
Abstract
Purpose
Lamivudine treatment of chronic hepatitis B (CHB) is associated with frequent resistance and loss of clinical benefit. We present outcomes of lamivudine-refractory Japanese patients treated with entecavir for 3 years.
Methods
Eighty-two patients refractory to lamivudine therapy received entecavir 0.5 or 1 mg daily for 52 weeks in phase II study ETV-052, directly entered rollover study ETV-060, and received entecavir 1 mg daily. Responses were evaluated among patients with available samples.
Results
After 96 weeks in ETV-060 (148 weeks total entecavir treatment time), 55% (36/65) of patients had hepatitis B virus (HBV) DNA of >400 copies/mL, 85% (52/61) had alanine aminotransferase (ALT) of ≥1 × upper limit of normal (ULN), and 14.6% (7/48) achieved HBe seroconversion. A subset of 42 patients received entecavir 1 mg from phase II baseline through 148 weeks: 54% (19/35) had HBV DNA of >400 copies/mL, 84% (27/32) had ALT of ≥1 × ULN, and 15% (4/27) achieved HBe seroconversion. Sixteen patients in the 1-mg subset had baseline and week 148 evaluable biopsy pairs: 81% (13/16) showed histologic improvement and 38% (6/16) showed improvement in fibrosis. Genotypic resistance to entecavir emerged in 31 patients for a 3-year cumulative resistance probability of 35.9%. Entecavir was generally well tolerated during ETV-060, with no on-treatment ALT flares.
Conclusions
Long-term entecavir treatment of lamivudine-refractory CHB resulted in virologic suppression, ALT normalization, and improvements in liver histology. Resistance was consistent with that observed in worldwide studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic hepatitis B (CHB) infection is a global public health problem that is estimated to cause between 500,000 and 1.2 million deaths annually [1–3]. Three-quarters of all chronically infected individuals live in the Asia–Pacific region, where hepatitis B virus (HBV) is the leading cause of chronic hepatitis, cirrhosis, and hepatocellular carcinoma (HCC) [4]. In Japan, the prevalence of HBV infection was estimated to be 0.8% in 2000, and the vast majority of individuals are infected with HBV genotype C [4–6]. Genotype C virus has been associated with high rates of progression to the complications of CHB, including cirrhosis and HCC [7–11]. In addition to genotype, the level of HBV DNA in the serum is strongly associated with liver disease progression [12, 13]. Persistently detectable and elevated viral loads predict the highest risk of progression to cirrhosis and HCC [12–14]. Suppression of HBV replication with antiviral therapy may reduce the risk of complications and improve the long-term outcomes of CHB patients [15].
Lamivudine has been widely used for the treatment of CHB since its development and initial approval 10 years ago [16, 17]. Lamivudine has demonstrated efficacy and long-term safety and was shown to result in histologic improvement when administered for up to 3 years [16, 18, 19]. However, resistance to lamivudine emerges at a rate of approximately 20% per year and is found in approximately 70% of patients after 4 years of therapy [20, 21]. The emergence of lamivudine resistance may be associated with increases in HBV DNA and alanine aminotransferase (ALT) levels, and loss of histologic response [16, 18, 22]. In patients with cirrhosis, lamivudine resistance may lead to hepatic decompensation and HCC [15, 23, 24]. Recently published CHB treatment guidelines no longer recommend lamivudine as first-line therapy for treatment-naive patients because of the problems that resistance introduces in the management of individual patients and the negative impact that lamivudine resistance has on the subsequent use of other antivirals [25].
Entecavir is a guanosine nucleoside analog that has demonstrated efficacy against nucleoside-naive and lamivudine-refractory CHB [26–29]. In global clinical studies, patients with lamivudine-refractory CHB treated with entecavir 1 mg daily for 48 weeks experienced reduction in HBV DNA levels of more than 5 log copies/mL and improvements in hepatic necroinflammation and fibrosis [28, 29]. Treatment for up to 96 weeks resulted in continued improvement of virologic, biochemical, and serologic end points [30]. In contrast to the nucleoside-naive population, emergence of resistance to entecavir occurred more frequently in the lamivudine-refractory population [30, 31]. To date, there are limited data on the efficacy of entecavir treatment beyond 96 weeks in the lamivudine-refractory patient population. A phase II study in Japan (ETV-052) demonstrated the efficacy and safety of entecavir in Japanese patients who were refractory to lamivudine therapy [32]. Immediately following completion of treatment in study ETV-052, patients were eligible to enroll in rollover study ETV-060 and receive entecavir 1 mg daily for up to 96 weeks. We present efficacy, safety, and resistance results for all patients treated in ETV-052 who rolled over into study ETV-060 for a total entecavir treatment time of up to 3 years (148 weeks). A subset of this cohort received the recommended dose of entecavir (1 mg daily) continuously from ETV-052 baseline, and results for this subset are also reported.
Materials and methods
Study design
Study ETV-060 was a long-term rollover study designed to provide open-label entecavir to lamivudine-refractory patients who completed treatment in the phase II study ETV-052 in Japan. In study ETV-052, 84 patients were randomized 1:1 to entecavir 0.5 mg (n = 41) or 1 mg (n = 43) daily for 52 weeks [32]. At baseline in this study, all patients had detectable lamivudine-resistance substitutions. Patients who completed 52 weeks of dosing in ETV-052 could enroll in ETV-060 and receive entecavir 1.0 mg daily in an open-label fashion. After completing 96 weeks of treatment in study ETV-060, patients could discontinue therapy or were eligible to receive commercially available entecavir that was approved by Japanese health authorities while ETV-060 was ongoing. The current analysis reports results for patients who completed ETV-052 and were subsequently treated in ETV-060 (n = 82) for a total entecavir treatment time (ETV-052 plus ETV-060) of up to 148 weeks. This cohort is termed the lamivudine-refractory, long-term treatment cohort (Fig. 1).

Lamivudine-refractory, long-term treatment cohort. Eighty-two patients completed 52 weeks of treatment in study ETV-052 and entered rollover study ETV-060, with no interruption or gap in treatment. Sixty-five patients remained on treatment (entecavir 1.0 mg daily) through 96 weeks in study ETV-060, for a total entecavir treatment time of 148 weeks
During study ETV-060, clinical and laboratory measurements (serum chemistries, hematology, prothrombin time/international normalized ratio, and urinalysis) were assessed at baseline, weeks 2 and 4, and every 4 weeks thereafter throughout the dosing period. HBV DNA by PCR and HBV serologies were assayed at baseline, weeks 12 and 24, and subsequently every 24 weeks until week 96 or end of dosing. Liver biopsy specimens were obtained and scored for all patients at baseline and end (48 weeks) of study ETV-052, and repeat biopsy specimens were obtained at week 96 of study ETV-060 (148 weeks total entecavir treatment time) for patients who consented. Biopsy specimens were evaluated using the Knodell necroinflammatory and fibrosis scores and the corresponding New Inuyama classifications [33, 34].
Written informed consent was obtained from all patients, and the study was conducted in compliance with the Declaration of Helsinki, Good Clinical Practice Guidelines, and Articles/Notifications of the Ministry of Health and Labor in Japan.
Patients
The inclusion criteria for study ETV-052 have been fully described elsewhere [32]. Eligible patients were adults with CHB infection and either evidence of active viral replication (HBV DNA ≥105 copies/mL) despite at least 24 weeks of lamivudine therapy that was ongoing at the time of randomization or documented evidence of infection with HBV expressing lamivudine-resistance mutations. Patients could be hepatitis B e antigen (HBeAg)-positive or -negative and were required to have elevated levels of ALT [(1.3–10) × upper limit of normal (ULN)] and compensated liver disease. Exclusion criteria included coinfection with hepatitis C virus, hepatitis D virus, or human immunodeficiency virus; other forms of liver disease; therapy with any anti-HBV medication other than lamivudine within 24 weeks prior to randomization; and more than 12 weeks of therapy with a nucleoside or nucleotide analog (other than lamivudine) with activity against HBV. Pregnant and breast-feeding women were also excluded. All patients who completed 52 weeks of dosing in study ETV-052 were eligible to enroll in study ETV-060.
Efficacy and safety end points
Efficacy end points included the proportion of patients who achieved undetectable HBV DNA by PCR assay (<400 copies/mL), the proportion achieving ALT normalization (ALT ≤ 1.0 × ULN) among those with abnormal ALT at baseline, and the proportion with HBeAg loss and HBe seroconversion among those who were HBeAg-positive at baseline. Histologic results are presented for the cohort of patients who received entecavir 1 mg daily from phase II baseline and had evaluable liver biopsy pairs. Histologic improvement was defined as a ≥2-point decrease in the Knodell necroinflammatory score and no worsening of fibrosis (worsening: ≥1-point increase in the Knodell fibrosis score). Improvement in fibrosis was defined as a ≥1-point decrease in the Knodell fibrosis score. Histologic results were also assessed by the New Inuyama classification [34].
Safety analyses included the incidence of adverse events, serious adverse events, laboratory abnormalities, and discontinuations due to adverse events of treatment during study ETV-060, including results for patients treated beyond 96 weeks. ALT flare was defined as an on-treatment ALT measurement of more than 2 × baseline and more than 10 × ULN.
Resistance assessment
Genotypic analysis was performed on serum samples from all patients at baseline of study ETV-052 for evidence of the lamivudine-resistance substitution M204V/I in the HBV polymerase/reverse transcriptase. During study ETV-052, genotypic analysis to detect substitutions associated with entecavir resistance (at residues L180, T184, S202, M204, or M250 in the HBV polymerase/reverse transcriptase) was performed for patients with virologic breakthrough, defined as an increase in HBV DNA of ≥1 log10 copies/mL from nadir in two consecutive measurements or the last on-treatment measurement. During study ETV-060, serum samples were subjected to genotypic analysis to detect substitutions associated with entecavir resistance for patients who had HBV DNA of more than 400 copies/mL at week 100 or 148 (from study ETV-052 baseline), or at the end of treatment (for patients who discontinued prior to week 148), and for patients who experienced virologic breakthrough.
Assay methods
All clinical laboratory tests, including HBV DNA levels, HBV serologies, and genotypic analyses, were performed at a central laboratory designated by the sponsor (SRL, Inc., Tokyo, Japan). Serum HBV DNA levels were determined by the Roche Amplicor™ PCR assay (limit of quantification = 400 copies/mL; Roche Diagnostics K.K., Tokyo, Japan). Lamivudine-resistance substitutions were identified using a PCR enzyme-linked minisequence assay (Medical & Biological Laboratories Co., Ltd., Aichi, Japan). On-treatment resistance testing was carried out by extraction of HBV DNA followed by PCR amplification and sequencing of codons 1–344 of the reverse transcriptase encoding region.
Statistical analysis
Descriptive summaries were performed. Analyses of efficacy and safety end points were based on patients who received at least one dose of study medication in study ETV-060. For binary end points, patients with missing on-treatment measurements were treated as missing (noncompleter = missing analysis). Parameters represented by continuous variables were summarized by means and standard errors. Analyses of HBV DNA as a continuous parameter were applied after log10 transformation.
Results
Study population
Eighty-four patients were treated with entecavir in phase II study ETV-052, and 82 patients entered ETV-060, constituting the lamivudine-refractory, long-term treatment cohort (Fig. 1). Seventeen patients discontinued treatment during ETV-060 for the following reasons: adverse event (n = 8), protocol violation (n = 1), loss to follow-up (n = 1), and insufficient effect in the judgment of the investigator (n = 7). Sixty-five patients completed 96 weeks of treatment in ETV-060 for a total of 148 weeks of entecavir from ETV-052 baseline through ETV-060 (Fig. 1). Baseline (pretreatment) demographics and disease characteristics of this cohort (n = 82) are shown in Table 1. Eighty-seven percent (71/82) of patients were men, and mean age was 44 years. Mean HBV DNA level was 7.69 log10 copies/mL, mean ALT level was 135 IU/L, and 76% (62/82) of patients were HBeAg positive. All patients had documented lamivudine-resistance substitutions at M204. Ninety-four percent (77/82) of patients were infected with HBV genotype C.
Virologic response
HBV DNA was suppressed and decreased rapidly during phase II study ETV-052 [32]. For the 82 patients who entered ETV-060 after completing ETV-052, mean HBV DNA level decreased from 7.69 log10 copies/mL at pretreatment baseline to 3.99 log10 copies/mL at ETV-060 entry (after 52 weeks of entecavir treatment). HBV DNA was further suppressed during 96 weeks of treatment in ETV-060. At baseline of study ETV-060, 33% of patients (27/82) had HBV DNA of >400 copies/mL (Fig. 2), and this proportion increased to 55% (36/65) by week 96 of ETV-060 (148 weeks total entecavir treatment time). Of the 17 patients who discontinued treatment during ETV-060, one patient had HBV DNA of >400 copies/mL at the last on-treatment measurement.

Distribution of HBV DNA over time in the lamivudine-refractory, long-term treatment cohort. The proportion of patients with HBV DNA of >400 copies/mL increased through ETV-060 week 96 (148 weeks of total entecavir treatment time)
Biochemical response
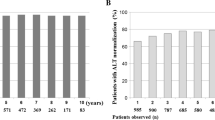
At pretreatment baseline, 95.1% (78/82) of patients had abnormal ALT (ALT > 1.0 × ULN; Table 1; Fig. 3). After 52 weeks of treatment in ETV-052, 79.5% (62/78) of patients had normalized ALT. After 96 weeks of further treatment in ETV-060 (148 weeks total entecavir treatment time), ALT had normalized in 85.2% (52/61) of patients.

Proportions of patients with normal ALT (ALT ≤ 1.0 × ULN) over time in the lamivudine-refractory, long-term treatment cohort. Seventy-eight patients had abnormal ALT (ALT > 1.0 × ULN) at pretreatment baseline. At week 96 of study ETV-060, patients had received a total of 148 weeks of entecavir therapy
Serologic response
Sixty-two patients (76%) were HBeAg-positive at pretreatment baseline (Table 1; Fig. 4). At ETV-060 entry, 16.1% (10/62) of these patients had achieved HBe seroconversion and the same number had lost HBeAg (Fig. 4). After 96 weeks in ETV-060 (148 weeks total entecavir treatment time), 33.3% of patients (16/48) had lost HBeAg and 14.6% (7/48) had undergone HBe seroconversion.

Proportions of patients with HBeAg loss and HBe seroconversion over time in the lamivudine-refractory, long-term treatment cohort. Sixty-two patients were HBeAg positive at pretreatment baseline. At week 96 of study ETV-060, patients had received a total of 148 weeks of entecavir therapy
Resistance analysis
No substitutions associated with entecavir resistance emerged during study ETV-052 [32]. Eighty-one of 82 patients were monitored for resistance from ETV-052 baseline through to the end of treatment in ETV-060 (1 patient refused consent for resistance testing). Thirty-one patients developed genotypic resistance to entecavir during the second or third year of treatment, of whom 21 experienced virologic breakthrough. The 3-year cumulative probability of resistance was 35.9% [35].
Safety
Mean exposure to entecavir during study ETV-060 was 101.3 weeks (range 7.1–148). All patients experienced at least one adverse event, and 11% (9/82) experienced serious adverse events (Table 2). One patient was diagnosed with HCC at week 57 of ETV-060. Eight patients (9.8%) discontinued treatment during ETV-060 because of adverse events, such as increased ALT, virologic breakthrough, and genotypic resistance emergence. Five of these eight patients had received entecavir 0.5 mg daily during phase II study ETV-052, and three received entecavir 1 mg from phase II baseline. There were no ALT flares during ETV-060, and no deaths were reported during the study.
Entecavir 1-mg cohort
A subset of 42 patients (42/82) received the recommended 1-mg dose of entecavir for lamivudine-refractory CHB from phase II baseline through to the end of treatment in study ETV-060. In this subset, among patients with available samples, 54% (19/35) had HBV DNA of >400 copies/mL, 84% (27/32) had ALT of ≥1 × ULN, and 15% (4/27) achieved HBe seroconversion after 3 years of continuous treatment with entecavir 1 mg daily. Genotypic resistance emerged in 13 patients in this cohort, and 9 of 13 patients experienced virologic breakthrough. The cumulative 3-year probability of resistance was 30.4%.
Sixteen (16/42) patients in the 1-mg cohort had paired evaluable liver biopsies from three time points: pretreatment (phase II) baseline, week 48, and week 148 total entecavir treatment time (ETV-060, week 96). Of these, 81% (13/16) demonstrated histologic improvement from baseline through week 148. The mean Knodell necroinflammatory score improved from 6.06 at baseline to 1.44 at week 148, and all patients (16/16) exhibited minimal necroinflammation (a Knodell necroinflammatory score of ≤3 points) at week 148 (Fig. 5a). Knodell fibrosis scores improved in 38% (6/16) of patients from baseline through week 148, and the mean Knodell fibrosis score decreased from 2.44 at baseline to 1.94 at week 148 (Fig. 5b). Liver biopsy assessments using the New Inuyama classification system confirmed the results obtained using the Knodell classification system (data not shown).

Distribution of Knodell necroinflammatory scores (a) and Knodell fibrosis scores (b) at baseline, year 1 (48 weeks), and year 3 (148 weeks) for the 16 patients who had evaluable liver biopsies at all 3 time points
Discussion
This report describes the results of 3 years of continuous entecavir therapy in a lamivudine-refractory patient population. All patients in the lamivudine-refractory, long-term treatment cohort had highly elevated levels of HBV DNA with documented lamivudine-resistance mutations at baseline, and 94% were infected with HBV genotype C. This represents a population with potentially poor long-term outcomes. Patients with lamivudine-resistant HBV may have cross-resistance to other antivirals, and genotype C infection is associated with low rates of HBe seroconversion and high rates of liver disease progression [7, 25, 36]. These results show that entecavir therapy for up to 3 years for this population resulted in durable HBV DNA suppression and ALT normalization. More than 50% of patients in the cohort achieved undetectable HBV DNA and almost 90% normalized ALT by year 3. Similar levels of HBV DNA suppression and ALT normalization were observed for the subset of patients who received entecavir 1 mg daily throughout the treatment period. Among patients with liver biopsies from three time points (all of whom received the recommended 1-mg dose of entecavir from phase II baseline), substantial improvements in liver histology were observed: more than 80% of patients demonstrated histologic improvement at year +++3 and slow improvements in fibrosis were observed in 38% of patients.
In previously published results of a multinational clinical trial, entecavir demonstrated potent inhibition of viral replication in HBeAg-positive, lamivudine-refractory patients infected with a variety of HBV genotypes (A–D) [28, 30]. In that trial, after 48 weeks of treatment with entecavir 1 mg daily, the mean change from baseline in HBV DNA was −5.11 log10 copies/mL, and 19% of patients achieved HBV DNA of >300 copies/mL. Among patients who continued to a second year of entecavir therapy, the mean change from baseline in HBV DNA increased to −5.9 log10 copies/mL, and 40% of patients achieved HBV DNA of >300 copies/mL. In the current study in Japanese patients, 54% achieved HBV DNA of >400 copies/mL. The higher proportion of Japanese patients suppressing HBV DNA to below the PCR limit of quantification in the current study likely reflects the effect of an additional year of entecavir therapy, as well as the lower baseline HBV DNA (7.69 log10 vs. 9.59 log10 copies/mL in the multinational study). The relatively low rate of HBe seroconversion observed in this study (15%) may be related to infection with genotype C virus. In studies in Japan and elsewhere in Asia, HBV genotype C has been associated with lower seroconversion rates than with other HBV genotypes [7, 36–38].
Achieving and maintaining HBV DNA suppression is a principal goal of CHB therapy [25, 39]. Data from prospective long-term studies have shown that elevated HBV DNA levels are associated with the development of long-term complications including cirrhosis and HCC [12–14]. Other research has correlated durable HBV DNA suppression with improved liver histology among antiviral-treated patients [19, 40]. Liaw et al. [15] showed that lamivudine therapy benefits CHB patients with advanced liver disease by reducing the risk of liver disease progression, including the development of HCC. In the present study, the reduction in hepatic necroinflammation and fibrosis observed in a subset of patients through 3 years, along with the durable virologic suppression observed in the larger cohort, suggests that entecavir helps halt or reverse liver disease progression that can lead to poor long-term outcomes.
The emergence of lamivudine resistance can lead to serious clinical consequences, including elevated levels of HBV DNA, exacerbations of hepatitis, and hepatic decompensation [18, 22, 23, 41]. While early studies of patients with lamivudine-resistant HBV suggested that switching to adefovir was efficacious, subsequent work demonstrated the rapid emergence of adefovir resistance in this patient population [42–44]. The emergence of adefovir resistance in this setting can be associated with viral rebound and hepatic decompensation [45]. Adding adefovir to ongoing lamivudine for patients who have developed lamivudine resistance has been recommended as a strategy to reduce the subsequent emergence of adefovir resistance [25, 46]. This strategy is most efficacious in patients with low HBV DNA levels and requires continued resistance surveillance [47, 48]. Studies evaluating the combination of entecavir with adefovir in lamivudine-resistant patients are currently in progress.
The rate of genotypic resistance to entecavir reported here is consistent with the rate that has been observed in multinational populations of lamivudine-refractory patients [49]. In nucleoside-naive patients, emergence of entecavir resistance is rare because of entecavir’s potent viral load reduction and high genetic barrier to resistance [49, 50]. Substitutions at M204 ± L180 were detected at baseline for all patients described in this report and have been shown in previous studies to reduce in vitro susceptibility to entecavir by approximately eightfold [51]. Resistance to entecavir requires the presence of the rtM204V/I lamivudine-resistance substitution plus at least one additional amino acid substitution at rtT184, rtS202, or rtM250. In the current study, for the subset of patients who received entecavir 1 mg daily throughout the treatment period, the cumulative rate of entecavir resistance was 30% through 3 years. This is consistent with the rate observed in the entire lamivudine-refractory, long-term treatment cohort and in multinational studies of lamivudine-refractory patients through 3 years (36%) [49]. Combining entecavir with an antiviral with a different resistance profile, such as tenofovir or adefovir, may result in less frequent resistance emergence.
Entecavir was well tolerated during treatment in study ETV-052, with no discontinuations due to adverse events and three early on-treatment flares that were transient and associated with declining levels of HBV DNA [32]. Throughout the extended treatment period during ETV-060, entecavir continued to be well tolerated with relatively few discontinuations and no ALT flares observed. There were no deaths during the study, and one patient was diagnosed with HCC at week 57 of ETV-060. The extent to which long-term treatment with entecavir may reduce development of HCC in CHB patients remains under investigation.
In summary, these results show that treatment with entecavir for up to 3 years in lamivudine-refractory CHB results in continued benefit beyond the first year, including durable HBV DNA suppression and progressive improvements in liver histology, with a resistance profile consistent with that observed in other studies. Entecavir at the recommended dose of 1 mg daily is an option for patients with lamivudine-refractory CHB. Additional research evaluating the combination of entecavir plus adefovir or tenofovir in this patient population is ongoing.
References
Lavanchy D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J Viral Hepat 2004;11:97–107
Lee WM. Hepatitis B virus infection. N Engl J Med 1997;337:1733–1745
World Health Organization. Hepatitis B Fact Sheet WHO/204. Geneva: World Health Organization; 2000 [cited 2008 June 30]. http://www.who.int/mediacentre/factsheets/fs204/en/.
Merican I, Guan R, Amarapuka D, Alexander MJ, Chutaputti A, Chien RN, et al. Chronic hepatitis B virus infection in Asian countries. J Gastroenterol Hepatol 2000;15:1356–1361
Usuda S, Okamoto H, Iwanari H, Baba K, Tsuda F, Miyakawa Y, et al. Serological detection of hepatitis B virus genotypes by ELISA with monoclonal antibodies to type-specific epitopes in the pre S2-region product. J Virol Methods 1999;80:97–112
Hou J, Liu X, Gu F. Epidemiology and prevention of hepatitis B virus infection. Int J Med Sci 2005;2:50–57
Orito E, Mizokami M, Sakugawa H, Michitaka K, Ishikawa K, Ichida T, et al. A case-control study of clinical and molecular biological differences between hepatitis B viruses of genotypes B and C. Hepatology 2001;33:218–223
Jang JW, Lee YC, Kim MS, Lee SY, Bae SH, Choi JY, et al. A 13-year longitudinal study of the impact of double mutations in the core promoter region of hepatitis B virus on HBeAg seroconversion and disease progression in patients with genotype C chronic active hepatitis. J Viral Hepat 2007;14:169–175
Yu MW, Yeh SH, Chen PJ, Liaw YF, Lin CL, Liu CJ, et al. Hepatitis B virus genotype and DNA level and hepatocellular carcinoma: a prospective study in men. J Natl Cancer Inst 2005;97:265–272
Orito E, Mizokami M. Differences of HBV genotypes and hepatocellular carcinoma in Asian countries. Hepatol Res 2007;37(Suppl 1):S33–S35
Chan HLY, Hui AY, Wong ML, Am Tse, Hung LC, Wong VW, et al. Genotype C hepatitis B virus infection is associated with an increased risk of hepatocellular carcinoma. Gut 2004;53:1494–1498
Iloeje UH, Yang HI, Su J, Jen CL, You SL, Chen CJ, et al. Predicting cirrhosis risk based on the level of circulating hepatitis B viral load. Gastroenterology 2006;130:678–686
Chen CJ, Yang HI, Su J, Jen CL, You SL, Lu SN, et al. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA 2006;295:65–73
Yuen MF, Yuan HJ, Wong DK, Yuen JC, Wong WM, Chan AO, et al. Prognostic determinants for chronic hepatitis B in Asians: therapeutic implications. Gut 2005;54:1610–1614
Liaw YF, Sung JJ, Chow WC, Farrell G, Lee CZ, Yuen H, et al. Lamivudine for patients with chronic hepatitis B and advanced liver disease. N Engl J Med 2004;351:1521–1531
Dienstag JL, Schiff ER, Wright TL, Perrillo RP, Hann HW, Goodman Z, et al. Lamivudine as initial treatment for chronic hepatitis B in the United States. N Engl J Med 1999;341:1256–1263
Lai CL, Chien RN, Leung NW, Chang TT, Guan R, Tai DI, et al. A one-year trial of lamivudine for chronic hepatitis B. N Engl J Med 1998;339:61–68
Lok AS, Lai CL, Leung N, Yao GB, Cui ZY, Schiff ER, et al. Long-term safety of lamivudine treatment in patients with chronic hepatitis B. Gastroenterology 2003;125:1714–1722
Dienstag JL, Goldin RD, Heathcote EJ, Hann HW, Woessner M, Stephenson SL, et al. Histological outcome during long-term lamivudine therapy. Gastroenterology 2003;124:105–117
Chang TT, Lai CL, Chien RN, Guan R, Lim SG, Lee CM, et al. Four years of lamivudine treatment in Chinese patients with chronic hepatitis B. J Gastroenterol Hepatol 2004;19:1276–1282
Lai CL, Dienstag J, Schiff E, Leung NW, Atkins M, Hunt C, et al. Prevalence and clinical correlates of YMDD variants during lamivudine therapy for patients with chronic hepatitis B. Clin Infect Dis 2003;36:687–696
Papatheodoridis GV, Dimou E, Laras A, Papadimitropoulos V, Hadziyannis SJ. Course of virologic breakthrough under long-term lamivudine in HBeAg-negative precore mutant HBV liver disease. Hepatology 2002;36:219–226
Di Marco V, Marzano A, Lampertico P, Andreone P, Santantonio T, Almasio PL, et al. Clinical outcome of HBeAg-negative chronic hepatitis B in relation to virological response to lamivudine. Hepatology 2004;40:883–891
Andreone P, Gramenzi A, Cursaro C, Biselli M, Cammà C, Trevisani F, et al. High risk of hepatocellular carcinoma in anti-HBe positive liver cirrhosis patients developing lamivudine resistance. J Viral Hepat 2004;11:439–442
Lok ASF, McMahon BJ. Chronic hepatitis B. Hepatology 2007;45:507–539
Chang TT, Gish RG, de Man R, Gadano A, Sollano J, Chao YC, et al. A comparison of entecavir and lamivudine for HBeAg-positive chronic hepatitis B. N Engl J Med 2006;354:1001–1010
Lai CL, Shouval D, Lok AS, Lai CL, Shouval D, Lok AS, et al. Entecavir versus lamivudine for patients with HBeAg-negative chronic hepatitis B. N Engl J Med 2006;354:1011–1020
Sherman M, Yurdaydin C, Sollano J, Silva M, Liaw YF, Cianciara J, et al. Entecavir for treatment of lamivudine-refractory, HBeAg-positive chronic hepatitis B. Gastroenterology 2006;130:2039–2049.
Chang TT, Gish RG, Hadziyannis SJ, Cianciara J, Rizetto M, Schiff ER, et al. A dose-ranging study of the efficacy and tolerability of entecavir in lamivudine-refractory chronic hepatitis B patients. Gastroenterology 2005;129:1198–1209
Sherman M, Yurdaydin C, Simsek H, Silva M, Liaw YF, Rustgi VK, et al. Entecavir therapy for lamivudine-refractory chronic hepatitis B: improved virologic, biochemical and serology outcomes through 96 weeks. Hepatology 2008;48:99–108.
Gish RG, Lok AS, Chang TT, de Man RA, Gadano A, Sollano J, et al. Entecavir therapy for up to 96 weeks in patients with HBeAg-positive chronic hepatitis B. Gastroenterology 2007;133:1437–1444.
Suzuki F, Toyoda J, Katano Y, Sata M, Moriyama M, Imazeki F, et al. Efficacy and safety of entecavir in lamivudine-refractory patients with chronic hepatitis B: randomized controlled trial in Japanese patients. J Gastroenterol Hepatol 2008;23(9):1320–1326.
Knodell RG, Ishak KG, Black WC, Chen TS, Craig R, Kaplowitz N, et al. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. Hepatology 1981;1:431–435.
Ichida F, Tsuji T, Omata M, Ichida T, Inoue K, Kamimura T, et al. New Inuyama classification: new criteria for histological assessment of chronic hepatitis. Int Hepatol Commun 1996;6:112–119
Yokosuka O, Kumada H, Toyota J, Takaguchi K, Kobashi H, Shindo M, et al. Three-year assessment of entecavir (ETV) resistance in nucleoside-naïve and lamivudine (LVD) refractory Japanese patients with chronic hepatitis B (CHB). Hepatol Int 2008;2:A161. Abstract No.: FP067
Nakayoshi T, Maeshiro T, Nakayoshi T, Nakasone H, Sakugawa H, Kinjo F, et al. Difference in prognosis between patients infected with hepatitis B virus with genotype B and those with genotype C in the Okinawa Islands: a prospective study. J Med Virol 2003;70:350–354
Duong TN, Horiike N, Michitaka K, Yan C, Mizokami M, Tanaka Y, et al. Comparison of genotypes C and D of the hepatitis B virus in Japan: a clinical and molecular biological study. J Med Virol 2004;72:551–557
Kao JH, Wu NH, Chen PJ, Lai MY, Chen DS. Hepatitis B genotypes and the response to interferon therapy. J Hepatol 2000;33:998–1002
Liaw YF, Leung N, Kao JH, Piratvisuth T, Gane E, Han KH, et al. Asian-Pacific consensus statement on the management of chronic hepatitis B: a 2008 update. Hepatol Int 2008;2:263–283
Mommeja-Marin H, Mondou E, Blum MR, Rousseau F. Serum HBV DNA as a marker of efficacy during therapy for CHB infection: analysis and review of the literature. Hepatology 2003;37:1309–1319.
Liaw YF, Chien RN, Yeh CT. No benefit to continue lamivudine therapy after emergence of YMDD mutations. Antivir Ther 2004;9:257–62
Peters MG, Hann Hw H, Martin P, Heathcote EJ, Buggisch P, Rubin R, et al. Adefovir dipivoxil alone or in combination with lamivudine in patients with lamivudine-resistant chronic hepatitis B. Gastroenterology 2004;126:91–101
Fung SK, Chae HB, Fontana RJ, Conjeevaram H, Marrero J, Oberhelman K, et al. Virologic response and resistance to adefovir in patients with chronic hepatitis B. J Hepatol 2006;44:283–290
Lee YS, Suh DJ, Lim YS, Jung SW, Kim KM, Lee HC, et al. Increased risk of adefovir resistance in patients with lamivudine-resistant chronic hepatitis B after 48 weeks of adefovir dipivoxil monotherapy. Hepatology 2006;43:1385–1391
Fung SK, Andreone P, Han SH, Rajender Reddy K, Regev A, Keeffe EB, et al. Adefovir-resistant hepatitis B can be associated with viral rebound and hepatic decompensation. J Hepatol 2005;43:937–943
Rapti I, Dimou E, Mitsoula P, Hadziyannis SJ. Adding-on versus switching-to adefovir therapy in lamivudine-resistant HBeAg-negative chronic hepatitis B. Hepatology 2007;45:307–313
Lampertico P, Vigano M, Manenti E, Iavarone M, Lunghi G, Colombo M. Adefovir rapidly suppresses hepatitis B in HBeAg-negative patients developing genotypic resistance to lamivudine. Hepatology 2005;42:1414–1419
Lampertico P, Marzano A, Levrero M, Santantonio T, Di Marco V, Brunetto M, et al. Adefovir and lamivudine combination therapy is superior to adefovir monotherapy for lamivudine-resistant patients with HBeAg-negative chronic hepatitis B. J Hepatol 2007;46(Suppl 1):S191
Tenney DJ, Rose RE, Baldick CJ, Pokornowski KA, Eggers BJ, Fang J, et al. Long-term monitoring shows hepatitis B virus resistance to entecavir in nucleoside-naïve patients is rare through 5 years of therapy. Hepatology 2009;49:1503–1514
Colonno RJ, Rose R, Baldick CJ, Levine S, Pokornowski K, Yu CF, et al. Entecavir resistance is rare in nucleoside-naïve patients with hepatitis B. Hepatology 2006;44:1656–1665
Baldick CJ, Tenney DJ, Mazzucco CE, Eggers BJ, Rose RE, Pokornowski KA, et al. Comprehensive evaluation of hepatitis B reverse transcriptase substitutions associated with entecavir resistance. Hepatology 2008;47:1473–1482
Acknowledgments
This work was carried out with a grant from Bristol-Myers Squibb. Taku Seriu, Hiroki Ishikawa, and Nobuyuki Masaki are employees of Bristol-Myers Squibb. Masao Omata serves as an advisor to Bristol-Myers Squibb. The authors thank Chitomi Hasebe, Teruaki Kawanishi, Kazuyuki Suzuki, Yoshiyuki Ueno, Satoshi Mochida, Osamu Yokosuka, Hidetsugu Saito, Naohiko Masaki, Keiko Tatemoto, Yoshiyuki Arakawa, Yasunobu Matsuda, Shunichi Okada, Eiji Tanaka, Etsuro Orito, Shinichi Kakumu, Noboru Hirashima, Eiichi Tomita, Takashi Kumada, Takeshi Okanoue, Norio Hayashi, Kazihiro Katayama, Michio Kato, Harumasa Yoshihara, Taizo Hijioka, Michiko Shindo, Kosaku Sakaguchi, Gotaro Yamada, Kazuaki Chayama, Keisuke Hino, Norio Horiike, Shotaro Sakisaka, Ryukichi Kumashiro, Keisuke Hamasaki, Hiroshi Yatsuhashi, Masataka Seike, Yutaka Sasaki, Katsuhiro Hayashi, Shinichi Fijioka, Koichi Takaguchi, Hiroshi Ikeda, Masanori Miyake, Yasuyuki Araki, Kozo Fujio, and Masaharu Ando, who were investigators in this study; Kazuyuki Suzuki, Osamu Yokosuka, Takeshi Okanoue, Norio Hayashi, Yasushi Shiratori, and Hirohito Tsubouchi, who were on the coordinating committee for this study; and Chifumi Sato, Kendo Kiyosawa, and Kyuichi Tanikawa, who were on the efficacy and safety committee for this study. The work was carried out at Sapporo-Kosei General Hospital, Hokkaido, Japan; Keiyukai Yoshida Hospital, Hokkaido, Japan; Iwate Medical University Hospital, Iwate, Japan; Tohoku University Hospital, Miyagi, Japan; Chiba University Hospital, Chiba, Japan; Musashino Red Cross Hospital, Tokyo, Japan; Tokyo Women’s Medical University Hospital, Tokyo, Japan; Toranomon Hospital, Tokyo, Japan; Nagoya University Hospital, Aichi, Japan; Nagoya City University Hospital, Aichi, Japan; Aichi Medical University Hospital, Aichi, Japan; Ogaki Municipal Hospital, Gifu, Japan; University Hospital, Kyoto Prefectural University of Medicine, Kyoto, Japan; Akashi Municipal Hospital, Hyogo, Japan; Okayama University Hospital, Okayama, Japan; and Kurume University Hospital, Fukuoka, Japan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Financial support for research provided by Bristol-Myers Squibb.
An erratum to this article is available at http://dx.doi.org/10.1007/s12072-010-9224-0.
Rights and permissions
About this article
Cite this article
Karino, Y., Toyota, J., Kumada, H. et al. Efficacy and resistance of entecavir following 3 years of treatment of Japanese patients with lamivudine-refractory chronic hepatitis B. Hepatol Int 4, 414–422 (2010). https://doi.org/10.1007/s12072-009-9162-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12072-009-9162-x