
Abstract
The absence of pentose-utilizing enzymes in Saccharomyces cerevisiae is an obstacle for efficiently converting lignocellulosic materials to ethanol. In the present study, the genes coding xylose reductase (XYL1) and xylitol dehydrogenase (XYL2) from Pichia stipitis were successfully engineered into S. cerevisae. As compared to the control transformant, engineering of XYL1 and XYL2 into yeasts significantly increased the microbial biomass (8.1 vs. 3.4 g/L), xylose consumption rate (0.15 vs. 0.02 g/h) and ethanol yield (6.8 vs. 3.5 g/L) after 72 h fermentation using a xylose-based medium. Interestingly, engineering of XYL1 and XYL2 into yeasts also elevated the ethanol yield from sugarcane bagasse hydrolysate (SUBH). This study not only provides an effective approach to increase the xylose utilization by yeasts, but the results also suggest that production of ethanol by this recombinant yeasts using unconventional nutrient sources, such as components in SUBH deserves further attention in the future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The yeast Saccharomyces cerevisiae is used universally for industrial ethanol production because of its ability to produce high concentrations of ethanol and high inherent ethanol tolerance [1]. However, there are two bottlenecks for industrial ethanol production by S. cerevisiae from lignocellulosic materials. The one is the fermentation inhibitors (such as weak acids, furfural and phenolic compounds) produced in the hydrolytic process [2], and the other is the inability of S. cerevisiae to utilize pentose sugars (mainly produced from hydrolysis of hemicelluloses). As a consequence, both the ethanol yield and productivity were significantly decreased [3].
Xylose is one of the major five-carbon sugars present in lignocellulosic materials such as sugarcane bagasse and other agricultural residues. It can, however, be fermented to ethanol by bacteria, yeasts and filamentous fungi [4]. In naturally xylose-utilizing yeasts such as Pichia stipitis, Pachysolen tannophilus and Candida shehatae, the main enzymatic steps in xylose metabolism are catalyzed by xylose reductase (XYL1) and xylitol dehydrogenase (XYL2). Xylose is first oxidized by XYL1 to xylitol, which is then oxidized by XYL2 to xylulose [5]. Since the S. cerevisiae cannot utilize xylose, but does utilize and ferment its isomer d-xylulose, the obvious first step to allow xylose metabolism is to introduce a heterologous pathway converting xylose to xylulose [6–8].
In the present study, we have engineered two xylose metabolic genes (XYL1 and XYL2) into S. cerevisiae from the xylose-utilizing yeast, P. stipitis. The influence of XYL1 and XYL2 activities on ethanol production during xylose fermentation was investigated by using strains overexpressing XYL1 and XYL2. Moreover, the ethanol production from sugarcane bagasse hydrolysate using the engineered yeast strains was also investigated.
Materials and methods
Strains and media
S. cerevisiae H158 (MATα leu2-3 leu2-112 ura3-52 trp1-289 his4-519 prb1 cir+) was cultivated in YPD medium (10 g/L yeast extract, 20 g/L peptone, 20 g/L glucose) or defined minimal medium (6.7 g/L yeast nitrogen base without amino acids). Glucose and xylose (30 g/L) was added to the minimal medium in fermentation experiments. P. stipitis CBS 6,054 was maintained on agar plate (10 g/L yeast extract, 20 g/L peptone, 20 g/L xylose, 15 g/L agar).
Cloning of P. stipitis XYL1 and XYL2 genes and construction of recombinant plasmids
The DNA fragment encoding the P. stipitis XYL1 and XYL2 were amplified from the genomic DNA by PCR with the two specific primers (Up1: 5′-ATAAAGCTTATGCCTTCTATTAAGTTGAACTCTGG-3′, Down1: 5′-TTAGGA-TCCTTAGACGAAGATAGGAATCTTGTCCC-3′; Up2: GTCGGATCCATGACTGC-TAACCCTTCCTTGGTGTTG-3′, Down2: CATGAATTCTTACTCAGGGCCGTCAA-TGAGACACTTG-3′) supplied with BamHI/HindIII and BamHI/EcoRI restriction sites, respectively. The primers were based on the published sequences of the P. stipitis XYL1 (GenBank accessioin no. X59465) and XYL2 (GenBank accessioin no. AF127801) [9, 10]. The PCR reaction was performed in 25-μl reaction mixtures (0.15 μM each primer, 1 μl of template DNA [about 10 ng of genomic DNA], 12.5 μl PCR premix [Invitrogen 403061]). Denaturation, annealing and polymerization were carried out for 1 min at 94 °C, 1 min at 59.5 °C, and 1 min at 72 °C, respectively for 35 cycles. An E. coli/S. cerevisiae shuttle vector, pYX212 (Novagen), was used for protein expression. The XYL1 and XYL2 genes obtained from PCR amplification were gel-purified and digested with restriction enzymes before cloning into pYX212. The digested fragments were ligated to pYX212 expression vector predigested with the same restriction enzymes (Fig. 1). To multiply the recombinant plasmids, competent E. coli cells (DH5α) were transformed with the two plasmids by using calcium chloride and heat-shock treatment [11]. Purification of the plasmids was carried out with a Mini Plasmid Purification Kit (Omega).

Construction of recombinant plasmids
Yeast transformation
Yeast transformation was performed by the electroporation methods as described by the manufacturer (Bio-Rad, USA). The positive transformants were selected from SC-Ura agar plates (20 g/L glucose, 6.7 g/L yeast nitrogen base without amino acids, 10 % amino acid supplement solution excluding uracil, 20 g/L agar) after incubation at 30 °C for 72 h.
Fermentation in shake flasks
After pre-cultivation of the positive transformants in 5 mL SC-Ura medium for 3 days, yeast cells were aerobically cultivated for 3 days at 30 °C in 50 mL minimal medium supplemented with glucose (10 g/L) and xylose (15 g/L) with shaking at 200 rpm. Cell pellets were harvested by centrifugation at 4 °C and washed with cold NaCl solution (0.9 %) and resuspended in 200 mL fermentation medium. The fermentation was carried out in a 500 mL flask (in triplicates) sealed with two layers of Saran wrap (under oxygen-limiting conditions) in an incubator at 150 rpm. Samples (yeast cells or culture supernatant) were collected at intervals and stored at −70 °C before analysis the enzyme activities, substrates and fermentation products.
Preparation of cell-free extract
After batch fermentation in flasks, the yeast cells were harvested by centrifugation (3,000×g) at 4 °C. The cell pellet was resuspended in 0.1 M sodium phosphate (pH 7.0), containing 1 mM MgCl2, 0.5 mM EDTA and 0.5 mM dithiothreitol, and vortexed together with an equal volume of glass beads (0.5 mm diameter). Cells were disrupted by vortexing six times 60 s. The samples were cooled on ice for 30 s in between the vortex steps. Cell debris and glass beads from the cell extract were separated by centrifugation and the remaining supernatant was used for enzyme determinations.
Enzyme assays
The activity of XYL1 was measured spectrophotometrically by monitoring the oxidation of NADPH at 340 nm in a reaction solution with following composition: 0.1 M sodium phosphate buffer (pH 7.0), 0.2 M xylose, and 0.15 mM NADPH [5]. The activity of XYL2 was determined by the method described previously [12]. The standard assay mixture for XYL2 activity contained 50 mM MgCl2 and 300 mM xylitol in 50 mM Tris–HCl (pH 9.0) buffer. All reactions were started by the addition of 0.1 mL of a 20 mM NAD(P)+ solution to a final volume of 1.0 mL. One unit of enzyme activity refers to 1 μmol of NAD(P)+ H produced/min. Protein concentrations were determined by the Bradford assay method using bovine serum albumin as the standard [13].
Fermentation with sugarcane bagasse hydrolysate (SUBH)
The pretreatment and enzymatic hydrolysis methods for the preparation of SUBH were described in previous reports [14, 15]. The fermentation experiments were carried out with a 5-L bioreactor (Roch Mechatronics Inc). The preparation of yeast cells was similar to the fermentation in shake flasks except that the culture volume was 500 mL. Cell pellets were harvested by centrifugation (3,000×g) and inoculated into 1.5 L SUBH supplemented with a nutrient solution, giving a final concentration of 0.5 g/L (NH4)2HPO4, 0.025 g/L MgSO4·7H2O, 1.38 g/L NaH2PO4·H2O, and 1 g/L yeast extract. The fermentation was maintained at 30 °C with an agitation speed at 500 rpm. Throughout the fermentation period (72 h), pH was controlled at 5.5 by automatic addition of 0.5 M NaOH and 1 M H2SO4. The fermentation was repeated three times and samples were collected and stored at −70 °C before analysis of the substrates and fermentation products.
Analysis of fermentation products
The cell growth was monitored spectrophotometrically at 600 nm. The glucose, xylose, xylitol, and ethanol concentrations were determined by high-performance liquid chromatography (HPLC) method as described by Walfridsson et al. [12]. The separation was carried out with the using of an Aminex ion-exclusion HPX-87H cation-exchange column (BioRad, USA) at 65 °C with 5 mM H2SO4 as the mobile phase. The separated compounds were detected by a 410 differential refractometer (Waters, Milford, MA, USA).
Results
Expression of XYL1 and XYL2 genes in S. cerevisiae
The specific XYL1 and XYL2 activities of the cell-free extracts from recombinant yeasts transformed with different vectors are summarized in Table 1. As compared to the control strain, the strain SXYL1 exhibited the highest specific activity of XYL1, whereas the strain (SXYL2) produced more XYL2 activity. The highest specific activities of XYL1 and XYL2 were 10.41 and 13.22 U/mg, respectively. Both the activities of XYL1 and XYL2 were simultaneously elevated after the two genes were co-transformed into S. cerevisiae. One of the co-transformants (SXYL12) which showed the highest XYL2/XYL1 ratio (about 6/1) was selected for further investigations.
Growth, products formation and sugar utilization by recombinant yeast strains
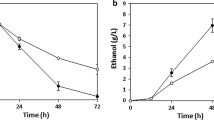
As shown in Fig. 2a, yeast strains transformed with either XYL1 or XYL2 genes grew faster than the control strain. However, the highest microbial biomass was achieved by the strain SXYL12. The ethanol production profile was in good agreement with the microbial growth rate (Fig. 2b). But the highest ethanol yield (6.9 g/L) was recorded in yeast co-expressing XYL1 and XYL2. Moreover, engineering of the XYL1 gene into S. cerevisiae (SXYL1 and SXYL12) significantly increased the xylitol production (Fig. 2c). As shown in Fig. 3a, engineering of XYL1 and XYL2 into yeasts did not affect the glucose consumption rate. However, the yeast strains expressing XYL1 (SXYL1 and SXYL12) significantly increased the xylose consumption rate (Fig. 3b).

Growth curves (a), ethanol (b) and xylitol (c) formation by different S. cerevisiae strains. Growth rate was monitored as OD at OD600. One OD600 was converted to 0.21 g cells/L (dry wt). For clarity purposes, standard deviations have not been added for this figure

Glucose (a) and xylsoe (b) utilization by different S. cerevisiae strains
Fermentation with sugarcane bagasse hydrolysate (SUBH)
As shown in Table 2, co-expressing of XYL1 and XYL2 in yeasts significantly increased ethanol (P < 0.05) and xylitol (<0.01) yield from SUBH. After 72 h fermentation, the glucose and mannose were both depleted by the two recombinant yeast strains. During the fermentation, the strain XYL12 consumed more xylose than H158 (P < 0.05), whereas the strain H158 tended to utilize more galactose (Table 2). No significant difference was observed for arabinose utilization between the two yeast strains (P > 0.05).
Discussion
Ethanol is the most widely used liquid biofuel and is fermented from sugars, starches or lignocellulosic materials [16]. Although the production of ethanol by fermentation of sugars has already been commercially established, the conversion technologies for producing of ethanol from lignocelluloses are still under development and have not been demonstrated commercially [17]. Since the pentose sugars (i.e. xylose) comprise a high percentage of the available sugars in lignocellulosic materials [4], its fermentation is essential for the economic conversion of lignocellulose to ethanol. In this study, we have successfully engineered two xylose metabolic genes (XYL1 and XYL2) into S. cerevisiae from P. stipitis. As a result, both the ethanol yield and xylose consumption rate were significantly elevated.
The yeast strain co-expressing XYL1 and XYL2 (SXYL12) consumed more xylose than other engineered strains (SC, SXYL1 and SXYL2) after 72 h fermentation with a xylose-containing medium. The highest ethanol yield obtained in shake flasks for the most efficient recombinant yeast strain (SXYL12) was 6.8 g/L, which is much higher than a reported yeast strain transformed only with the XYL1 gene [1]. As shown in Table 3, there are many reports about the ethanol yield in cultures with glucose and xylose by recombinant yeast strains. However, most of them showed quite low production levels, except in one study a higher level was obtained [21]. Actually, it is difficult to compare the production levels, since the carbon sources (glucose/xylsoe) in different studies are variable, and optimization is likely to further improve the production efficiency. It is note worthy that the xylose consumption rate in the early stage (before 24 h) was not significantly improved (Fig. 3b). This is probably due to the fact that yeast maintains a strict hierarchy in terms of sugar utilization and glucose is at the top [23]. In the present study, the glucose consumption rate was not affected by the engineering of XYL1 or XYL2, and free glucose in the fermentation medium was depleted after 30 h fermentation.
The xylitol is an intermediate metabolite and is produced in the first enzymatic step during xylose metabolism. Previous study indicated that most xylose-utilizing fungi produce considerable amounts of xylitol from xylose, and only species containing also the NADH-dependent XYL1 activity are capable of producing ethanol from it [4, 24]. We observed that yeast transformed with XYL1 gene (SXYL1 and SXYL12) significantly elevated the xylitol concentration. As compared to SXYL12, the strain SXYL1 produced more xylitol during the fermentation (Fig. 2c). This is due to the highest XYL1 activity (10.41 U/mg) produced in SXYL1. Moreover, the absence of XYL2 activity may also result in the accumulating of xylitol. In most naturally xylose-utilizing fungi, xylitol formation is a consequence of the inability of the cells to oxidize reduced cofactors (i.e. NADH) in the absence of oxygen [25]. The intracellular redox imbalance facilitates the unfavorable excretion of xylitol and reduced the substrates for ethanol formation [26]. Since the ratio of XYL1 to XYL2 plays an important role in maintaining yeast cellular redox balance [1], and a higher level of XYL2 is believed to be necessary to drive the xylose toward central metabolism (ethanol formation) [27, 28], the strain SXYL12 showing the highest XYL2/XYL1 ratio (about 6/1) has therefore, been selected for further investigation.
The ethanol production from lignocellulosic materials such as the sugarcane bagasse hydrolysate was investigated. As compared to the control strain (H158), the SXYL12 consumed more xylose (P < 0.01) and produced more ethanol (P < 0.05) from SUBH. To our astonishment, the strain H158 consumed more galactose than SXYL12 during the fermentation. It is a well known fact that of the many carbon sources, yeast prefers glucoses and enzymes of the galactose metabolic pathway are not expressed in the presence of glucose [29]. Carbon sources such as raffinose and glycerol neither induce nor repress the ability of galactose to activate the GAL gene [30]. In this study, the free glucose was completely depleted during the fermentation. Coupled with its inability of S. cerevisiae H158 to efficiently utilize the xylose, the expression of GAL may be activated in the presence of free galactose. Moreover, our results also validated the existence of a strict hierarchy for sugar utilization in yeasts and the xylose cannot prevent the galactose utilization after the free glucose is completely depleted.
In summary, the engineering of exogenous xylose-metabolizing genes into S. cerevisiae not only elevated the xylose consumption rate, but also increased the yield of ethanol from a synthetic medium. Interestingly, engineering of these genes in yeasts also increased the yield of ethanol from lignocellulosic materials, such as the SUBH. This study not only provides an attractive approach to increase the xylose utilization by yeasts, but the results also suggest that production of ethanol by this recombinant yeasts using unconventional nutrient sources, such as components in SUBH deserves further attention in the future.
References
Watanabe S, Saleh AA, Pack SP, Annaluru N, Kodaki T, Makino K (2007) Ethanol production from xylose by recombinant Saccharomyces cerevisiae expressing protein engineered NADP+-dependent xylitol dehydrogenase. J Biotechnol 130:316–319
Palmqvist E, Hahn-Hägerdal B (2000) Fermentation of lignocellulosic hydrolysates. II: inhibitors and mechanisms of inhibition. Bioresour Technol 74:25–33
Hahn-Hägerdal B, Galbe M, Gorwa-Grauslund G, Liden G, Zacchi G (2006) Bio-ethanol. the fuel of tomorrow from the residues of today. Trends Biotechnol 24:549–556
McMillan JD, Boynton BL (1994) Arabinose utilization by xylose-fermenting yeasts and fungi. Appl Biochem Biotehcnol 45–46:569–584
Smiley KL, Bolen PL (1982) Demonstration of d-xylose reductase and d-xylitol dehydrogenase in Pachysolen tannophilus. Biotechnol Lett 4:607–610
Aristidou A, Penttilä M (2000) Metabolic engineering applications to renewable resource utilization. Curr Opin Biotechnol 11:187–198
Hahn-Hägerdal B, Wahlbom CF, Gardonyi M, Van Zyl WH, Cordero Otero RR, Jönsson LJ (2001) Metabolic engineering of Saccharomyces cerevisiae for xylose utilization. Adv Biochem Eng Biotechnol 73:53–84
Hahn-Hägerdal B, Karhumaa K, Jeppsson M, Gorwa-Grauslund MF (2007) Metabolic engineering for pentose utilization in Saccharomyces cerevisiae. Adv Biochem Eng Biotechnol 108:147–177
Amore R, Kötter P, Küster C, Ciriacy M, Hollenberg CP (1991) Cloning and expression in Saccharomyces cerevisiae of the NAD(P)H-dependent xylose reductase-encoding gene (XYL1) from the xylose-assimilating yeast Pichia stipitis. Gene 109:89–97
Shi NQ, Prahl K, Hendrick J, Cruz J, Lu P, Cho JY, Jones S, Jeffries T (2000) Characterization and complementation of a Pichia stipitis mutant unable to grow on d-xylose or l-arabinose. Appl Biochem Biotechnol 84–86:201–216
Sambrook J, Russel DW (2001) Molecular cloning—a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, New York
Watanabe S, Kodaki T, Makino K (2005) Complete reversal of coenzyme specificity of xylitol dehydrogenase and increase of thermostability by the introduction of structural zinc. J Biol Chem 280:10340–10349
Bradford MM (1976) A rapid sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Martin C, Alriksson B, Sjöde A, Nilvebrant NO, Jönsson LJ (2007) Dilute-sulphuric acid prehydrolysis of agricultural and agro-industrial residues for ethanol production. Appl Biochem Biotechnol 137–140:339–352
Alriksson B, Rose SH, Van Zyl WH, Sjöde A, Nilvebrant NO, Jönsson LJ (2009) Cellulase production from spent lignocellulose hydrolysates by recombinant Aspergillus niger. Appl Environ Microbiol 75:2366–2374
Lang X, Macdonald DG, Hill GA (2001) Recycle bioreactor for bioethanol production from wheat starchII. Fermentation and economics. Energy Sour 23:427–436
Demirbas A (2005) Bioethanol from cellulosic materials: a renewable motor fuel from biomass. Energy Sour 27:327–337
Eliasson A, Christensson C, Wahlbom CF, Hahn-Hägerdal B (2009) Anaerobic xylose fermentation by recombinant Saccharomyces cerevisiae carrying XYL1, XYL2, and XKS1 in mineral medium chemostat cultures. Appl Environ Microbiol 66:3381–3386
Roca C, Nielsen J, Olsson L (2003) Metabolic engineering of ammonium assimilation in xylose fermenting Saccharomyces cerevisiae improves ethanol production. Appl Environ Microbiol 69:4732–4736
Pitkänen JP, Aristidou A, Salusjärvi L, Ruohonen L, Penttilä M (2003) Metabolic flux analysis of xylose metabolism in recombinant Saccharomyces cerevisiae using continuous culture. Metab Eng 5:16–31
Kuyper M, Harhangi HR, Stave AK, Winkler AA, Jetten MS, de Laat WT, den Ridder JJ, Op den Camp HJ, van Dijken JP, Pronk JT (2003) High-level functional expression of a fungal xylsoe isomerase: the key to efficient ethanolic fermentation of xylose by Saccharomyces cerevisiae. FEMS Yeast Res 4:69–78
Zaldivar J, Borges A, Johansson B, Smits HP, Villas-Boas SG, Nielsen J, Olsson L (2002) Fermentation performance and intracellular metabolite patterns in laboratory and industrial xylose-fermenting Saccharomyces cerevisiae. Appl Microbiol Biotechnol 59:436–442
Sellick CA, Campbell RN, Reece RJ (2008) Galactose metabolism in yeast-structure and regulation of the leloir pathway enzymes and the genes encoding them. Int Rev Cell Mol Biol 269:111–150
Bruinenberg PM, De Bot PHM, Van Dijken JP, Scheffers TW (1984) NADH-linked aldose reductase: the key to anaerobic alcoholic fermentation of xylose by yeasts. Appl Microbiol Biotechnol 19:256–260
Bruinenberg PM, De Bot PHM, Van Dijken JP, Scheffers WA (1983) The role of redox balances in the anaerobic fermentation of xylose by yeasts. Eur J Appl Microbiol Biotechnol 18:287–292
Jeffries TW, Jin YS (2004) Metabolic engineering for improved fermentation of pentoses by yeasts. Appl Microbiol Biotechnol 63:495–509
Cornish-Bowden A, Hofmeyr JHS, Cardenas ML (1995) Strategies for manipulating metabolic fluxes in biotechnology. Bioorg Chem 23:439–449
Eliasson A, Hofmeyr JHS, Pedler S, Hahn-Hägerdal B (2001) The xylose reductase/xylitol dehydrogenase/xylulokinase ratio affects product formation in recombinant xylose-utilizing Saccharomyces cerevisiae. Enzyme Microb Technol 29:288–297
De Robichion-Szulmajster H (1958) Induction of enzymes of galactose pathway in mutants of Saccharomyces cerevisiae. Science 127:19–28
Bhat PJ, Murthy TVS (2001) Transcriptional control of the GAL/MEL regulation of yeast Saccharomyces cerevisiae: mechanism of galactose mediated signal transduction. Mol Microbiol 40:1059–1066
Acknowledgments
This work was granted by Prominent Youthfund Project of Sichuan Agricultural University with grant. No. 00924201.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jun, H., Jiayi, C. Metabolic engineering of Saccharomyces cerevisiae for increased bioconversion of lignocellulose to ethanol. Indian J Microbiol 52, 442–448 (2012). https://doi.org/10.1007/s12088-012-0259-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12088-012-0259-x