
Abstract
Pitaya (Hylocereus polyrhizus) is a highly valued functional food that is widely planted in Southeast Asia. However, with increased soil salinization in cultivation areas, a better understanding of the molecular mechanisms associated with salt stress responses in red pitaya is necessary. Herein, RNA-Seq was used to de novo assemble and characterize the transcriptomic profiles of red pitaya roots in response to salt stress. A total of 73,589 transcripts were obtained and the average sequence length was 1308 bp. From these transcripts, 26,878 unique transcripts were successfully matched to 13,519 Swiss-prot proteins. Gene ontology (GO) annotations showed that within the categories of molecular function, biological function, and cell component, catalytic activity (GO:0003824), metabolic process (GO:0008152), and cell part (GO:0044464) were the most enriched, respectively. 2624 transcripts were significantly differentially expressed among three time points (3 h, 7 h, and 30 h) following exposure to 450 mM NaCl. Furthermore, 261 genes were up-regulated and 61 down-regulated in all three of the time points. Glycolysis/gluconeogenesis was one of the most significantly modulated pathways. The findings presented herein provide further insight into salt stress responses in pitaya and will provide a valuable resource for future functional studies examining salt adaptations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Salt tolerance is an important quantitative trait (Gu et al. 2018a, b; Quesada et al. 2002), with approximately 7% of the world’s total land area affected by salt (Ghassemi et al. 1995; Szabolcs and Salinisation 1994) and these areas are on the rise due to irrigation or land clearing (FAO Statistics). Soil salt limits agricultural production by affecting plant growth, limiting crop yields, and reducing crop quality (Yang 2008). Thus, improved crop salt tolerance is needed to improve growth in areas at risk of salinization.
The identification of plant genes involved in salinization responses is required to unravel the molecular basis of agronomical important traits and aid in the selection of salt-tolerant lines. Genetic and molecular studies have indicated that salt tolerance is complex in crop plants. Studies utilizing quantitative trait loci (QTL) analysis have been utilized to scan natural variations related to salt tolerance traits in Arabidopsis (DeRose-Wilson and Gaut 2011) and other crops (Collins et al. 2008), with genes modulating these QTLs identified. In rice, Streptomyces lividans K+ channel (SKC1), which encodes an HKT-type Na+-selective transporter involved in unloading Na+ from the xylem (Ren et al. 2005). In durum wheat, the Na+ exclusion 1 (Nax1) and the Na+ exclusion 2 (Nax2) function in Na+ exclusion (Byrt et al. 2007; Huang et al. 2006). In Arabidopsis, Salt 1 (RAS1) has been shown to be an ABA and a salt stress-inducible gene (Ren et al. 2010). Furthermore, SOS2 ser/thr protein kinase (Qiu et al. 2003; Shi et al. 2000), the high-affinity K+ transporter (HKT) (Rus et al. 2001), and CCCH zinc finger proteins (CCCH-ZFP) (Mazumdar et al. 2017) have been shown to be associated with salt tolerance responses.
Red pitaya (Hylocereus polyrhizus) is a tropical fruit that is a highly valued functional food. This fruit is popular in Southeast Asia where soil resources are limited, thus culturing in areas with salinization is of interest. In previous studies examining pitaya, a catalase gene, HuCAT3, was found to be associated with abiotic stress responses to peroxide (H2O2), drought, cold, and salt stresses (Nie et al. 2015). Also in pitaya, a total of 309 expressed sequence tags (ESTs) and 138 unique ESTs were identified in response to drought stress (Fan et al. 2014). Herein, to better characterize red pitaya gene expression associated with salt stress responses, the root transcriptome was de novo assembled using RNA sequencing (RNA-Seq) and differential expression associated with salt stress was examined. This study provides a valuable gene resource for future functional studies pertaining to salt tolerance in pitaya.
Results
Sample Sequencing and De Novo Assembly
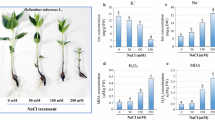
To investigate salt tolerance in red pitaya, seedlings were treated with 300, 450, or 600 mM NaCl for 1 week (Fig. 1a). In the seedling treated with 600 mM NaCl, most of the roots experienced rot and the seedlings looked wilted. However, at the lower salt concentrations, the seedling grew well (Fig. 1a), thus showing that a short-term NaCl concentration of 450 mM NaCl can be tolerated. During drought and salt stress, some plants showed an increase in soluble sugar contents to confer tolerance (Mark et al. 2012), thus levels were examined herein. Under450 mM NaCl concentration, the soluble sugar content increased at all of the time points, but with the lowest elevation at 7 h and the highest at 30 h (Fig. 1b).

a Plant growth in different salt concentrations for 1 week. b Soluble sugar content in seedling treated with 450 mM NaCl
To identify genes associated with salt stress responses in the roots, samples were treated with 450 mM NaCl and collected at four time points (0 h, 3 h, 7 h, and 30 h) for RNA-Seq, with the 0 h sample used as a control. Using the Illumina HiSeq2500 platform, an average of 23,306,060 paired-end raw reads with an average length of 150 bp were generated from each of the 12 root sample datasets (20,225,559–25,097,895 reads; three replicates for each time point). In the de novo transcriptome, 73,589 total transcripts, with a total sequence length of 96.3 Mb, were obtained. The average sequence length was 1308 bp, with a range of 200 bp to 22,390 bp, and the N50 length was 1883 bp (Table 1).
Sanger Sequencing and qRT-PCR Validations
To confirm the sequence assembly, nine transcripts were randomly selected and sequenced. The average CDS length was found to be 1179 bp ± 228 bp, with a range of 771–1662 bp. The amplified CDS regions showed a 99–100% identity to their associated red pitaya transcripts (Table 2). To validate the RNA-Seq expressional data, 12 contigs (genes) were randomly selected from the transcriptome and examined via qRT-PCR. The qRT-PCR results showed fold changes and time associated patterns that were consistent with the RNA-Seq dataset (Fig. 2). These findings confirm the reliability of the obtained data.

Twelve DEG contigs (x-axis) were randomly selected from the RNA-Seq dataset and validated by qRT-PCR. The y-axis displays natural log (ln)- transformed fold changes for both datasets
Functional Annotations
A total of 26,878 unique transcripts were successfully mapped to 13,519 protein sequences in Swiss-Prot, with the sequences associated with 960 species. The species with the most sequences in common was Arabidopsis thaliana (5473 genes), followed by Mus musculus (616 genes), Oryza sativa subsp. japonica (608 genes), and Caenorhabditis elegans (597 genes). Similar results were seen when performing a query against the NCBI refseq RNA database, with 24,569 transcripts successfully mapped to 16,060 genes (Supplementary Tables 2 and 3).
GO annotations were obtained using the PANTHER database (Fig. 3). Within the categories of molecular functions, biological functions, and cell component, the most enriched terms were associated with catalytic activity (GO:0003824), metabolic process (GO:0008152), and cell part (GO:0044464), respectively (Supplementary Table 4). Pathway analysis was performed using the KEGG database, with 405 pathways associated with the obtained annotated DEGs. The most enriched pathways were associated with metabolic pathways (926 genes), biosynthesis of secondary metabolites (407 genes), and biosynthesis of antibiotics (231 genes; Fig. 4 and Supplementary Table 5).

The obtained 13,519 annotated DEGs were classified into three GO categories: molecular function (a), biological process (b), and cellular component (c). Displayed percentages are based on gene numbers for a given GO category

A display of the gene numbers (y-axis) for the top 20 most enriched pathways (x-axis)
Identifying DEGs Associated with Salt Stress Responses
When comparing the experimental group that was treated with 450 mM NaCl to the control, a total of 2624 significant DEGs were identified at 3 h, 7 h, and 30 h after the start of the treatment. Of these, 540 genes were up-regulated at 3 h post-treatment, 445 genes at 7 h, and 994 at 30 h. Down-regulated genes were also identified at 3 h (549 genes), 7 h (257 genes), and 30 h (869 genes) post-treatment (Table 3 and Supplementary Table 6). The DEG numbers were correlated with the soluble sugar content levels, with the highest levels in both noted at 30 h. Interestingly, 261 genes were shown to be up-regulated across all three of the time points, while 61 genes were down-regulated (Fig. 5 and Table 4).

Numbers represent deduplicated UniProtKB IDs included in each subset
Functional Annotations of DEGs Associated with Salt Stress Responses
A total of 1617 DEGs were successfully mapped to 1284 protein sequences in Swiss-Prot, while 1007 DEGs were uncharacterized transcripts (Supplementary Table 7). The DEGs found to be in common with all three of the time points in samples exposed to 450 mM NaCl were further analyzed using GO and KEGG analyses. GO analysis revealed that the DEGs were mainly associated with catalytic activity (GO:0003824), metabolic process (GO:0008152), and cell part (GO: 0044464) in the categories of molecular function, biological process, and cellular component (Fig. 6).

Annotated DEGs were classified into three GO categories (molecular function (a), biological process (b), and cellular component (c)). Displayed percentages are based on gene numbers for a given GO category
KEGG pathway analysis showed that the DEGs associated with salt stress were involved in 86 pathways, and they were mainly enriched in secondary metabolite, carbohydrate, and amino acid metabolic pathways (Supplementary Table 8). The secondary metabolite pathway was comprised of 15 essential pathways and contained 33 DEGs. Of those, pathways associated with secondary metabolite biosynthesis were the most prevalent and contained 18 DEGs (Supplementary Table 9). Among the 18 DEGs, 15 were up-regulated and 3 were down-regulated. The down-regulated DEGs included Contig31365 (PORA), Contig6213 (TYRA2), and Contig13954 (COMT1; Supplementary Table 10). The carbohydrate metabolic pathway was comprised of 13 essential pathways and contained 33 DEGs, with the glycolysis/gluconeogenesis pathway being the most significant pathway of the 13 and containing 8 up-regulated DEGs (Supplementary Tables 11 and 12). The amino acid metabolic pathway was comprised of 9 essential pathways and contained 23 DEGs, with the biosynthesis of amino acids pathway being the most significant of the 9 and containing 10 DEGs (Supplementary Table 13). Among the 10 DEGs, 9 DEGs were up-regulated and 1 DEG was down-regulated, Contig6213 (TYRA2; Supplementary Table 14).
When comparing DEGs associated with secondary metabolite biosynthesis, glycolysis/gluconeogenesis, and amino acid biosynthesis, 5 up-regulated genes were found to be in common. These genes included Contig25524, Contig7784, Contig29387, Contig29388, and Contig9566, which encode glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 6-phosphofructokinase 1 (pfkA), phosphoglycerate kinase (PGK), PGK, and fructose-bisphosphate aldolase, class I (ALDO), respectively.
Discussion
Salt exposure activates a complex response that inhibits plant growth and other physiological processes (Albacete et al. 2008; Naeem et al. 2012; Radwan et al. 2015). When compared with crop plants, few genomic studies have focused on stress response molecular markers in the economically important pitaya plant. When examining stress responses, RNA-Seq analysis is an effective strategy compared to EST or microarray analyses. Herein, de novo transcriptome sequencing and assembly was performed using RNA-Seq and differential expression associated with red pitaya salt stress responses was identified. These identified DEGs were then annotated and further examined via GO and KEGG analyses. The findings presented herein will provide valuable insights into the molecular mechanisms of salt stress responses in red pitaya.
The de novo assembly identified a total of 73,589 transcripts (Table 1), with 26,878 (36.5%) unique transcripts functionally characterized and annotated (Supplementary Tables 2 and 3). In a previous study utilizing RNA-Seq, 122,677 transcripts associated with red pitaya pulp coloration were identified, with approximately 99.99% of all of the transcripts annotated (Hua et al. 2016). However, the annotation rate herein was much lower, with other reports also showing much lower annotation rates. For example, only 25% to 50% of the assembled sequences in the Jamaican fruit bat (Artibeus jamaicensis) (Shaw et al. 2012), the carrot transcriptome (Iorizzo et al. 2011), and the European abalone (Haliotistuberculata) (Xia et al. 2015) were functionally annotated. Therefore, our data are comparable to the latter studies, with the uncharacterized transcripts possibly attributed to non-coding RNAs (ncRNAs), transcribed transposable elements (TEs), pitaya specific genes, or pseudogenes.
In previous studies, many different pathways have been implicated in response to salt stress in different plants.. In Aspergillus oryzae, the DEGs were mainly enriched in the areas of fatty acid biosynthesis and degradation, linoleic acid metabolism, arginine biosynthesis, and arginine and proline metabolism (He et al. 2018). In Arabidopsis, they were mainly associated with transcription factors, cell wall remodeling, ethylene biosynthesis, and signaling pathways (Shen et al. 2014). Tomato DEGs were mainly enriched in amino acid and carbohydrate metabolic pathways (Zhang et al. 2017). In this study, DEGs were involved in 86 pathways, which suggested that there are complex biological processes trigged by saline stress in pitaya. The DEGs were mainly enriched in metabolic pathways including secondary, carbohydrate, and amino acid metabolic pathways (Supplementary Table 8). These pathways appear to play an important role in pitaya salt tolerance.
Secondary metabolites, such as steroids, alkaloids, and phenylpropanoids, have been closely associated with abiotic stress tolerance (Ewas et al. 2017; Hua et al. 2016; Qiao et al. 2017; Resmi et al. 2015). In red pitaya,, 15 essential pathways comprised the secondary metabolite pathway and included 33 DEGs (26 up-regulated and 7 down-regulated genes; Supplementary Table 8). These pathways included the biosynthesis of isoquinoline alkaloid, sesquiterpenoid, triterpenoid, porphyrin, steroid, tropane, piperidine, pyridine alkaloid, phenylpropanoid, and chlorophyll metabolism. This finding further supports that secondary metabolites play important roles in abiotic stress tolerance.
Carbohydrate metabolism is a fundamental metabolic process in living organisms and is one of the most sensitive to salt stress in red pitaya (Table 4). The carbohydrate metabolic process category is comprised of 13 essential pathways, including glycolysis/gluconeogenesis, carbon fixation in photosynthetic organisms, fructose and mannose metabolism, and starch and sucrose metabolism. Within the glycolysis/gluconeogenesis pathway, 8 up-regulated DEGs were identified after short and continuous salt stress. Glycolysis is the process of converting glucose into pyruvate and generating small amounts of ATP (energy) and NADH (reducing power) (Berg et al. 2010). It is a central pathway that produces important precursor metabolites that are either six-carbon compounds (glucose-6-phosphate and fructose-6-phosphate) or three-carbon compounds (glycerone-phosphate, glyceraldehyde-3-phosphate, glycerate-3-phosphate, phosphoenolpyruvate, and pyruvate). Glycolysis/gluconeogenesis has been implicated in many stress responses, such as a resistance to aluminum stress in wheat (Hamilton et al. 2001) and rice (Wang et al. 2014), drought stress in ramie and rice (An et al. 2016; Lenka et al. 2011), and freezing stress in loquat defense (Xu et al. 2017).
During the salt stress response in red pitaya, the carbon fixation in the photosynthetic organism pathway was the second most significant pathway. Salt stress has been reported to limit photosynthesis and CO2 diffusion by decreasing stomatal and mesophyll conductance (Flexas et al. 2004). In photosynthetic organisms, carbon fixation has been shown to be impacted during salt stress in rice (Lee et al. 2016) and cotton (Chen et al. 2016). Collectively, these findings suggest that the glycolysis/gluconeogenesis and carbon fixation in photosynthetic organism pathways play an important role in red pitaya salt stress tolerance.
Amino acid metabolism is crucial to the salt stress response (Zhang et al. 2017). In red pitaya, 9 biosynthetic and metabolic pathways associated with amino acid metabolism were identified following salt stress and included 9 DEGs, 8 up-regulated and 1 down-regulated. This may indicate that nitrogen distributions have been altered in response to salt stress. Two of the up-regulated genes, prolyl 4-hydroxylase (P4H) and aspartate aminotransferase (GOT1), are associated with arginine and proline metabolism (Supplementary Table 7). Proline can protect plants during stress and aids in plant growth when exposed to salinization (de Carvalho et al. 2013; Razavizadeh and Ehsanpour 2009; Werner and Finkelstein 1995). Prolyl 4-hydroxylasegene (P4H1) is induced by low oxygen stress (Asif et al. 2009). GOT1 is important in the modulation of carbon and nitrogen distributions that are needed for amino acid metabolism by catalyzing the reversible transamination of an amino group from aspartate to α-ketoglutarate to yield glutamate and oxaloacetate (Boutet et al. 2005). Therefore, P4H and GOT1 up-regulation during salt stress may have an important role in enhancing salt tolerance in red pitaya.
Futher more, 5 of the up-regulated DEGs were found in all of the three key pathways (biosynthesis of secondary metabolites, glycolysis/gluconeogenesis, and biosynthesis of amino acids), namely, Contig25524 (GAPDH), Contig7784 (pfkA), Contig29387 (PGK), Conting29388 (PGK), and Contig9566 (ALDO). In plants, pfkA is a key enzyme in glycolysis that catalyzes the conversion of fructose 6-phosphate to fructose 1,6-diphosphate; which can then be converted to phosphodihydroxy acetone and glyceraldehyde 3-phosphate, which is catalyzed by ALDO. GAPDH is an oxidative phosphorylation enzyme, which further catalyzes glyceraldehyde 3-phosphate into 1,3-bisphosphoglycerate. This step is the only oxidization reaction in glycolysis. PGK catalyzes 1,3-bisphosphoglycerate into 3-phosphoglycerate and ATP. PDC catalyzes pyruvate into acetaldehyde and CO2. During an infection or environmental stress, such as osmotic, salt, heat, ABA, or methyl viologen stress, GAPDH has been seen to be induced (Zhang et al. 2011). Furthermore, GAPDH overexpression was shown to enhance salt stress tolerance in Oryza sativa (Zhang et al. 2011). In plants exposed to butanol toxicity, pfkA and pyruvate kinase (pykA) overexpression increased the stress tolerance (Ventura et al. 2013). Pfk has also been shown to play an adaptive role during an infection in Rhizoctonia solani (Mutuku and Nose 2012). During thermal stress in Saccharomyces cerevisiae, PGK was identified as a DEG following transcriptional analysis (Piper et al. 1986). In Arabidopsis, PGK2 overexpression increased salt stress tolerance (Nie et al. 2015), while in transgenic tobacco, PGK2a-P overexpression improved yields during salt stress (Joshi et al. 2016). Moreover, ALDO has been associated with abiotic stress responses to heat stress (Traylor-Knowles et al. 2017), hypoxic stress (Abe et al. 2007), drought stress (Khueychai et al. 2015), and salt stress (Fan et al. 2009). These findings suggested that these genes play an important role in the salt stress response in red pitaya.
Interestingly, we found that 38.4% DEGs (1007 DEGs) were functionally uncharacterized and unannotated, which possibly attributed to ncRNAs, TEs, or pitaya specific genes (Supplementary Table 7). ncRNAs are sequences with very low or no potential for encoding protein and are functional in developmental processes or stress responses (Borah et al. 2018; Brant and Budak 2018; Zhao et al. 2016). In Arabidopsis, long noncoding RNA (lncRNA) SVALKA affected plant freezing tolerance, while overexpressing lncRNA DRIR enhanced drought and salt stress tolerance (Kindgren et al. 2018; Qin et al. 2017). In Zea mays, miR159, miR164, miR167, miR393, miR408 and miR528 were involved in root development and stress responses (Liu et al. 2012). TEs are abundant in plants that play an important role in genome and gene evolution (Vicient 2010). In plants, TEs contribute to the origin of ncRNAs and generate novel gene regulatory networks in stress response and development (Cho 2018; Wang et al. 2016, 2017). Epigenetic regulation of TEs affected gene expression (Le et al. 2015). Thus, we convinced that these uncharacterized DEGs will contribute to better understand salt stress response in pitaya.
Material and Methods
Plants and Materials
Red pitaya seeds were geminated and grown in a greenhouse under controlled conditions (14 h photoperiod, 28 °C ± 1 °C, and 60% ± 5% relative humidity). Three-month-old seedlings were treated with 300, 450, or 600 mM NaCl for 1 h–36 h. The treated seedlings were collected, the soluble sugar content was measured, and RNA was extracted from the roots.
Sample Collection and Preparation
Total RNA was extracted using Trizol (Invitrogen, USA) according to the manufacturer’s protocol. RNA quality was assessed using 1% agarose gel electrophoresis and a RNA Nano 6000 Assay Kit with an Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). A total of 1.5 μg of total RNA per sample was utilized as the starting material. Sequencing libraries were generated using a NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (New England Biolabs, USA) according to the manufacturer’s instructions, and index codes were added to attribute sequences to each sample. Finally, paired-end cluster generation and sequencing were performed using an Illumina Hiseq platform.
Quality Control
Raw data in a fastq format were processed using in-house perl scripts. In this step, clean data were obtained by removing reads containing adapters or ploy-Ns or low quality reads from the raw data. All of the downstream analyses were based on the obtained clean high-quality data.
De Novo Transcriptome Assembly
Two programs were used to assemble the transcriptome. De novo assembly of the RNA-Seq reads was first conducted for each sample separately using the Trinity program (Haas et al. 2013) with default parameters. The program assembled the high-quality RNA-Seq reads into sequences, clustered the sequences, and then reported full-length transcripts. Next, de novo assembly of the 12 transcriptomes was performed using a contig assembly program (CAP3; https://www.msi.umn.edu/sw/cap3-contig) with parameters -p 99 -f 2 -o 100. Statistical analysis was performed using the NGS QC Toolkit v.2.3.3 with default parameters (Trapnell et al. 2012).
Identification of Differential Gene Expression
The TopHat program (Trapnell et al. 2012) was utilized to map the clean RNA-Seq library reads to the de novo assembled reference transcriptome. The program ‘multicov’ in the bed tools was then applied to count the number of alignments for each transcript per sample. To identify differentially expressed genes (DEGs) between two datasets, a pairwise comparison was performed using the ‘run_DE_analysis.pl’ function in the trinityrnaseq-2.0.6 program (Haas et al. 2013) with the parameter setting of ‘--method edgeR’. For data to be considered high quality, a log2 FC (fold change) > 2, a count value for each gene in the two datasets >20 and an adjusted p value FDR (False Discovery Rate) ≤ 0.001 had to be obtained.
Transcriptome Annotations and Classifications
The generated transcripts were queried against the Swiss-Prot protein database (http://www.uniprot.org/) using BLASTX and the NCBI refseq RNA database using BLASTN with an E-value cut-off of 0.00000001. Gene ontology (GO) annotations were obtained using the PANTHER database (http://www.pantherdb.org/) and KEGG pathway analysis was performed using KASS (KEGG Automatic Annotation Server v.2.1; http://www.genome.jp/kaas-bin/kaas_main?).
Transcriptome Assembly Validation Via Sanger Sequencing and DEG Validations Via qRT-PCR
To validate the assembly accuracy, 9 randomly selected transcripts from the de novo transcriptome were selected. PCR primers were designed using primer premier 5 (Premier Biosoft, USA) (Supplementary Table 1) and whole coding sequence (CDS) regions were amplified. PCR reactions were performed using a thermo-cycler (Sensquest, Germany) with a PCR kit (DSMix, Dongsheng Biotech, China) and red pitaya cDNA was obtained according to the manufacturer’s instructions. Amplifications were performed in a total reaction volume of 20 μl [20 ng cDNA and 0.5 μl of each primer (10 μM)]. The PCR reaction protocol was as follows: an initial denaturation at 95 °C for 4 min, followed by 35 cycles of denaturation at 95 °C for 30 s, annealing at 57 °C for 30 s and extension at 72 °C for 1–2 min, with an additional extension at 72 °C for 10 min. PCR products were sequenced and analyzed using vector NTI 9.1 software (Invitrogen corporation, USA).
To validate the identified RNA-Seq based DEGs, quantitative real-time PCR (qRT-PCR) was employed with 12 randomly selected transcripts evaluated (Supplementary Table 1). Prior to performing qRT-PCR, total RNA was extracted and cDNA was synthesized (Xia et al. 2015). The qRT-PCR reaction was performed on a Roche Light Cycler 480 Real-time PCR System (Roche, Switzerland) using SYBR Green qPCR mix (Dongsheng Biotech, China) according to the manufacturer’s instructions. The amplifications were performed in a total volume of 10 μl [5 μl of 2X SYBR Green Master Mix reagent (Promega, USA), 1 μl of 1:10 diluted cDNA, and 0.4 μl of each primer (10 μM)]. The thermal cycling protocol was as follows: an initial denaturation at 95 °C for 3 min, followed by 40 cycles of denaturation at 95 °C for 15 s, annealing at 60 °C for 15 s, and an extension at 72 °C for 20 s. An additional temperature-ramping step from 95 °C to 65 °C was used to produce a melting curve. Three technical replicates were performed for each sample and EF1α was used as an internal control. Expression levels were normalized to three housekeeping genes using the ΔΔCt method. Expressional differences between the control and experimental samples were evaluated using a two-tailed t test.
Quantification of Soluble Sugars
The soluble sugar content was quantified as previously described (Wei et al. 2014).
Abbreviations
- FC:
-
Fold change
- GO:
-
Gene ontology
- FDR:
-
False Discovery Rate
- CDS:
-
Coding sequence
- DEG:
-
Differentially expressed gene
- QTL:
-
Quantitative trait loci
- ABA:
-
Abscisic acid
- pfkA:
-
6-phosphofructokinase 1
- PGK:
-
Phosphoglycerate kinase
- GOT1:
-
Aspartate aminotransferase
- ALDO:
-
Fructose-bisphosphate aldolase, class I
- SQLE:
-
Squalene monooxygenase
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- qRT-PCR:
-
Quantitative real-time PCR
- ncRNAs:
-
Non-coding RNAs
- TEs:
-
Transposable elements
- RAS1 :
-
Salt 1
- SKC1 :
-
Streptomyces lividans K+ channel
- Nax1 :
-
Na + exclusion 1
- Nax2 :
-
Na + exclusion 2
- P4H :
-
Prolyl 4-hydroxylase
- HKT :
-
High-affinity K+ transporter
- CCCH-ZFP :
-
CCCH zinc finger protein
References
Abe H, Hirai S, Okada S (2007) Metabolic responses and arginine kinase expression under hypoxic stress of the kuruma prawn Marsupenaeus japonicus. Comp Biochem Physiol A Mol Integr Physiol 146(1):40–46
Albacete A, Ghanem ME, Martinez-Andujar C, Acosta M, Sanchez-Bravo J, Martinez V, Lutts S, Dodd IC, Perez-Alfocea F (2008) Hormonal changes in relation to biomass partitioning and shoot growth impairment in salinized tomato (Solanum lycopersicum L.) plants. J Exp Bot 59(15):4119–4131
An X, Zhang JY, Dai LJ, Deng G, Liao Y, Liu L, Wang B, Peng D (2016) Isobaric tags for relative and absolute quantitation (iTRAQ)-based comparative proteome analysis of the response of ramie under drought stress. Int J Mol Sci 17(10):1607
Asif MH, Trivedi PK, Misra P, Nath P (2009) Prolyl-4-hydroxylase (AtP4H1) mediates and mimics low oxygen response in Arabidopsis thaliana. Funct Integr Genomics 9(4):525–535
Berg JM, Tymoczko JL, Stryer L (2010) Glycolysis and gluconeogenesis. In: Freeman WH (ed) Biochemistry, 7th edn. W. H. Freeman and Company, New York, pp 433–469
Borah P, Das A, Milner MJ, Ali A, Bentley A, Pandey R (2018) Long non-coding RNAs as endogenous target mimics and exploration of their role in low nutrient stress tolerance in plants. Genes 9(9):459
Boutet I, Meistertzheim AL, Tanguy A, Thébault MT, Moraga D (2005) Molecular characterization and expression of the gene encoding aspartate aminotransferase from the Pacific oyster Crassostrea gigas exposed to environmental stressors. Comp Biochem Physiol C Toxicol Pharmacol 140(1):69–78
Brant EJ, Budak H (2018) Plant small non-coding RNAs and their roles in biotic stresses. Front Plant Sci 9:1038
Byrt CS, Platten JD, Spielmeyer W, James RA, Lagudah ES, Dennis ES, Tester M, Munns R (2007) HKT1;5-like cation transporters linked to Na+ exclusion loci in wheat, Nax2 and Kna1. Plant Physiol 143(4):1918–1928
Carvalho DK, Campos DMKF, Domingues DS et al (2013) The accumulation of endogenous proline induces changes in gene expression of several antioxidant enzymes in leaves of transgenic Swingle citrumelo. Mol Biol Rep 40(4):3269–3279
Chen TT, Zhang L, Shang HH, Liu S, Peng J, Gong W, Shi Y, Zhang S, Li J, Gong J, Ge Q, Liu A, Ma H, Zhao X, Yuan Y (2016) iTRAQ-based quantitative proteomic analysis of cotton roots and leaves reveals pathways associated with salt stress. PLoS One 11(2):e0148487
Cho J (2018) Transposon-derived non-coding RNAs and their function in plants. Front Plant Sci 9:600
Collins NC, Tardieu F, Tuberosa R (2008) Quantitative trait loci and crop performance under abiotic stress: where do we stand? Plant Physiol 147(2):469–486
DeRose-Wilson L, Gaut BS (2011) Mapping salinity tolerance during Arabidopsis thaliana germination and seedling growth. PLoS One 6(8):e22832
Ewas M, Gao Y, Ali F et al (2017) RNA-seq reveals mechanisms of SlMX1 for enhanced carotenoids and terpenoids accumulation along with stress resistance in tomato. Sci Bull 62(7):476–485
Fan W, Zhang Z, Zhang Y (2009) Cloning and molecular characterization of fructose-1,6-bisphosphate aldolase gene regulated by high-salinity and drought in Sesuvium portulacastrum. Plant Cell Rep 28(6):975–984
Fan QJ, Yan FX, Qiao G, Zhang BX, Wen XP (2014) Identification of differentially-expressed genes potentially implicated in drought response in pitaya (Hylocereus undatus) by suppression subtractive hybridization and cDNA microarray analysis. Gene 533(1):322–331
Flexas J, Bota J, Loreto F, Cornic G, Sharkey TD (2004) Diffusive and metabolic limitations to photosynthesis under drought and salinity in C(3) plants. Plant Biol (Stuttg) 6(3):269–279
Ghassemi F, Jakeman AJ, Nix HA (1995) Salinisation of land and water resources: human causes, extent, management and case studies. UNSW Press, Sydney, Australia, and CAB International, Wallingford, UK
Gu XH, Jiang DL, Huang Y, Li BJ, Chen CH, Lin HR, Xia JH (2018a) Identifying a major QTL associated with salinity tolerance in Nile tilapia using QTL-seq. Mar Biotechnol 20(1):98–107
Gu XH, Li BJ, Lin HR, Xia JH (2018b) Unraveling the associations of the tilapia DNA polymerase delta subunit 3 (POLD3) gene with saline tolerance traits. Aquaculture 485:53–58
Haas BJ et al (2013) De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat Protoc 8(8):1494–1512
Hamilton CA, Good AG, Taylor GJ (2001) Induction of vacuolar ATPase and mitochondrial ATP synthase by aluminum in an aluminum-resistant cultivar of wheat. Plant Physiol 125(4):2068–2077
He B, Ma L, Hu ZH, Li H, Ai M, Long C, Zeng B (2018) Deep sequencing analysis of transcriptomes in Aspergillus oryzae in response to salinity stress. Appl Microbiol Biotechnol 102(2):897–906
Hua QZ, Chen CJ, Chen Z et al (2016) Transcriptomic analysis reveals key genes related to betalain biosynthesis in pulp coloration of Hylocereus polyrhizus. Front Plant Sci 6(499):1179
Huang SB, Spielmeyer W, Lagudah ES, James RA, Platten JD, Dennis ES, Munns R (2006) A sodium transporter (HKT7) is a candidate for Nax1, a gene for salt tolerance in durum wheat. Plant Physiol 142(4):1718–1727
Iorizzo M, Senalik DA, Grzebelus D, Bowman M, Cavagnaro PF, Matvienko M, Ashrafi H, van Deynze A, Simon PW (2011) De novo assembly and characterization of the carrot transcriptome reveals novel genes, new markers, and genetic diversity. BMC Genomics 12(1):389
Joshi R, Karan R, Singla-Pareek SL, Pareek A (2016) Ectopic expression of Pokkali phosphoglycerate kinase-2 (OsPGK2-P) improves yield in tobacco plants under salinity stress. Plant Cell Rep 35(1):27–41
Khueychai S, Jangpromma N, Daduang S, Jaisil P, Lomthaisong K, Dhiravisit A, Klaynongsruang S (2015) Comparative proteomic analysis of leaves, leaf sheaths, and roots of drought-contrasting sugarcane cultivars in response to drought stress. Acta Physiol Plant 37(4):88
Kindgren P, Ard R, Lvanov M et al (2018) Transcriptional read-through of the long non-coding RNA SVALKA governs plant cold acclimation. Nat Commun 9:4561
Le TN, Miyazaki Y, Takuno S et al (2015) Epigenetic regulation of intragenic transposable elements impacts gene transcription in Arabidopsis thaliana. Nucleic Acids Res 43(8):3911–3921
Lee KJ, Kwon SJ, Hwang JE, Han SM, Jung I, Kim JB, Choi HI, Ryu J, Kang SY (2016) Genome-wide expression analysis of a rice mutant line under salt stress. Genet Mol Res 15
Lenka SK, Katiyar A, Chinnusamy V, Bansal KC (2011) Comparative analysis of drought-responsive transcriptome in Indica rice genotypes with contrasting drought tolerance. Plant Biotechnol J 9:315–327
Liu ZJ, Kumari S, Zhang LF, Zheng Y, Ware D (2012) Characterization of miRNAs in response to short-term waterlogging in three inbred lines of Zea mays. PLoS One 7(6):e39786
Mark CFRR, Su-Hyun P, Lee JW et al (2012) Accumulation of trehalose increases soluble sugar contents in rice plants conferring tolerance to drought and salt stress. Plant Biotechnol Rep 6:89–96
Mazumdar P, Lau S, Wee WY (2017) Genome-wide analysis of the CCCH zinc-finger gene family in banana (Musa acuminata): An insight into motif and gene structure arrangement, evolution and salt stress responses. Trop Plant Biol 10(4):177–193
Mutuku JM, Nose A (2012) High activities and mRNA expression of pyrophosphate-fructose-6-phosphate-phosphotransferase and 6-phosphofructokinase are induced as a response to Rhizoctonia solani infection in rice leaf sheaths. Physiol Mol Plant Pathol 77:41–51
Naeem MS, Warusawitharana H, Liu H, Liu D, Ahmad R, Waraich EA, Xu L, Zhou W (2012) 5-aminolevulinic acid alleviates the salinity-induced changes in Brassica napus as revealed by the ultrastructural study of chloroplast. Plant Physiol Biochem 57:84–92
Nie Q, Gao GL, Fan QJ, Qiao G, Wen XP, Liu T, Peng ZJ, Cai YQ (2015) Isolation and characterization of a catalase gene “HuCAT3” from pitaya (Hylocereus undatus) and its expression under abiotic stress. Gene 563:63–71
Piper PW, Curran B, Davies MW et al (1986) Transcription of the phosphoglycerate kinase gene of Saccharomyces cerevisiae increases when fermentative cultures are stressed by heat-shock. Eur J Biochem 161:525–531
Qiao Y, Zhang J, Zhang J, Wang Z, Ran A, Guo H, Wang D, Zhang J (2017) Integrated RNA-seq and sRNA-seq analysis reveals miRNA effects on secondary metabolism in Solanum tuberosum L. Mol Gen Genomics 292:37–52
Qin T, Zhao HY, Cui P, Albesher N, Xiong L (2017) A nucleus-localized long non-coding RNA enhances drought and salt stress tolerance. Plant Physiol 175(3):1321–1336
Qiu QS, Guo Y, Quintero FJ et al (2003) Regulation of vacuolar Na+/H+ exchange in Arabidopsis thaliana by the salt-overly-sensitive (SOS) pathway. J Biol Chem 279:207–215
Quesada V, Garcia-Martinez S, Piqueras P et al (2002) Genetic architecture of NaCl tolerance in Arabidopsis. Plant Physiol 130:951–963
Radwan O, Arro J, Keller C (2015) RNA-Seq transcriptome analysis in date palm suggests multi-dimensional responses to salinity stress. Trop Plant Biol 8(3–4):74–86
Razavizadeh R, Ehsanpour A (2009) Effects of salt stress on proline content, expression of delta-1-pyrroline-5-carboxylate synthetase, and activities of catalase and ascorbate peroxidase in transgenic tobacco plants. Biol Lett 46:63–75
Ren ZH, Gao JP, Li LG, Cai XL, Huang W, Chao DY, Zhu MZ, Wang ZY, Luan S, Lin HX (2005) A rice quantitative trait locus for salt tolerance encodes a sodium transporter. Nat Genet 37:1141–1146
Ren Z, Zheng Z, Chinnusamy V, Zhu J, Cui X, Iida K, Zhu JK (2010) RAS1, a quantitative trait locus for salt tolerance and ABA sensitivity in Arabidopsis. Proc Natl Acad Sci USA 107:5669–5674
Resmi MS, Vivek PJ, Soniya EV (2015) Over-expression of bael quinolone synthase in tobacco improves plant vigor under favorable conditions, drought, or salt stress. FEBS Lett 589:332–341
Rus A, Yokoi S, Sharkhuu A, Reddy M, Lee BH, Matsumoto TK, Koiwa H, Zhu JK, Bressan RA, Hasegawa PM (2001) AtHKT1 is a salt tolerance determinant that controls Na+ entry into plant roots. Proc Natl Acad Sci U S A 98:14150–14155
Shaw TI, Srivastava A, Chou WC, Liu L, Hawkinson A, Glenn TC, Adams R, Schountz T (2012) Transcriptome sequencing and annotation for the Jamaican fruit bat (Artibeus jamaicensis). PLoS One 7:e48472
Shen X, Wang Z, Song X, Xu J, Jiang C, Zhao Y, Ma C, Zhang H (2014) Transcriptomic profiling revealed an important role of cell wall remodeling and ethylene signaling pathway during salt acclimation in Arabidopsis. Plant Mol Biol 86:303–317
Shi HZ, Ishitani M, Kim CS, Zhu JK (2000) The Arabidopsis thaliana salt tolerance gene SOS1 encodes a putative Na+/H+ antiporter. Proc Natl Acad Sci USA 97:6896–6901
Szabolcs IS, Salinisation (1994) In handbook of plant and crop stress. Marcel Dekker, New York
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578
Traylor-Knowles N, Rose NH, Palumbi SR (2017) The cell specificity of gene expression in the response to heat stress in corals. J Exp Biol 220:1837–1845
Ventura JR, Hu H, Jahng D (2013) Enhanced butanol production in Clostridium acetobutylicum ATCC 824 by double overexpression of 6-phosphofructokinase and pyruvate kinase genes. Appl Microbiol Biotechnol 97:7505–7016
Vicient CM (2010) Transcriptional activity of transposable elements in maize. BMC Genomics 11:601
Wang ZQ, Xu XY, Gong QQ, Xie C, Fan W, Yang JL, Lin QS, Zheng SJ (2014) Root proteome of rice studied by iTRAQ provides integrated insight into aluminum stress tolerance mechanisms in plants. J Proteome 98:189–205
Wang X, Ai G, Zhang CL (2016) Expression and diversification analysis reveals transposable elements play important roles in the origin of Lycopersicon-specific lncRNAs in tomato. New Phytol 209(4):1442–1455
Wang D, Qu ZP, Yang L, Zhang Q, Liu ZH, Do T, Adelson DL, Wang ZY, Searle I, Zhu JK (2017) Transposable elements (TEs) contribute to stress-related long intergenic noncoding RNAs in plants. Plant J 90(1):133–146
Wei J, Wu CY, Jiang Y, Wang HL (2014) Sample preparation optimization for determination of soluble sugar in red jujube fruits by anthrone method. Food Sci 37:168–176
Werner JE, Finkelstein RR (1995) Arabidopsis mutants with reduced response to NaCl and osmotic-stress. Physiol Plant 93:659–666
Xia K, Ou X, Gao C, Tang H, Jia Y, Deng R, Xu X, Zhang M (2015) OsWS1 involved in cuticular wax biosynthesis is regulated by Osa-miR1848. Plant Cell Environ 38:2662–2673
Xu HX, Li XY, Chen JW (2017) Comparative transcriptome profiling of freezing stress responses in loquat (Eriobotrya japonica) fruitlets. J Plant Res 130:893–907
Yang J (2008) Development and prospect of the research on salt-affected soils in China. Acta Pedol Sin 45:837–845
Zhang X, Rao X, Shi H et al (2011) Overexpression of a cytosolic glyceraldehyde-3-phosphate dehydrogenase gene OsGAPC3 confers salt tolerance in rice. Plant Cell Tissue Organ Cult 107:1–11
Zhang Z, Mao C, Shi Z, Kou X (2017) The amino acid metabolic and carbohydrate metabolic pathway play important roles during salt-stress response in tomato. Front Plant Sci 8:1231
Zhao Z, Xue YD, Yang HL, Li H, Sun G, Zhao X, Ding D, Tang J (2016) Genome-wide identification of miRNAs and their targets involved in the developing internodes under maize ears by responding to hormone signaling. PLoS One 11(10):e0164026
Acknowledgements
This project was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA13020500), the National Natural Science Foundation of China (31671659 and 31772384), the National Key Research and Development Program of China (2016YFC1403002), and the Guangzhou Science and Technology Project (201804010409).
Availability of Data and Materials Section
All the read data were available at DDBJ database (BioSample Accession no.: SAMD00115774- SAMD00115785).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Yuan Qin
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary Table 1
(DOCX 25 kb)
Supplementary Table 2
(XLSX 2127 kb)
Supplementary Table 3
(XLSX 3062 kb)
Supplementary Table 4
(XLSX 1365 kb)
Supplementary Table 5
(XLSX 338 kb)
Supplementary Table 6
(XLSX 271 kb)
Supplementary Table 7
(XLSX 201 kb)
Supplementary Table 8
(XLSX 15 kb)
Supplementary Table 9
(DOCX 16 kb)
Supplementary Table 10
(DOCX 19 kb)
Supplementary Table 11
(DOCX 16 kb)
Supplementary Table 12
(DOCX 17 kb)
Supplementary Table 13
(DOCX 16 kb)
Supplementary Table 14
(DOCX 18 kb)
Rights and permissions
About this article

Cite this article
Nong, Q., Zhang, M., Chen, J. et al. RNA-Seq De Novo Assembly of Red Pitaya (Hylocereus polyrhizus) Roots and Differential Transcriptome Analysis in Response to Salt Stress. Tropical Plant Biol. 12, 55–66 (2019). https://doi.org/10.1007/s12042-019-09217-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12042-019-09217-3