, Kerecuk L, Hegde S, Feather S, Gilbert RD, Krischock L, Jones C, Sinha MD, Webb NJ, Christian M, Williams MM, Marks S, Koziell A, Welsh GI, Saleem MA; RADAR the UK SRNS Study Group. Simultaneous sequencing of 24 genes associated with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2013;8:637–48. The investigators used next generation sequencing analysis to screen 446 genes for mutations in 36 consecutive pediatric patients with steroid-resistant nephrotic syndrome enrolled in the United Kingdom renal registry. Analysis revealed novel and known disease associations in 19% patients, suggesting that comprehensive genetic evaluation using next generation sequencing is useful but requires evaluating more that the 24 known genes associated with childhood onset steroid resistance.
Mele C, Iatropoulos P, Donadelli R, et al. MYO1E mutations and childhood familial focal segmental glomerulosclerosis. N Engl J Med. 2011;365:295–306.
Ovunc B, Otto EA, Vega-Warner V, et al. Exome sequencing reveals cubilin mutation as a single-gene cause of proteinuria. J Am Soc Nephrol. 2011;22:1815–20.
Gupta IR, Baldwin C, Auguste D, et al. ARHGDIA: a novel gene implicated in nephrotic syndrome. J Med Genet. 2013;50:330–8.
Boyer O, Nevo F, Plaisier E, et al. INF2 mutations in Charcot–Marie–Tooth disease with glomerulopathy. N Engl J Med. 2011;365:2377–88.
Genovese G, Tonna SJ, Knob AU, et al. A risk allele for focal segmental glomerulosclerosis in African Americans is located within a region containing APOL1 and MYH9. Kidney Int. 2010;78:698–704.
Colares VS, Titan SM, Pereira Ada C, Malafronte P, Cardena MM, Santos S, Santos PC, Fridman C, Barros RT, Woronik V. MYH9 and APOL1 gene polymorphisms and the risk of CKD in patients with lupus nephritis from an admixture population. PLoS ONE. 2014;9:e87716.
Buscher AK, Kranz B, Buscher R, et al. Immunosuppression and renal outcome in congenital and pediatric steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2010;5:2075–84.
Sinha A, Sharma S, Gulati A, Sharma A, Agarwala S, Hari P, Bagga A. Frasier syndrome: early gonadoblastoma and cyclosporine responsiveness. Pediatr Nephrol. 2010;25:2171–4.
Holmberg C, Jalanko H. Congenital nephrotic syndrome and recurrence of proteinuria after renal transplantation. Pediatr Nephrol. 2014. doi:10.1007/s00467-014-2781-z.
Zhang SY, Audard V, Fan Q, Pawlak A, Lang P, Sahali D. Immunopathogenesis of idiopathic nephrotic syndrome. Contrib Nephrol (Basel, Karger). 2011;169:94–106.
Liu LL, Qin Y, Cai JF, Wang HY, Tao JL, Li H, Chen LM, Li MX, Li XM, Li XW. Th17/Treg imbalance in adult patients with minimal change nephrotic syndrome. Clin Immunol. 2011;139:314–20.
Wang L, Li Q, Wang LJ, Li X. Level of Th17 cell and CD4(+);CD25(+); Foxp3(+); regulatory T cell in peripheral blood mononuclear cells of primary nephrotic syndrome in children. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2010;26:783–6.
Shao XS, Yang XQ, Zhao XD, Li Q, Xie YY, Wang XG, Wang M, Zhang W. The prevalence of Th17 cells and FOXP3 regulate T cells (Treg) in children with primary nephrotic syndrome. Pediatr Nephrol. 2009;24:1683–90.
Luo XD, Liu QF, Zhang Y, Sun J, Wang GB, Fan ZP, Yi ZS, Ling YW, Wei YQ, Liu XL, Xu B. Nephrotic syndrome after allogeneic hematopoietic stem cell transplantation: etiology and pathogenesis. Blood Cells Mol Dis. 2011;46:182–7.
Araya C, Diaz L, Wasserfall C, Atkinson M, Mu W, Johnson R, Garin E. T regulatory cell function in idiopathic minimal lesion nephrotic syndrome. Pediatr Nephrol. 2009;24:1691–8.
Bertelli R, Trivelli A, Magnasco A, Cioni M, Bodria M, Carrea A, Montobbio G, Barbano G, Ghiggeri GM. Failure of regulation results in an amplified oxidation burst by neutrophils in children with primary nephrotic syndrome. Clin Exp Immunol. 2010;161:151–8.
Le Berre L, Bruneau S, Naulet J, Renaudin K, Buzelin F, Usal C, Smit H, Condamine T, Soulillou JP, Dantal J. Induction of T regulatory cells attenuates idiopathic nephrotic syndrome. J Am Soc Nephrol. 2009;20:57–67.
Sellin CI, Jégou JF, Renneson J, Druelle J, Wild TF, Marie JC, et al. Interplay between virus-specific effector response and Foxp3 regulatory T cells in measles virus immunopathogenesis. PLoS ONE. 2009;4:e4948.
Zhao Y, Lutalo PM, Thomas JE, Sangle S, Choong LM, Tyler JR, Tree T, Spencer J, D’Cruz DP. Circulating T follicular helper cell and regulatory T cell frequencies are influenced by B cell depletion in patients with granulomatosis with polyangiitis. Rheumatology (Oxford). 2014;53:621–30.
Coppo R, Camilla R, Porcellini MG, Peruzzi L, Gianoglio B, Amore A, Daprà V, Loiacono E, Fonsato V, Dal Canton A, Esposito C, Esposito P, Tovo PA. Saquinavir in steroid-dependent and -resistant nephrotic syndrome: a pilot study. Nephrol Dial Transplant. 2012;27:1902–10.
Ishimoto T, Cara-Fuentes G, Wang H, Shimada M, Wasserfall CH, Winter WE, Rivard CJ, Araya CE, Saleem MA, Mathieson PW, Johnson RJ, Garin EH. Serum from minimal change patients in relapse increases CD80 expression in cultured podocytes. Pediatr Nephrol. 2013;28:1803–12.
Ishimoto T, Shimada M, Gabriela G, Kosugi T, Sato W, Lee PY, Lanaspa MA, Rivard C, Maruyama S, Garin EH, Johnson RJ. Toll-like receptor 3 ligand, polyIC, induces proteinuria and glomerular CD80, and increases urinary CD80 in mice. Nephrol Dial Transplant. 2013;28:1439–46.
Shimada M, Araya C, Rivard C, Ishimoto T, Johnson RJ, Garin EH. Minimal change disease: a “two-hit” podocyte immune disorder? Pediatr Nephrol. 2011;26:645–9.
• Ling C, Liu X, Shen Y, Chen Z, Fan J, Jiang Y, Meng Q. Urinary CD80 levels as a diagnostic biomarker of minimal change disease. Pediatr Nephrol. 2014. doi:10.1007/s00467-014-2915-3. Estimation of urinary excretion of CD80 in 94 patients with minimal change disease, focal segmental glomerulosclerosis and other glomerular diseases and 71 healthy volunteers, suggested that this non-invasive biomarker discriminates well between patients with minimal change disease and other conditions. And the authors propose a cutoff of 328.98 ng/g creatinine as providing an accurate prediction of histology as minimal change disease.
Garin EH, Diaz LN, Mu W, Wasserfall C, Araya C, Segal M, Johnson RJ. Urinary CD80 excretion increases in idiopathic minimal change disease. J Am Soc Nephrol. 2009;20:260–6.
•• Garin EH, Mu W, Arthur JM, Rivard CJ, Araya CE, Shimada M, Johnson RJ. Urinary CD80 is elevated in minimal change disease but not in focal segmental glomerulosclerosis. Kidney Int. 2010;78:296–302. The authors reported significantly higher levels of CD80 in the urine samples of subjects with minimal change disease than in those with focal segmental glomerulosclerosis, patients with minimal change disease in remission, and patients with other glomerulopathies. They also demonstrated that increased excretion of CD80 in urine was associated with positive staining for CD80 on immunohistochemical staining.
Yu CC, Fornoni A, Weins A, Hakroush S, Maiguel D, Sageshima J, Chen L, Ciancio G, Faridi MH, Behr D, Campbell KN, Chang JM, Chen HC, Oh J, Faul C, Arnaout MA, Fiorina P, Gupta V, Greka A, Burke GW III, Mundel P. Abatacept in B7-1–positive proteinuric kidney disease. N Engl J Med. 2013;369:2416–23.
Garin EH, Reiser J, Cara-Fuentes G, Wei C, Matar D, Wang H, Alachkar N, Johnson RJ. CTLA4-IgG1 therapy in minimal change disease and focal segmental glomerulosclerosis. Pediatr Nephrol. 2014. doi:10.1007/s00467-014-2957-6.
Leca N. Focal segmental glomerulosclerosis recurrence in the renal allograft. Adv Chronic Kidney Dis. 2014;2:448–52.
McCarthy ET, Sharma M, Savin VJ. Circulating permeability factor in idiopathic nephrotic syndrome and focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2010;5:2115–21.
Lai KW, Wei CL, Tan LK, Tan PH, Chiang GSC, Lee CGL, et al. Overexpression of interleukin-13 induces minimal-change-like nephropathy in rats. J Am Soc Nephrol. 2007;18:1476–85.
Audard V, Zhang SY, Copie-Bergman C, Rucker-Martin C, Ory V, Candelier M, Baia M, Lang P, Pawlak A, Sahali D. Occurrence of minimal change nephrotic syndrome in classical Hodgkin lymphoma is closely related to the induction of c-mip in Hodgkin-Reed Sternberg cells and podocytes. Blood. 2010;115:3756–62.
•• Wei C, El Hindi S, Li J, et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat Med. 2011;17:952–60. Levels of soluble urokinase plasminogen activating receptor (suPAR) were demonstrated to be elevated in a majority of patients with focal segmental glomerulosclerosis but not in other glomerular diseases, were markedly increased in patients with recurrent than non-recurrent FSGS, and a value above 3000 pg/ml was useful in distinguishing patients with active disease compared to other conditions. Further, experimental evidence confirmed circulating suPAR induces foot process effacement through activation of podocyte β3 integrin. This work raised hopes that the putative circulating factor in patients with FSGS had been found and may be amenable to therapeutic interventions.
Wei C, Trachtman H, Li J, et al. Circulating suPAR in two cohorts of primary FSGS. J Am Soc Nephrol. 2012;23:2051–9.
Segarra A, Jatem E, Quiles MT, Arbós MA, Ostos H, Valtierra N, Carnicer C, Salcedo MT. Value of soluble urokinase receptor serum levels in the differential diagnosis between idiopathic and secondary focal segmental glomerulosclerosis. Nefrologia. 2014;34:53–61.
Gellermann J, Schaefer F, Querfeld U. Serum suPAR levels are modulated by immunosuppressive therapy of minimal change nephrotic syndrome. Pediatr Nephrol. 2014. doi:10.1007/s00467-014-2913-5.
Morath C, Wei C, Macher-Goeppinger S, Schwenger V, Zeier M, Reiser J. Management of severe recurrent focal segmental glomerulosclerosis through circulating soluble urokinase receptor modification. Am J Ther. 2013;20:226–9.
•• Sinha A, Bajpai J, Saini S, Bhatia D, Gupta A, Puraswani M, Dinda AK, Agarwal SK, Sopory S, Pandey RM, Hari P, Bagga A. Serum-soluble urokinase receptor levels do not distinguish focal segmental glomerulosclerosis from other causes of nephrotic syndrome in children. Kidney Int. 2014;85:649–58. Findings of this study, including 469 pediatric patients with nephrotic syndrome and other glomerular diseases and 83 controls, showed that levels of serum suPAR were similar in patients with steroid resistant and sensitive nephrotic syndrome, did not vary between disease relapse and remission, and failed to discriminate FSGS from minimal change disease. Further, suPAR levels correlated inversely with eGFR and directly with C-reactive protein.
Bock ME, Price HE, Gallon L, Langman CB. Serum soluble urokinase-type plasminogen activator receptor levels and idiopathic FSGS in children: a single-center report. Clin J Am Soc Nephrol. 2013;8:1304–11.
Meijers B, Maas RJ, Sprangers B, Claes K, Poesen R, Bammens B, Naesens M, Deegens JK, Dietrich R, Storr M, Wetzels JF, Evenepoel P, Kuypers D. The soluble urokinase receptor is not a clinical marker for focal segmental glomerulosclerosis. Kidney Int. 2014;85:636–40.
Harita Y, Ishizuka K, Tanego A, Sugawara N, Chikamoto H, Akioka Y, Tsurumi H, Miura K, Gotoh Y, Tsujita M, Yamamoto T, Horike K, Takeda A, Oka A, Igarashi T, Hattori M. Decreased glomerular filtration as the primary factor of elevated circulating suPAR levels in focal segmental glomerulosclerosis. Pediatr Nephrol. 2014;29:1553–60.
Clement LC, Avila-Casado C, Macé C, Soria E, Bakker WW, Kersten S, Chugh SS. Podocyte-secreted angiopoietin-like-4 mediates proteinuria in glucocorticoid-sensitive nephrotic syndrome. Nat Med. 2011;17:117–22.
Clement LC, Macé C, Avila-Casado C, Joles JA, Kersten S, Chugh SS. Circulating angiopoietin-like 4 links proteinuria with hypertriglyceridemia in nephrotic syndrome. Nat Med. 2014;20:37–46.
Kirk R. Nephrotic syndrome: negative feedback loop reveals novel potential therapy. Nat Rev Nephrol. 2014;10:63.
Chugh SS, Macé C, Clement LC, Del Nogal Avila M, Marshall CB. Angiopoietin-like 4 based therapeutics for proteinuria and kidney disease. Front Pharmacol. 2014;5:23.
Luo Y, Wang C, Chen X, Zhong T, Cai X, Chen S, Shi Y, Hu J, Guan X, Xia Z, Wang J, Zen K, Zhang CY, Zhang C. Increased serum and urinary microRNAs in children with idiopathic nephrotic syndrome. Clin Chem. 2013;59:658–66.
Zhang C, Zhang W, Chen HM, Liu C, Wu J, Shi S, Liu ZH. Plasma microRNA-186 and proteinuria in focal segmental glomerulosclerosis. Am J Kidney Dis. 2014. doi:10.1053/j.ajkd.2014.07.013.
Cai X, Xia Z, Zhang C, Luo Y, Gao Y, Fan Z, Liu M, Zhang Y. Serum microRNAs levels in primary focal segmental glomerulosclerosis. Pediatr Nephrol. 2013;28:1797–801.
Coppo R. Different targets for treating focal segmental glomerular sclerosis. Contrib Nephrol. 2013;181:84–90.
Yu-Shengyou Li Y. Dexamethasone inhibits podocyte apoptosis by stabilizing the PI3K/Akt signal pathway. Biomed Res Int. 2013;2013:326986.
Wu J, Zheng C, Fan Y, Zeng C, Chen Z, Qin W, Zhang C, Zhang W, Wang X, Zhu X, Zhang M, Zen K, Liu Z. Downregulation of microRNA-30 facilitates podocyte injury and is prevented by glucocorticoids. J Am Soc Nephrol. 2014;25:92–104.
Li X, Zhang X, Li X, Wang X, Wang S, Ding J. Cyclosporine A protects podocytes via stabilization of cofilin-1 expression in the unphosphorylated state. Exp Biol Med (Maywood). 2014;239:922–36.
Fornoni A, Sageshima J, Wei C, Merscher-Gomez S, Aguillon-Prada R, Jauregui AN, Li J, Mattiazzi A, Ciancio G, Chen L, Zilleruelo G, Abitbol C, Chandar J, Seeherunvong W, Ricordi C, Ikehata M, Rastaldi MP, Reiser J, Burke GW 3rd. Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci Transl Med. 2011;3:85ra46.
Sinha A, Bagga A. Rituximab therapy in nephrotic syndrome: implications for patients’ management. Nat Rev Nephrol. 2013;9:154–69.
Sinha A, Bagga A. Nephrotic syndrome. Indian J Pediatr. 2012;79:1045–55.
Tarshish P, Tobin JN, Bernstein J, et al. Prognostic significance of the early course of minimal change nephrotic syndrome: report of the International Study of Kidney Disease in Children. J Am Soc Nephrol. 1997;8:769–76.
Sinha A, Hari P, Sharma PK, et al. Disease course in steroid sensitive nephrotic syndrome. Indian Pediatr. 2012;49:881–7.
Kidney Disease Improving Global Outcomes (KDIGO) Workgroup on clinical practice guideline for glomerulonephritis. Steroid-sensitive nephrotic syndrome in children. Kidney Int Suppl. 2012;2:163–71.
Indian Society of Pediatric Nephrology, Gulati A, Bagga A, Gulati S, Mehta KP, Vijayakumar M. Management of steroid resistant nephrotic syndrome. Indian Pediatr. 2009;46:35–47.
Kidney Disease Improving Global Outcomes (KDIGO) Glomerulonephritis Work Group. KDIGO Clinical Practice Guideline for Glomerulonephritis. Steroid-resistant nephrotic syndrome in children. Kidney Int. 2012;2:172–6.
Samuel S, Bitzan M, Zappitelli M, et al. Canadian Society of Nephrology Commentary on the 2012 KDIGO clinical practice guideline for glomerulonephritis: management of nephrotic syndrome in children. Am J Kidney Dis. 2014;63:354–62.
Ehrich JH, Brodehl J. Long versus standard prednisone therapy for initial treatment of idiopathic nephrotic syndrome in children. Arbeitsgemeinschaft fur Padiatrische Nephrologie. Eur J Pediatr. 1993;152:357–61.
Hodson EM, Willis NS, Craig JC. Corticosteroid therapy for nephrotic syndrome in children. Cochrane Database Syst Rev. 2007;4:CD001533.
Pravitsitthikul N, Willis NS, Hodson EM, Craig JC. Non-corticosteroid immunosuppressive medications for steroid-sensitive nephrotic syndrome in children. Cochrane Database Syst Rev. 2013;10:CD002290.
Kidney Disease Improving Global Outcomes (KDIGO) Workgroup on clinical practice guideline for glomerulonephritis. Methods for guideline development. Kidney Int Suppl. 2012;2:243–51.
Bagga A, Ali U, Banerjee S, Indian Pediatric Nephrology Group, Indian Academy of Pediatrics, et al. Management of steroid sensitive nephrotic syndrome: revised guidelines. Indian Pediatr. 2008;45:203–14.
Sinha A, Saha A, Kumar M, et al. Extending initial prednisolone treatment in a randomized control trial from 3- to 6-months did not significantly influence the course of illness in children with steroid sensitive nephrotic syndrome. Kidney Int. 2014. doi:10.1038/ki.2014.240.
Yoshikawa N, Nakanishi K, Sako M, et al. A multicenter randomized trial indicates initial prednisolone treatment for childhood nephrotic syndrome for two months is not inferior to six-month treatment. Kidney Int. 2014. doi:10.1038/ki.2014.260.
Teeninga N, Kist-van Holthe J, van Rijskwijk N, et al. Extending prednisolone therapy does not reduce relapse in childhood nephrotic syndrome. J Am Soc Nephrol. 2012;24:149–59.
Gulati A, Sinha A, Sreenivas V, Math A, Hari P, Bagga A. Daily corticosteroids reduce infection-associated relapses in frequently relapsing nephrotic syndrome: a randomized controlled trial. Clin J Am Soc Nephrol. 2011;6:63–9.
Gellermann J, Weber L, Pape L, Tönshoff B, Hoyer P, Querfeld U. for the Gesellschaft für Pädiatrische Nephrologie (GPN). Mycophenolate mofetil versus cyclosporin A in children with frequently relapsing nephrotic syndrome. J Am Soc Nephrol. 2013;24:1689–97.
Iijima K, Sako M, Nozu K, et al. Rituximab for childhood-onset, complicated, frequently relapsing nephrotic syndrome or steroid-dependent nephrotic syndrome: a multicentre, double-blind, randomized, placebo controlled trial. Lancet. 2014. doi:10.1016/S0140-6736(14)60541-9.
Ravani P, Magnasco A, Edefonti A, et al. Short-term effects of rituximab in children with steroid- and calcineurin-dependent nephrotic syndrome: a randomized controlled trial. Clin J Am Soc Nephrol. 2011;6:1308–15.
Gipson DS, Trachtman H, Kaskel FJ, et al. Clinical trial of focal segmental glomerulosclerosis in children and young adults. Kidney Int. 2011;80:868–78.
Gulati A, Sinha A, Gupta A, Kanitkar M, Sreenivas V, Sharma J, et al. Treatment with tacrolimus and prednisolone is preferable to intravenous cyclophosphamide as the initial therapy for children with steroid-resistant nephrotic syndrome. Kidney Int. 2012;82:1130–5.
Magnasco A, Ravani P, Edefonti A, et al. Rituximab in children with resistant idiopathic nephrotic syndrome. J Am Soc Nephrol. 2012;23:1117–24.
Saadeh SA, Baracco R, Jain A, Kapur G, Mattoo TK, Valentini RP. Weight or body surface area dosing of steroids in nephrotic syndrome: is there an outcome difference? Pediatr Nephrol. 2011;26:2167–71.
Andersen RF, Thrane N, Noergaard K, Rytter L, Jespersen B, Rittig S. Early age at debut is a predictor of steroid dependent and frequent relapsing nephrotic syndrome. Pediatr Nephrol. 2010;25:1299–304.
Mishra OP, Abhinay A, Mishra RN, Prasad R, Pohl M. Can we predict relapses in children with idiopathic steroid-sensitive nephrotic syndrome? J Trop Pediatr. 2013;59:343–9.
Mattoo TK, Mahmoud MA. Increased maintenance corticosteroids during upper respiratory infection decrease the risk of relapse in nephrotic syndrome. Nephron. 2000;85:343–5.
Abeyagunawardena A, Trompeter RS. Increasing the dose of prednisolone during viral infections reduces the risk of relapse in nephrotic syndrome: a randomized controlled trial. Arch Dis Child. 2008;93:226–8.
Leisti S, Hallman N, Koskimies O, et al. Association of postmedication hypocortisolism with early first relapse of idiopathic nephrotic syndrome. Lancet. 1977;2:795–6.
Leisti S, Koskimies O. Risk of relapse in steroid-sensitive nephrotic syndrome: effect of stage of post-prednisolone adrenocortical suppression. J Pediatr. 1983;103:553–7.
Arun S, Bhatnagar S, Menon S, Saini S, Hari P, Bagga A. Efficacy of zinc supplements in reducing relapses in steroid-sensitive nephrotic syndrome. Pediatr Nephrol. 2009;24:1583–6.
Sherali AR, Moorani KN, Chishty SH, Khan SI. Zinc supplement in reduction of relapses in children with steroid sensitive nephrotic syndrome. J Coll Phys Surg Pak. 2014;24:110–3.
Bagga A, Hari P, Moudgil A, Jordan SC. Mycophenolate mofetil and prednisolone therapy in children with steroid dependent nephrotic syndrome. Am J Kidney Dis. 2003;4:1114–20.
Fujinaga S, Ohtomo Y, Umino D, Takemoto M, Shimizu T, Yamashiro Y, Kaneko K. A prospective study on the use of mycophenolate mofetil in children with cyclosporine-dependent nephrotic syndrome. Pediatr Nephrol. 2007;22:71–6.
Baudouin V, Alberti C, Lapeyraque AL, Bensman A, André JL, Broux F, Cailliez M, Decramer S, Niaudet P, Deschênes G, Jacqz-Aigrain E, Loirat C. Mycophenolate mofetil for steroid-dependent nephrotic syndrome: a phase II Bayesian trial. Pediatr Nephrol. 2012;27:389–96.
Ishikura K, Ikeda M, Hattori S, Yoshikawa N, Sasaki S, Iijima K, Nakanishi K, Yata N, Honda M. Effective and safe treatment with cyclosporine in nephrotic children: a prospective, randomized multicenter trial. Kidney Int. 2008;73:1167–73.
Sinha MD, MacLeod R, Rigby E, Clark AGB. Treatment of severe steroid-dependent nephrotic syndrome in children with tacrolimus. Nephrol Dial Transplant. 2006;21:1848–54.
• Sinha A, Bhatia D, Gulati A, Rawat M, Dinda AK, Hari P, Bagga A. Efficacy and safety of rituximab in children with difficult-to-treat nephrotic syndrome. Nephrol Dial Transplant 2014. doi: 10.1093/ndt/gfu267. This report on 193 patients with nephrotic syndrome treated with rituximab at a single centre shows that therapy was effective and safe in maintaining remission, reducing the frequency of relapses and ensuring steroid sparing in difficult-to-treat steroid dependent nephrotic syndrome and calcineurin inhibitor dependent steroid resistance. However, less than one-third of patients with steroid and CNI resistance treated with rituximab showed remission.
• Ruggenenti P, Ruggiero B, Cravedi P et al. for the Rituximab in Nephrotic Syndrome of Steroid-Dependent or Frequently Relapsing Minimal Change Disease Or Focal Segmental Glomerulosclerosis (NEMO) Study Group. Rituximab in steroid-dependent or frequently relapsing idiopathic nephrotic syndrome. J Am Soc Nephrol 2014;25:850–63. This single limb randomized study, in 22 patients, including 10 children, with steroid dependent or frequently relapsing nephrotic syndrome, demonstrated that therapy with two doses of rituximab enables sustained remission at one year in the majority, reduces relapse frequency and the need for corticosteroid and other immunosuppressive medication, resulting in improved standard deviation scores for height and body mass index.
Sinha A, Bagga A, Gulati A, et al. Short-term efficacy of rituximab versus tacrolimus in steroid-dependent nephrotic syndrome. Pediatr Nephrol. 2012;27:235–41.
Gipson DS, Chin H, Presler TP, Jennette C, Ferris ME, Massengill S, Gibson K, Thomas DB. Differential risk of remission and ESRD in childhood FSGS. Pediatr Nephrol. 2006;21:344–9.
Troyanov S, Wall CA, Miller JA, Scholey JW, Cattran DC. Focal and segmental glomerulosclerosis: definition and relevance of a partial remission. J Am Soc Nephrol. 2005;16:1061–8.
Hodson EM, Willis NS, Craig JC. Interventions for idiopathic steroid-resistant nephrotic syndrome in children. Cochrane Database Syst Rev. 2010;11:CD003594.
Choudhry S, Bagga A, Hari P, Sharma S, Kalaivani M, Dinda A. Efficacy and safety of tacrolimus versus cyclosporine in children with steroid-resistant nephrotic syndrome: a randomized controlled trial. Am J Kidney Dis. 2009;53:760–9.
Sinha A, Sharma A, Mehta A, Gupta R, Gulati A, Hari P, Dinda AK, Bagga A. Calcineurin inhibitor induced nephrotoxicity in steroid resistant nephrotic syndrome. Indian J Nephrol. 2013;23:41–6.
Ren H, Shen P, Li X, Pan X, Zhang W, Chen N. Tacrolimus versus cyclophosphamide in steroid-dependent or steroid-resistant focal segmental glomerulosclerosis: a randomized controlled trial. Am J Nephrol. 2013;37:84–90.
Gellermann J, Ehrich JH, Querfeld U. Sequential maintenance therapy with cyclosporin A and mycophenolate mofetil for sustained remission of childhood steroid-resistant nephrotic syndrome. Nephrol Dial Transplant. 2012;27:1970–8.
Bagga A, Sinha A, Moudgil A. Rituximab in patients with the steroid-resistant nephrotic syndrome. New Engl J Med. 2007;356:2751–2.
Hibino S, Uemura O, Nagai T, Yamakawa S, Iwata N, Ito H, Nakano M, Tanaka K. Three-year outcome of children with idiopathic nephrotic syndrome under a unified immunosuppressive protocol. Pediatr Int. 2014. doi:10.1111/ped.12498.
Muso E, Mune M, Hirano T, Hattori M, Kimura K, Watanabe T, Yokoyama H, Sato H, Uchida S, Wada T, Shoji T, Yuzawa Y, Takemura T, Sugiyama S, Nishizawa Y, Ogahara S, Yorioka N, Sakai S, Ogura Y, Yukawa S, Iino Y, Imai E, Matsuo S, Saito T. Immediate therapeutic efficacy of low-density lipoprotein apheresis for drug-resistant nephrotic syndrome: evidence from the short-term results from the POLARIS Study. Clin Exp Nephrol. 2014. doi:10.1007/s10157-014-0996-8.
Sgambat K, Banks M, Moudgil A. Effect of galactose on glomerular permeability and proteinuria in steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2013;28:2131–5.
Bitzan M, Babayeva S, Vasudevan A, Goodyer P, Torban E. TNFα pathway blockade ameliorates toxic effects of FSGS plasma on podocyte cytoskeleton and β3 integrin activation. Pediatr Nephrol. 2012;27:2217–26.
Hogan J, Bomback AS, Mehta K, Canetta PA, Rao MK, Appel GB, Radhakrishnan J, Lafayette RA. Treatment of idiopathic FSGS with adrenocorticotropic hormone gel. Clin J Am Soc Nephrol. 2013;8:2072–81.
Webb NJ, Shahinfar S, Wells TG, Massaad R, Gleim GW, Santoro EP, Sisk CM, Lam C. Losartan and enalapril are comparable in reducing proteinuria in children. Kidney Int. 2012;82:819–26.
•• Persson F, Rossing P. Sequential RAAS blockade: is it worth the risk? Adv Chronic Kidney Dis 2014;1:159–65. This review discussed the benefits and potential adverse effects of dual blockade of the renin angiotensin aldosterone system in patients with proteinuric kidney diseases, as demonstrated in findings from uncontrolled as well as randomized studies and results of meta-analyses.
• de Borst MH, Hajhosseiny R, Tamez H, Wenger J, Thadhani R, Goldsmith DJ. Active vitamin D treatment for reduction of residual proteinuria: a systematic review. J Am Soc Nephrol 2013;24:1863–71. This meta-analysis included findings from 6 randomized controlled trials of efficacy of vitamin D analogs in patients with proteinuric chronic kidney disease and demonstrated that therapy with calcitriol or paracalcitol reduces proteinuria by an average of 16% over and above the reduction achieved with ACE inhibitors or aldosterone receptor agonists.
Xu L, Wan X, Huang Z, Zeng F, Wei G, Fang D, Deng W, Li Y. Impact of vitamin D on chronic kidney diseases in non-dialysis patients: a meta-analysis of randomized controlled trials. PLoS ONE. 2013;8:e61387.
Rüth EM, Kemper MJ, Leumann EP, Laube GF, Neuhaus TJ. Children with steroid-sensitive nephrotic syndrome come of age: long-term outcome. J Pediatr. 2005;147:202–7.
Niaudet P. Long-term outcome of children with steroid-sensitive idiopathic nephrotic syndrome. Clin J Am Soc Nephrol. 2009;4:1547–8.
• Ishikura K, Yoshikawa N, Nakazato H, Sasaki S, Nakanishi K, Matsuyama T, Ito S, Hamasaki Y, Yata N, Ando T, Iijima K, Honda M; for Japanese Study Group of Renal Disease in Children. Morbidity in children with frequently relapsing nephrosis: 10-year follow-up of a randomized controlled trial. Pediatr Nephrol. 2014. doi:10.1007/s00467-014-2955-8. Ten year follow up in 46 of 56 patients previously enrolled in a study of the efficacy of cyclosporine for frequently relapsing nephrotic syndrome demonstrated that half the patients continue to relapse into adulthood and/or require immunosuppression.
Skrzypczyk P, Panczyk-Tomaszewska M, Roszkowska-Blaim M, Wawer Z, Bienias B, Zajgzkowska M, Kilis-Pstrusinska K, Jakubowska A, Szczepaniak M, Pawlak-Bratkowska M, Tkaczyk M. Long-term outcomes in idiopathic nephrotic syndrome: from childhood to adulthood. Clin Nephrol. 2014;81:166–73.
Zagury A, Oliveira AL, Montalvão JA, Novaes RH, Sá VM, Moraes CA, Tavares Mde S. Steroid-resistant idiopathic nephrotic syndrome in children: long-term follow-up and risk factors for end-stage renal disease. J Bras Nefrol. 2013;35:191–9.
Hamasaki Y, Yoshikawa N, Nakazato H, Sasaki S, Iijima K, Nakanishi K, Matsuyama T, Ishikura K, Ito S, Kaneko T, Honda M, for Japanese Study Group of Renal Disease in Children. Prospective 5-year follow-up of cyclosporine treatment in children with steroid-resistant nephrosis. Pediatr Nephrol. 2013;28:765–71.
Straatmann C, Ayoob R, Gbadegesin R, Gibson K, Rheault MN, Srivastava T, Tran CL, Gipson DS, Greenbaum LA, Smoyer WE, Vehaskari VM. Treatment outcome of late steroid-resistant nephrotic syndrome: a study by the Midwest Pediatric Nephrology Consortium. Pediatr Nephrol. 2013;28:1235–41.
Ding WY, Koziell A, McCarthy HJ, Bierzynska A, Bhagavatula MK, Dudley JA, Inward CD, Coward RJ, Tizard J, Reid C, Antignac C, Boyer O, Saleem MA. Initial steroid sensitivity in children with steroid-resistant nephrotic syndrome predicts post-transplant recurrence. J Am Soc Nephrol. 2014;25:1342–8.
Delville M, Sigdel TK, Wei C, Li J, Hsieh SC, Fornoni A, Burke GW, Bruneval P, Naesens M, Jackson A, Alachkar N, Canaud G, Legendre C, Anglicheau D, Reiser J, Sarwal MM. A circulating antibody panel for pretransplant prediction of FSGS recurrence after kidney transplantation. Sci Transl Med. 2014;6:256ra136.
Vinai M, Waber P, Seikaly MG. Recurrence of focal segmental glomerulosclerosis in renal allograft: an in-depth review. Pediatr Transplant. 2010;14:314–25.
Straatmann C, Kallash M, Killackey M, Iorember F, Aviles D, Bamgbola O, Carson T, Florman S, Vehaskari MV. Success with plasmapheresis treatment for recurrent focal segmental glomerulosclerosis in pediatric renal transplant recipients. Pediatr Transplant. 2014;18:29–34.
• Araya CE, Dharnidharka VR. The factors that may predict response to rituximab therapy in recurrent focal segmental glomerulosclerosis: a systematic review. J Transplant 2013;2011:374213. This systematic review of 39 reports included 39 patients treated with rituximab for recurrence of focal segmental glomerulosclerosis in the allograft and demonstrated, on multivariate analysis, that a younger age at transplant and normal serum albumin level at diagnosis of recurrence may predict response to rituximab.
Disclosure
Aditi Sinha, Shina Menon, and Arvind Bagga declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Renal.
Rights and permissions
About this article

Cite this article
Sinha, A., Menon, S. & Bagga, A. Nephrotic Syndrome: State of the Art. Curr Pediatr Rep 3, 43–61 (2015). https://doi.org/10.1007/s40124-014-0066-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40124-014-0066-4