
Abstract
MYC is a transcription factor that regulates many critical genes for cell proliferation, differentiation, and biomass accumulation. MYC is one of the most prevalent oncogenes found to be altered in human cancer, being deregulated in about 50 % of tumors. Although MYC deregulation has been more frequently associated to lymphoma and lymphoblastic leukemia than to myeloid malignancies, a body of evidence has been gathered showing that MYC plays a relevant role in malignancies derived from the myeloid compartment. The myeloid leukemogenic activity of MYC has been demonstrated in different murine models. Not surprisingly, MYC has been found to be amplified or/and deregulated in the three major types of myeloid neoplasms: acute myeloid leukemia, myelodysplastic syndromes, and myeloproliferative neoplasms, including chronic myeloid leukemia. Here, we review the recent literature describing the involvement of MYC in myeloid tumors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Myelopoiesis and MYC impact on myeloid cell proliferation and differentiation
Hematopoietic stem cells differentiate along two major lineages. The lymphoid lineages give rise to B- and T-lymphocytes, as well as natural killer cells. The myeloid lineage is much more complex, with at least six major differentiated circulating cells (erythrocytes, monocyte/macrophages, eosinophils, neutrophils, basophils, and megakaryocyte/platelets) each of them very different from the other cell types in its function in the organism, its number in peripheral blood, and its morphology and gene expression profile [1–3]. Myeloid lineage commitment relies on timely activation of appropriate transcription factors and silencing of others, which is the result from a network of extracellular signals [4, 5].
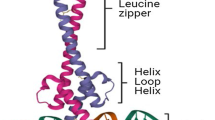
MYC is a transcription factor of the helix-loop-helix-leucine zipper family. MYC forms dimers with MAX protein and binds to E-box sequences in the regulatory regions of around one thousand target genes (Fig. 1a) [6]. MYC is involved in a number of fundamental functions for the cell biology, such as control of proliferation and differentiation, energy production, cell size, and many others (Fig. 1b) [7–9]. Not surprisingly, deregulation of MYC activities contributes to the tumor phenotype. MYC is one of the most prevalent oncogenes in human cancer, and particularly in hematopoietic neoplasias [10, 11]. In fact, in human cancer, MYC deregulation was first observed in Burkitt lymphoma, as well as in chemically induced murine plasmacytomas [12]. Virtually all cases of these lymphoma show a translocation affecting the MYC locus [13]. From then on, MYC translocations have been found in a significant fraction of high-grade lymphomas and multiple myeloma, and MYC overexpression has also been reported in numerous types of lymphoma and lymphoblastic leukemia [14].

MYC structure and functions. a Scheme showing the main features of MYC transcription factor: the N-terminal transactivation domain (TAD), conserved MYC box-I, II, and III, nuclear localization signal (NLS), and the basic-helix-loop-helix-leuzine zipper (B-HLH-LZ) DNA binding and dimerization domain. Heterodimers MYC-MAX bind E-box sequences at the regulatory regions of its target genes (the most representative indicated in the square). b MYC is involved in many cellular functions through the regulation (activation or repression) of its target genes. MYC favors the cell cycle progression by activating cyclin-dependent kinases (CDKs) and repressing CDK inhibitors; MYC regulates metabolic enzymes involved in the ATP production; MYC has a role in protein synthesis and cell size through the control of ribosomal genes among others; MYC induces genomic instability and cell immortalization; the inhibition of cell differentiation, maintenance of pluripotency, and induction of angiogenesis are important MYC functions. Elevated MYC levels (by gene amplification, chromosomal translocation, overexpression, and protein stabilization) deregulate such functions, contributing to tumorigenesis
In contrast to lymphoid tumors, MYC involvement in myeloid cancer has received much less attention. Nonetheless, MYC oncogene was first discovered as the oncogene carried by retrovirus that induced a myeloid neoplasm in chicken, i.e., myelocytomatosis, and MYC was named after this tumor [15]. Moreover, the inhibition of myeloid cell differentiation was one of the first biological effects described for MYC (reviewed in Ref. [9]). In 1986, three reports showed that MYC blocked the chemically induced erythroid differentiation of Friend murine erythroleukemia (MEL) cells [16–18]. We also showed that MYC inhibited the erythroid differentiation of human myeloid cells such as K562 derived from chronic myeloid leukemia (CML) [19], and found that MYC blocks erythroid differentiation in genetically-defined models where erythroid differentiation is induced by p27KIP1 [20]. In this model, MYC impairs the up-regulation of many erythroid-specific genes, as well as that of transcription factors that determine erythroid lineage differentiation (including GATA1 and NFE2) but, strikingly, it does not reverse the proliferation arrest and the repression of CDK activity mediated by p27KIP1. In a complementary approach, using the leukemia K562 and U937 cells, it has been shown that MYC is not down-regulated when cells are growth-arrested but not differentiated [21, 22]. Enforced MYC expression also blocks the monocytic differentiation of the AML-derived cell line U937 [23]. The fact that MYC can block differentiation of myeloid leukemia cells models in culture is consistent with its involvement in myeloid leukemia. However, MYC activity is not universally linked to differentiation inhibition of myeloid cells, as MYC enhances the retinoic acid-induced differentiation of a promyelocytic leukemia cell line (NB4) [24].
MYC roles in the determination of myeloid lineages in vivo
The results described above are consistent with the in vivo observed data. The effects of MYC deletion in the myeloid compartment have been recently reported in the MYC conditional knock-out mice. Myc −/− mice show significant thrombocytosis, severe anemia, and grossly decreased neutrophil/monocyte numbers [25]. Thus MYC induces opposite effects in the differentiation of megakaryocytic versus monocyte and erythroid lineages. Moreover, these effects of MYC deletion in vivo on hematopoietic lineages correlates well with MYC expression in mouse hematopoietic cells: cells expressing higher levels of MYC such as granulocyte/monocyte precursors, common lymphocyte progenitors, and erythrocytic blasts are significantly reduced while cells expressing lower MYC levels (hematopoietic stem cells (HSC), megakaryocyte/erythroid precursors, and megakaryocytes) are less affected or are increased in number [25].
Megakaryocytes from Myc −/− mice are significantly smaller in size and are lower in ploidy than those of control mice; as a result, fewer platelets are produced by each megakaryocyte. However, due to the increase in megakaryocytic number, a significant increase in platelet number was observed in Myc −/− mice. The involvement of MYC in megakaryocytic differentiation is confirmed in transgenic mice with MYC overexpression in the megakaryocytic lineage, where a Myc transgene is under the control of the platelet factor-4 promoter. These mice show an increase in low-ploidy megakaryocytes due to enhanced proliferation and survival, along with the blocking of differentiation [26].
In conclusion, MYC plays a pivotal role in regulating the normal hematopoiesis, and thus, it is not surprising that MYC is frequently deregulated in human leukemia and lymphoma. Such deregulation would destroy this balance and transform hematopoietic cells by stimulating proliferation and blocking terminal differentiation.
In vivo models for MYC-induced myeloid leukemia: engraftment of retrovirally mediated expression of MYC in hematopoietic precursors
Several reports illustrate the leukemogenic effects of MYC in hematopoietic cells after the enforced expression in murine hematopoietic precursors via retroviral infection, although the induced neoplasia varies depending on the virus and the cells type targeted by the virus. Unfractionated bone marrow cells retrovirally transduced with Myc, using different vectors and experimental settings, resulted in development of acute myeloid leukemia (AML) [27–29]. In one study, the MYC-transduced fetal liver cells transduced with another MYC vector induced a long-latency lymphoma [30].
The infection of bone marrow progenitors in a p53−/− background also resulted in lymphoma [31]. Interestingly, upon culturing in vitro, cells derived from these lymphomas underwent myeloid differentiation, but then switched from myeloid to lymphoid lineage and induced B cell lymphomas when returning to in vivo conditions [32]. In another study, mice bone marrow cells transduced with another member of the MYC family, MYCN (also called N-Myc), developed monoclonal and transplantable AML [33]. In a parallel report, it was shown that when the bone marrow from lethally irradiated cells was repopulated with bone marrow cells expressing Myc, the mice developed an AML-like disease [29]. In this model, the co-expression of several antiapoptotic genes of the BCL2 family accelerated leukemogenesis but did not change the myeloid phenotype of the leukemia [29]. In conclusion, these experimental models indicate that high MYC levels switch normal hematopoiesis into myeloid leukemia. The analysis of the AML induced in some of these models showed high expression of the antiapoptotic genes of the BCL2 family including the MCL1 gene. Moreover, MCL1 haploinsufficiency abrogated AML development in this model [28]. The results suggest that abrogation of apoptosis in MYC-targeted cells is probably necessary for the development of an aggressive AML. This has been recently confirmed in another report, in which retroviral transduction of MYC in purified (Lin−) murine HSC results in the development of both myeloid and T-lymphoid tumors within 2 months after transplantation. Interestingly, co-expression of MYC with BCLXL or BCL2 resulted in almost immediate development of AML-like disease, but not lymphoma (at the expense of other hematopoietic lineages) [34].
In vivo models for MYC-induced myeloid leukemia: transgenic mice developing myeloid neoplasia
Although most of the available literature on murine models of MYC-induced tumors is focused on MYC-induced lymphoma, there are also transgenic models that demonstrate the carcinogenic potential of MYC in the myeloid compartment. Mice carrying the human MYC proto-oncogene under the control of the murine GATA-1 promoter (an erythroid-specific gene) developed an early onset erythroleukemia [35]. Several transgenic mice lines have been generated carrying the MYC gene under the control of the Vav promoter (which is active in all hematopoietic cells lineages and hematopoietic precursors) showing different MYC expression levels [36]. Interestingly, the tumor lineage varied with the different MYC expression levels achieved in the transgenic mice. For instance, aggressive T-cell lymphomas were the predominant tumor arising in transgenic mice lines showing the highest MYC expression [36]. In contrast, most tumors in the mouse lines expressing lower MYC levels were late-onset monocytic tumors [37]. It is noteworthy that just a two-fold decrease in MYC levels switched the phenotype from T-cell tumors to monocytic tumors. Thus, relatively low MYC levels can transform monocyte-macrophages but are insufficient to transform T-lymphocytes [37]. These data and other gathered in the literature strongly suggest that relatively small increases in MYC are sufficient to promote leukemia.
MYC in human myeloproliferative neoplasms
Consistent with the data gathered in animal models (see above), MYC deregulation has been found in human myeloid neoplasias. Three major groups of clonal myeloid diseases have been described: acute myeloid leukemia, myelodysplastic syndromes, and myeloproliferative neoplasms [38]. Myeloproliferative neoplasms (MPN) are characterized by an enhanced proliferation of one or more of myeloid, erythroid, or megakaryocytic lineages. Chronic myeloid leukemia (CML) affects annually 1–2 cases per 100,000 individuals. CML is a proliferative clonal disorder of hematopoietic stem cells that results primarily in the expansion of mature myeloid cells that retains a capacity for differentiation. Untreated CML progress from the initial stage, termed chronic phase, to a blastic crisis phase which is similar to acute leukemia. Other frequent MPNs are essential thrombocythemia (ET), polycythemia vera (PV), and primary myelofibrosis (PMF).
The Philadelphia chromosome and the resulting fusion Bcr-Abl oncogene is the universal molecular marker of CML and has a central role in the disease etiology [39]. Bcr-Abl up-regulates MYC expression [40, 41] and MYC cooperates with Bcr-Abl in transformation [42–44]. Moreover, Bcr-Abl provokes the phosphorylation of MYC in Ser-62. This phosphorylation renders a more stable MYC protein. A proteomic analysis revealed that MYC-Ser62 was largely dephosphorylated after dasatinib treatment [45]. This result suggests that these kinase inhibitors would reduce the levels of MYC protein in the leukemic cell.
Studies performed with a small number of cases, showed that MYC mRNA levels are elevated in CML-blastic crisis [46, 47] and in chronic phase CML versus healthy bone marrow samples [48, 49]. We have recently observed the up-regulation of MYC expression during CML progression [50]. Moreover, high levels of MYC along the disease correlate with poorer response to imatinib, and higher MYC mRNA levels at diagnosis tend to correlate with a worse response to imatinib [50]. Essentially the same results have been recently reported in a smaller cohort of CML patients, showing that MYC protein is elevated at diagnosis in patients destined to progress to blastic crisis [51]. It is also of note that trisomy 8 and gain at 8q24 (where MYC maps) are among the most frequent cytogenetic alterations in CML [52, 53], although their correlation with MYC expression is yet unknown.
It has been recently shown that at the time of diagnosis of CML, patients who will later progress to blast crisis have significantly higher levels of CIP2A protein (cancerous inhibitor of PP2A) [51]. CIP2A inhibits PP2A-mediated dephosphorylation of MYC at Ser-62, as pSer62-MYC is stabilized against degradation [54]. Although this should be confirmed in bigger cohorts of patients, these results indicate that CML progression is not only a consequence of elevated mRNA levels but also of MYC protein stabilization [51].
Misregulation of the activity of a specific group of ATP-binding cassette transporters (ABC) is responsible for reducing the intracellular concentration of drugs in leukemic cells. CD34+ hematopoietic cell precursors of CML patients overexpress ABC transporters, and this overexpression is, at least in part, due to MYC, as ABC is a direct target of MYC [55]. This MYC activity could also contribute to CML transformation.
Why MYC should be deregulated during the CML progression? CML progression to blastic crisis is associated to cell survival, genomic instability, and differentiation arrest [39, 56]. We have shown that enforced MYC expression in CML-derived cells as K562, results in aberrant DNA synthesis under imatinib stress [50] and blocks imatinib-mediated differentiation [57], suggesting that MYC may contribute to CML transformation acting at those two levels (Fig. 2).

MYC mRNA is overexpressed in bone marrow cells from ET [58]. On the contrary, MYC overexpression is not found in the PV [58]. This is striking as more than 95 % of PV patients carry an activating mutation in the tyrosine kinase JAK2, and JAK2 has been reported to induce MYC expression in cell lines [40, 59]. Clearly, an analysis of STAT activation and the transcription factors phosphorylated by JAKs, is lacking in PV, making it difficult to obtain the interpretation of these results.
MYC in human myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML)
Acute myeloid leukemia (AML) is characterized by the proliferation and accumulation of immature hematopoietic cells in the bone marrow and blood. AML is a heterogeneous group of neoplasms affecting the myeloid lineage. Altogether, their incidence worldwide is about 2–3 cases in 100,000 individuals per year, and is reported to be the highest in Australia, Western Europe, and United States. The former FAB (French-American-British) classification system of AML (M0 to M7 subtypes, attending to the differentiation type and stage) has been superseded by the World Health Organization (WHO) classification, that identifies 15 diseases characterized by clinical presentation and recurrent chromosomal aberrations [38]. Understandably, most molecular studies to date are carried out with a relatively small number of cases of each myeloid disease or do not make distinctions between the different entities. Therefore, the information on MYC expression in myeloid leukemia actually refers to a range of related diseases, and therefore its involvement in a particular myeloid neoplasm has not been properly addressed.
MYC amplification and overexpression have been found in AML [62] (Table 1). The overexpression of MYC mRNA in bone marrow and peripheral blood in sporadic AML cases patients (as compared with normal bone marrow) was observed early on [60, 61]. Subsequent studies bases on microarray hybridizations, and RT-qPCR analysis confirmed this finding and showed that not only MYC but also MYCN is overexpressed in AML patients [33]. These data are in agreement with the observation that MYC is up-regulated in radiation-induced AML in mice [62]. MYC expression also appeared elevated in a microarray-based study in 5 AML samples, and was validated by RT-qPCR [63]. In contrast, MYC has not been detected as a major overexpressed gene in other microarray-based studies on AML samples [64–66]. However, it must be noted, first, that microarray-based studies are done at the mRNA level, and thus changes in MYC protein level are not evaluated. Second, if MYC up-regulation is moderate (e.g., two-fold with respect to controls), it might be filtered out by the statistical analysis of microarray data. It should be mentioned, however, that a two-fold expression change of MYC may be relevant. For example, as noted above, transgenic mice lines with low MYC expression in the hematopoietic precursors develop myeloid leukemia; whereas, high MYC expression results in T cell lymphomas [37]. Also, a mere two-fold change means a major difference for MYC-dependent transformation of fibroblasts and mouse embryonic stem cells [67, 68] as well as in transgenic animals, where MYC dosage can be modulated [69]. This finding reveals that the impact of MYC in human leukemia is difficult to evaluate in the clinical setting, as two-fold increase in gene expression is usually not considered as significant when measuring mRNA expression in the clinical laboratories; whereas, as commented above, it could mean a difference in cell transformation. Finally, the great diversity of AML commented above makes the analysis of homogeneous sample cohorts difficult.
The mechanisms that lead to MYC overexpression in these cases are essentially unknown. Recurrent translocations in AML generate fusion proteins that are leukemogenic transcription factors, [38] and at least three of these (RUNX1-ETO, PML-RARα, and PLZF-RARα) induce MYC expression. Thus, MYC could be a downstream target of these oncogenes [70, 71]. On the other side, MYC might be mediating the effects of some of these oncogenic translocation products. For example, it has been shown that MYC mediates the block in granulocytic differentiation brought about by MLL-ENL [72].
Cytogenetic hallmarks of gene amplification in tumors are double minute (dmin) chromosomes and homogeneously staining regions (hsr). In contrast to other tumors, both double minute chromosomes and homogeneously staining regions containing amplified regions are rare in AML. Although MYC amplification in AML is infrequent (less than 1 % of all cytogenetically abnormal leukemias, http://cgap.nci.nih.gov/Chromosomes/Mitelman), hsr and dmin including the region of 8q24 where MYC maps have been consistently described in AML and 8q24 is one of the most common amplified regions in dmin [73–82]. It has been suggested that a high MYC gene copy number does not necessarily result in higher MYC expression [79, 83]. However, recent reports show that MYC amplification is associated with higher MYC expression, with disease progression and poor prognosis [75]. Also, CD34+ cells from MDS patients with trisomy 8 showed up-regulation of MYC mRNA [84]. This situation is in agreement with the correlation between MYC amplification and expression observed in lymphoma [85].
The myelodysplastic syndromes (MDS) are also a heterogeneous group of clonal hematologic disorders characterized by both an aberrant differentiation process with morphologic evidence of marrow dysplasia in one or more of the three major myeloid cell types and an increased ineffective proliferation of the myeloid precursors in bone marrow, leading to cytopenia(s) and to an enhanced risk of transformation to an AML. Gene expression profiles of CD34+ cells from MDS patients showed MYC as one of the most up-regulated genes in these patients [86]. Many patients with MDS, especially those with increased myeloblasts counts and adverse cytogenetic prognostic markers, will develop AML. MYC amplification has been reported with low frequencies in MDS [77, 78, 81, 84] (Table 1) but its prognostic impact is uncertain.
MYC as a pharmacological target in leukemia
As most transcription factors, MYC is not an easy druggable target. However, using dominant negative forms (OmoMyc), it has been shown that in vivo OmoMyc expression abolishes the MYC-mediated skin carcinogenesis in animal models [87]. Moreover, it has been shown that MYC blocks the K-Ras and T-antigen-dependent carcinogenesis in lung and pancreas, respectively [88–90]. This result suggests that MYC inhibition would be a sensible antitumor approach even for tumors that show no MYC deregulation. Several small molecules have been described as MYC inhibitors, although none of them are in clinical use at the moment. Most of them impair the MYC-MAX interaction, thus inhibiting the transcriptional activity of MYC [11]. This has the caveat that some of the MYC activities such as DNA replication, RNA pol II elongation, or CAP addition do not depend on the DNA binding activity of MYC [91, 92]. On the other hand, MYC requires other proteins for transformation, which may not be essential in normal cell physiology. Thus, a number of secondary targets capable of synthetic lethal interaction with MYC have been described, and thus proposed as putative pharmacological targets for MYC-driven tumors. This is the case of proteins as WRN [93], aurora kinase [94], CHK1 kinase [95] or CSKN1e kinase [96]. A different and recent approach is to block MYC expression by impairing BET bromodomain protein, a strategy shown to be effective in AML [97]. In summary, in vitro and in vivo models demonstrate a critical role of MYC in normal and malignant myelopoiesis. Therefore, the pharmacological targeting of MYC in myeloid leukemias is nowadays a challenge.
References
Iwasaki H, Akashi K (2007) Myeloid lineage commitment from the hematopoietic stem cell. Immunity 26:726–740
Orkin SH, Zon LI (2008) Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132:631–644
Tsiftsoglou AS, Bonovolias ID, Tsiftsoglou SA (2009) Multilevel targeting of hematopoietic stem cell self-renewal, differentiation and apoptosis for leukemia therapy. Pharmacol Ther 122:264–280
Miranda-Saavedra D, Gottgens B (2008) Transcriptional regulatory networks in haematopoiesis. Curr Opin Genet Dev 18:530–535
Kim SI, Bresnick EH (2007) Transcriptional control of erythropoiesis: emerging mechanisms and principles. Oncogene 26:6777–6794
Dang CV, O’Donnell KA, Zeller KI, Nguyen T, Osthus RC et al (2006) The c-Myc target gene network. Semin Cancer Biol 16:253–264
Oster SK, Ho CS, Soucie EL, Penn LZ (2002) The myc oncogene: marvelouslY complex. Adv Cancer Res 84:81–154
Eilers M, Eisenman RN (2008) Myc’s broad reach. Genes Dev 22:2755–2766
Leon J, Ferrandiz N, Acosta JC, Delgado MD (2009) Inhibition of cell differentiation: a critical mechanism for MYC-mediated carcinogenesis? Cell Cycle 8:1148–1157
Nesbit CE, Tersak JM, Prochownik EV (1999) MYC oncogenes and human neoplastic disease. Oncogene 18:3004–3016
Vita M, Henriksson M (2006) The Myc oncoprotein as a therapeutic target for human cancer. Semin Cancer Biol 16:318–330
Varmus HE (1984) The molecular genetics of cellular oncogenes. Annu Rev Genet 18:553–612
Sanchez-Beato M, Sanchez-Aguilera A, Piris MA (2003) Cell cycle deregulation in B-cell lymphomas. Blood 101:1220–1235
Delgado MD, Leon J (2010) Myc roles in hematopoiesis and leukemia. Genes Cancer 1:605–616
Sheiness D, Bishop JM (1979) DNA and RNA from uninfected vertebrate cells contain nucleotide sequences related to the putative transforming gene of avian myelocytomatosis virus. J Virol 31:514–521
Coppola JA, Cole MD (1986) Constitutive c-myc oncogene expression blocks mouse erythroleukaemia cell differentiation but not commitment. Nature 320:760–763
Prochownik EV, Kukowska J (1986) Deregulated expression of c-myc by murine erythroleukaemia cells prevents differentiation. Nature 322:848–850
Dmitrovsky E, Kuehl WM, Hollis GF, Kirsch IR, Bender TP et al (1986) Expression of a transfected human c-myc oncogene inhibits differentiation of a mouse erythroleukaemia cell line. Nature 322:748–750
Delgado MD, Lerga A, Canelles M, Gomez-Casares MT, Leon J (1995) Differential regulation of Max and role of c-Myc during erythroid and myelomonocytic differentiation of K562 cells. Oncogene 10:1659–1665
Acosta JC, Ferrandiz N, Bretones G, Torrano V, Blanco R et al (2008) Myc inhibits p27-induced erythroid differentiation of leukemia cells by repressing erythroid master genes without reversing p27-mediated cell cycle arrest. Mol Cell Biol 28:7286–7295
Ryan KM, Birnie GD (1997) Cell-cycle progression is not essential for c-Myc to block differentiation. Oncogene 14:2835–2843
Gomez-Casares MT, Delgado MD, Lerga A, Crespo P, Quincoces AF et al (1993) Down-regulation of c-myc gene is not obligatory for growth inhibition and differentiation of human myeloid leukemia cells. Leukemia 7:1824–1833
Bahram F, Wu S, Oberg F, Luscher B, Larsson LG (1999) Posttranslational regulation of Myc function in response to phorbol ester/interferon-gamma-induced differentiation of v-Myc-transformed U-937 monoblasts. Blood 93:3900–3912
Uribesalgo I, Buschbeck M, Gutierrez A, Teichmann S, Demajo S et al (2011) E-box-independent regulation of transcription and differentiation by MYC. Nat Cell Biol 13:1443–1449
Guo Y, Niu C, Breslin P, Tang M, Zhang S et al (2009) c-Myc-mediated control of cell fate in megakaryocyte-erythrocyte progenitors. Blood 114:2097–2106
Thompson A, Zhang Y, Kamen D, Jackson CW, Cardiff RD et al (1996) Deregulated expression of c-myc in megakaryocytes of transgenic mice increases megakaryopoiesis and decreases polyploidization. J Biol Chem 271:22976–22982
Luo H, Li Q, O’Neal J, Kreisel F, Le Beau MM et al (2005) c-Myc rapidly induces acute myeloid leukemia in mice without evidence of lymphoma-associated antiapoptotic mutations. Blood 106:2452–2461
Xiang Z, Luo H, Payton JE, Cain J, Ley TJ et al (2010) Mcl1 haploinsufficiency protects mice from Myc-induced acute myeloid leukemia. J Clin Invest 120:2109–2118
Beverly LJ, Varmus HE (2009) MYC-induced myeloid leukemogenesis is accelerated by all six members of the antiapoptotic BCL family. Oncogene 28:1274–1279
Hemann MT, Bric A, Teruya-Feldstein J, Herbst A, Nilsson JA et al (2005) Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature 436:807–811
Yu D, Thomas-Tikhonenko A (2002) A non-transgenic mouse model for B-cell lymphoma: in vivo infection of p53-null bone marrow progenitors by a Myc retrovirus is sufficient for tumorigenesis. Oncogene 21:1922–1927
Yu D, Allman D, Goldschmidt MH, Atchison ML, Monroe JG et al (2003) Oscillation between B-lymphoid and myeloid lineages in Myc-induced hematopoietic tumors following spontaneous silencing/reactivation of the EBF/Pax5 pathway. Blood 101:1950–1955
Kawagoe H, Kandilci A, Kranenburg TA, Grosveld GC (2007) Overexpression of N-Myc rapidly causes acute myeloid leukemia in mice. Cancer Res 67:10677–10685
Hogstrand K, Hejll E, Sander B, Rozell B, Larsson LG et al (2012) Inhibition of the intrinsic but not the extrinsic apoptosis pathway accelerates and drives MYC-driven tumorigenesis towards acute myeloid leukemia. PLoS ONE 7:e31366
Skoda RC, Tsai SF, Orkin SH, Leder P (1995) Expression of c-MYC under the control of GATA-1 regulatory sequences causes erythroleukemia in transgenic mice. J Exp Med 181:1603–1613
Smith DP, Bath ML, Harris AW, Cory S (2005) T-cell lymphomas mask slower developing B-lymphoid and myeloid tumours in transgenic mice with broad haematopoietic expression of MYC. Oncogene 24:3544–3553
Smith DP, Bath ML, Metcalf D, Harris AW, Cory S (2006) MYC levels govern hematopoietic tumor type and latency in transgenic mice. Blood 108:653–661
Vardiman JW, Brunning RD, Arber DA, LeBeau MM, Porwit A et al (2008) Introduction and overview of the classification of the myeloid neoplasms. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA et al (eds) WHO classification of tumours of haematopoietic and lymphoid tissues, 4th edn. International Agency for Research on Cancer, Lyon, pp 18–37
Melo JV, Barnes DJ (2007) Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer 7:441–453
Xie S, Lin H, Sun T, Arlinghaus RB (2002) Jak2 is involved in c-Myc induction by Bcr-Abl. Oncogene 21:7137–7146
Gomez-Casares MT, Vaque JP, Lemes A, Molero T, Delgado MD et al (2004) C-myc expression in cell lines derived from chronic myeloid leukemia. Haematologica 89:241–243
Lugo TG, Witte ON (1989) The BCR-ABL oncogene transforms Rat-1 cells and cooperates with v-myc. Mol Cell Biol 9:1263–1270
Sawyers CL, Callahan W, Witte ON (1992) Dominant negative MYC blocks transformation by ABL oncogenes. Cell 70:901–910
Afar DE, Goga A, McLaughlin J, Witte ON, Sawyers CL (1994) Differential complementation of Bcr-Abl point mutants with c-Myc. Science 264:424–426
Pan C, Olsen JV, Daub H, Mann M (2009) Global effects of kinase inhibitors on signaling networks revealed by quantitative phosphoproteomics. Mol Cell Proteomics 8:2796–2808
Handa H, Hegde UP, Kotelnikov VM, Mundle SD, Dong LM et al (1997) Bcl-2 and c-myc expression, cell cycle kinetics and apoptosis during the progression of chronic myelogenous leukemia from diagnosis to blastic phase. Leuk Res 21:479–489
Beck Z, Bacsi A, Kovacs E, Kiss J, Kiss A et al (1998) Changes in oncogene expression implicated in evolution of chronic granulocytic leukemia from its chronic phase to acceleration. Leuk Lymphoma 30:293–306
Nowicki MO, Pawlowski P, Fischer T, Hess G, Pawlowski T et al (2003) Chronic myelogenous leukemia molecular signature. Oncogene 22:3952–3963
Diaz-Blanco E, Bruns I, Neumann F, Fischer JC, Graef T et al (2007) Molecular signature of CD34(+) hematopoietic stem and progenitor cells of patients with CML in chronic phase. Leukemia 21:494–504
Albajar M, Gomez-Casares MT, Llorca J, Mauleon I, Vaque JP et al (2011) MYC in chronic myeloid leukemia: induction of aberrant DNA synthesis and association with poor response to imatinib. Mol Cancer Res 9:564–576
Lucas CM, Harris RJ, Giannoudis A, Copland M, Slupsky JR et al (2011) Cancerous inhibitor of PP2A (CIP2A) at diagnosis of chronic myeloid leukemia is a critical determinant of disease progression. Blood 117:6660–6668
Johansson B, Fioretos T, Mitelman F (2002) Cytogenetic and molecular genetic evolution of chronic myeloid leukemia. Acta Haematol 107:76–94
Brazma D, Grace C, Howard J, Melo JV, Holyoke T et al (2007) Genomic profile of chronic myelogenous leukemia: imbalances associated with disease progression. Genes Chromosomes Cancer 46:1039–1050
Sears RC (2004) The life cycle of C-myc: from synthesis to degradation. Cell Cycle 3:1133–1137
Porro A, Iraci N, Soverini S, Diolaiti D, Gherardi S et al (2011) c-MYC oncoprotein dictates transcriptional profiles of ATP-binding cassette transporter genes in chronic myelogenous leukemia CD34 + hematopoietic progenitor cells. Mol Cancer Res 9:1054–1066
Quintas-Cardama A, Cortes J (2009) Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood 113:1619–1630
Gomez-Casares MT, Garcia-Alegria E, Lopez-Jorge CE, Ferrandiz N, Blanco R, et al. (2012) MYC antagonizes the differentiation induced by imatinib in chronic myeloid leukemia cells through downregulation of p27KIP1. Oncogene. doi:10.1038/onc.2012.246
Theophile K, Buesche G, Kreipe H, Bock O (2008) The expression levels of telomerase catalytic subunit hTERT and oncogenic MYC in essential thrombocythemia are affected by the molecular subtype. Ann Hematol 87:263–268
Watanabe S, Itoh T, Arai K (1996) JAK2 is essential for activation of c-fos and c-myc promoters and cell proliferation through the human granulocyte-macrophage colony-stimulating factor receptor in BA/F3 cells. J Biol Chem 271:12681–12686
Ferrari S, Narni F, Mars W, Kaczmarek L, Venturelli D et al (1986) Expression of growth-regulated genes in human acute leukemias. Cancer Res 46:5162–5166
Calabretta B, Venturelli D, Kaczmarek L, Narni F, Talpaz M et al (1986) Altered expression of G1-specific genes in human malignant myeloid cells. Proc Natl Acad Sci USA 83:1495–1498
Hirouchi T, Takabatake T, Yoshida K, Nitta Y, Nakamura M et al (2008) Upregulation of c-myc gene accompanied by PU.1 deficiency in radiation-induced acute myeloid leukemia in mice. Exp Hematol 36:871–885
Court EL, Smith MA, Avent ND, Hancock JT, Morgan LM et al (2004) DNA microarray screening of differential gene expression in bone marrow samples from AML, non-AML patients and AML cell lines. Leuk Res 28:743–753
Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, Barjesteh van Waalwijk van Doorn-Khosrovani S et al (2004) Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med 350:1617–1628
Ross ME, Mahfouz R, Onciu M, Liu HC, Zhou X et al (2004) Gene expression profiling of pediatric acute myelogenous leukemia. Blood 104:3679–3687
Stirewalt DL, Meshinchi S, Kopecky KJ, Fan W, Pogosova-Agadjanyan EL et al (2008) Identification of genes with abnormal expression changes in acute myeloid leukemia. Genes Chromosomes Cancer 47:8–20
Bazarov AV, Adachi S, Li SF, Mateyak MK, Wei S et al (2001) A modest reduction in c-myc expression has minimal effects on cell growth and apoptosis but dramatically reduces susceptibility to Ras and Raf transformation. Cancer Res 61:1178–1186
Baudino TA, McKay C, Pendeville-Samain H, Nilsson JA, Maclean KH et al (2002) c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev 16:2530–2543
Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K et al (2008) Distinct thresholds govern Myc’s biological output in vivo. Cancer Cell 14:447–457
Muller-Tidow C, Steffen B, Cauvet T, Tickenbrock L, Ji P et al (2004) Translocation products in acute myeloid leukemia activate the Wnt signaling pathway in hematopoietic cells. Mol Cell Biol 24:2890–2904
Rice KL, Hormaeche I, Doulatov S, Flatow JM, Grimwade D et al (2009) Comprehensive genomic screens identify a role for PLZF-RARalpha as a positive regulator of cell proliferation via direct regulation of c-MYC. Blood 114:5499–5511
Schreiner S, Birke M, Garcia-Cuellar MP, Zilles O, Greil J et al (2001) MLL-ENL causes a reversible and myc-dependent block of myelomonocytic cell differentiation. Cancer Res 61:6480–6486
Sait SN, Qadir MU, Conroy JM, Matsui S, Nowak NJ et al (2002) Double minute chromosomes in acute myeloid leukemia and myelodysplastic syndrome: identification of new amplification regions by fluorescence in situ hybridization and spectral karyotyping. Genes Chromosomes Cancer 34:42–47
Slovak ML, Ho JP, Pettenati MJ, Khan A, Douer D et al (1994) Localization of amplified MYC gene sequences to double minute chromosomes in acute myelogenous leukemia. Genes Chromosomes Cancer 9:62–67
Rayeroux KC, Campbell LJ (2009) Gene amplification in myeloid leukemias elucidated by fluorescence in situ hybridization. Cancer Genet Cytogenet 193:44–53
O’Malley F, Rayeroux K, Cole-Sinclair M, Tong M, Campbell LJ (1999) MYC amplification in two further cases of acute myeloid leukemia with trisomy 4 and double minute chromosomes. Cancer Genet Cytogenet 109:123–125
Thomas L, Stamberg J, Gojo I, Ning Y, Rapoport AP (2004) Double minute chromosomes in monoblastic (M5) and myeloblastic (M2) acute myeloid leukemia: two case reports and a review of literature. Am J Hematol 77:55–61
Mathew S, Lorsbach RB, Shearer P, Sandlund JT, Raimondi SC (2000) Double minute chromosomes and c-MYC amplification in a child with secondary myelodysplastic syndrome after treatment for acute lymphoblastic leukemia. Leukemia 14:1314–1315
Storlazzi CT, Fioretos T, Surace C, Lonoce A, Mastrorilli A et al (2006) MYC-containing double minutes in hematologic malignancies: evidence in favor of the episome model and exclusion of MYC as the target gene. Hum Mol Genet 15:933–942
Bruyere H, Sutherland H, Chipperfield K, Hudoba M (2010) Concomitant and successive amplifications of MYC in APL-like leukemia. Cancer Genet Cytogenet 197:75–80
Bajaj R, Xu F, Xiang B, Wilcox K, Diadamo AJ et al (2011) Evidence-based genomic diagnosis characterized chromosomal and cryptic imbalances in 30 elderly patients with myelodysplastic syndrome and acute myeloid leukemia. Mol Cytogenet 4:3
Micale L, Augello B, Daniele G, Macchia G, L’Abbate A et al (2011) Amplification of the G allele at SNP rs6983267 in 8q24 amplicons in myeloid malignancies as cause of the lack of MYC overexpression? Blood Cells Mol Dis 47:259–261
Paulsson K, Lassen C, Kuric N, Billstrom R, Fioretos T et al (2003) MYC is not overexpressed in a case of chronic myelomonocytic leukemia with MYC-containing double minutes. Leukemia 17:813–815
Sloand EM, Pfannes L, Chen G, Shah S, Solomou EE et al (2007) CD34 cells from patients with trisomy 8 myelodysplastic syndrome (MDS) express early apoptotic markers but avoid programmed cell death by up-regulation of antiapoptotic proteins. Blood 109:2399–2405
Stasik CJ, Nitta H, Zhang W, Mosher CH, Cook JR et al (2010) Increased MYC gene copy number correlates with increased mRNA levels in diffuse large B-cell lymphoma. Haematologica 95:597–603
Vasikova A, Belickova M, Budinska E, Cermak J (2010) A distinct expression of various gene subsets in CD34 + cells from patients with early and advanced myelodysplastic syndrome. Leuk Res 34:1566–1572
Soucek L, Nasi S, Evan GI (2004) Omomyc expression in skin prevents Myc-induced papillomatosis. Cell Death Differ 11:1038–1045
Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ et al (2008) Modelling Myc inhibition as a cancer therapy. Nature 455:679–683
Fukazawa T, Maeda Y, Matsuoka J, Yamatsuji T, Shigemitsu K et al (2010) Inhibition of Myc effectively targets KRAS mutation-positive lung cancer expressing high levels of Myc. Anticancer Res 30:4193–4200
Sodir NM, Swigart LB, Karnezis AN, Hanahan D, Evan GI et al (2011) Endogenous Myc maintains the tumor microenvironment. Genes Dev 25:907–916
Meyer N, Penn LZ (2008) Reflecting on 25 years with MYC. Nat Rev Cancer 8:976–990
Cole MD, Cowling VH (2008) Transcription-independent functions of MYC: regulation of translation and DNA replication. Nat Rev Mol Cell Biol 9:810–815
Moser R, Toyoshima M, Robinson K, Gurley KE, Howie HL et al (2012) MYC-driven tumorigenesis is inhibited by WRN syndrome gene deficiency. Mol Cancer Res 10:535–545
Yang D, Liu H, Goga A, Kim S, Yuneva M et al (2010) Therapeutic potential of a synthetic lethal interaction between the MYC proto-oncogene and inhibition of aurora-B kinase. Proc Natl Acad Sci USA 107:13836–13841
Murga M, Campaner S, Lopez-Contreras AJ, Toledo LI, Soria R, Montaña MF, D'Artista L, Schleker T, Guerra C, Garcia E, Barbacid M, Hidalgo M, Amati B, Fernandez-Capetillo O (2011) Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat Struct Mol Biol 18:1331–1335
Toyoshima M, Howie HL, Imakura M, Walsh RM, Annis JE et al (2012) Functional genomics identifies therapeutic targets for MYC-driven cancer. Proc Natl Acad Sci USA 109:9545–9550
Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J et al (2011) BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146:904–917
Larramendy ML, Niini T, Elonen E, Nagy B, Ollila J et al (2002) Overexpression of translocation-associated fusion genes of FGFRI, MYC, NPMI, and DEK, but absence of the translocations in acute myeloid leukemia. A microarray analysis. Haematologica 87:569–577
Qian Z, Fernald AA, Godley LA, Larson RA, Le Beau MM (2002) Expression profiling of CD34+ hematopoietic stem/progenitor cells reveals distinct subtypes of therapy-related acute myeloid leukemia. Proc Natl Acad Sci USA 99:14925–14930
Acknowledgments
This work, in the laboratory of the authors, was provided by grants SAF11-23796 and ISCIII-RETIC RD06/0020/0017 to JL, and FIS 11/00397 to MDD.
Conflict of interest
The authors declare that they have no conflict of interest regarding the publication of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Delgado, M.D., Albajar, M., Gomez-Casares, M.T. et al. MYC oncogene in myeloid neoplasias. Clin Transl Oncol 15, 87–94 (2013). https://doi.org/10.1007/s12094-012-0926-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-012-0926-8