
Abstract
Loquat (Eriobotrya japonica Lindl.) is an important subtropical, commercial fruit in China. It blossoms during autumn and winter in most areas of China and its fruitlets usually suffer from freezing stress. However, studies about the mechanisms underlying freezing stress in loquat are very limited. The gene expression profiles of loquat fruitlets subjected to freezing (G2 library) versus non-treated ones (G1 library) were investigated using Illumina sequencing technology to elucidate the molecular mechanisms and identify the genes that play vital roles in the freezing stress response. The results showed that approximately 157.63 million reads in total were obtained from freeze-treated and non-treated loquat fruitlets. These reads were assembled into 87,379 unigenes with an average length of 710 bp and an N50 of 1,200 bp. After comparing the profiles obtained from the G1 and G2 libraries, 2,892 differentially expressed genes were identified, of which 1,883 were up-regulated and 1,009 were down-regulated in the treated samples compared to non-treated ones. These unigenes showed significant differences in expression for carbohydrate transport and metabolism, amino acid metabolism, energy metabolism, and lipid metabolism, which are involved in defense against freezing stress. Glycolysis/gluconeogenesis was one of the most significantly regulated pathways. Freezing also significantly damaged the membrane system of loquat fruitlets, and several defense mechanisms were induced. Some selected genes related to low temperature resistance were validated by quantitative real-time PCR (qRT-PCR). The results revealed many genes and pathways that are part of freezing resistance processes and expand our understanding of the complex molecular events involved in freezing stress.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Low temperature stress is a critical environmental factor that affects plant survival, growth, and agricultural productivity. It decreases the physiological activities of plants, inhibits their growth, injures their organs, and can cause death (Thomashow 1999; Wisniewski et al. 2004). However, plants can alter their physiological and biochemical statuses to cope with low temperature stress, such as triggering relative signaling pathways (Peleg and Blumwald 2011), inducing antioxidative systems (Tao et al. 1998), and accumulating compatible osmolytes (Hare et al. 1998; Koster and Lynch 1992). To elucidate the mechanism underling low temperature stress, it is important to determine how plants alter gene expressions in response to these biological processes. The changes may include the reprogramming of a large number of relative gene expressions (Chinnusamy et al. 2007; Thomashow 1998). Therefore, it is difficult to reveal the complex network underlying the low temperature response system by studying one metabolic pathway.
In recent years, developments in RNA-sequencing (RNA-Seq) have provided us opportunities to perform genome-wide profiling and to comprehensively quantify the transcripts involved in plant defense responses. Many studies have utilized this technology to provide valuable insights into the molecular and cellular mechanisms used by plants when they respond to low temperature (Die and Rowland 2014; Zheng et al. 2015; Zhu et al. 2015). Numerous cold-regulated (COR) genes have been identified by transcriptome analysis. For example: transcriptome analysis of Chinese wild Vitis species showed that a total of 6,850 transcripts were involved in cold regulation (Xu et al. 2014). In Arabidopsis thaliana, 1,248 and 1,747 genes are up-regulated and down-regulated, respectively, under low temperature stress (Sham et al. 2014), while in cassava apical shoots, 508 transcripts were identified as early cold-responsive genes (An et al. 2012).
Loquat (Eriobotrya japonica Lindl.) is a typical subtropical fruit, which is native to China, but is commercially cultivated worldwide (Lin et al. 1999). Although it is a perennial, evergreen woody crop, its flowers and fruitlets are sensitive to low temperature. Loquat blooms continuously from fall to winter in southeast China, but this is also the period when the fruitlets experience the coldest time of the year. When the temperature drops below −3 °C, low temperature frost can seriously damage the fruitlets, especially at about 40 days after full bloom (DAFB). Therefore, freezing stress usually dramatically reduces loquat yield in the following year, and is an important environmental constraint that limits the extent of loquat cultivation. An understanding of the biochemical and molecular characteristics under freezing stress in loquat fruitlets will improve the breeding of low temperature tolerant cultivars. However, in loquat, most reports about low temperature stress have focused on chilling injury to ripe fruits during low temperature storage (Cai et al. 2006; Ding et al. 2002). There have been very few studies on the mechanisms affecting freezing injury in loquat fruitlets (Xu et al. 2014), and the whole transcriptome-level changes that occur during the freezing response in loquat fruitlets are far from being fully understood.
In this study, we used Illumina sequencing to perform deep transcriptome sequencing to compare the genome-wide differential gene expressions between fruitlets subjected to freezing and under normal conditions. Then, any transcripts that were significantly differentially expressed in response to freezing were explored. This information provided a better understanding of the freezing stress molecular mechanism and the potential genetic resources that could improve freeze tolerant characteristics in loquat.
Materials and methods
Plant materials
The loquat (Eriobotrya japonica Lindl.) cv. Ninghaibai was planted in the Base Orchard of the Zhejiang Academy of Agricultural Sciences (Jiaxing, Zhejiang Province, China). Branches with young fruit at 40 days after full bloom (DAFB) were collected and transferred to an environmental chamber (Model LI15-2, Sheldon Manufacturing Inc., Cornelius, OR, USA) and maintained at −3 °C with a 16/8 h (day/night) photoperiod. Fruitlets grown in another chamber maintained at 25 °C with the same photoperiod were used as controls. After 24 h, the fruitlets from three independent control or freeze-treated branches were harvested, immediately frozen in liquid nitrogen, and stored at −80 °C until needed.
RNA extraction and Illumina HiSeq™ 2000 sequencing
Fruitlets from each biological replicate of the samples were ground to a fine powder in liquid nitrogen. For all of the subsequent procedures, three biological replicates of each sample were managed individually until a pooling step is indicated. Total RNA was extracted using a modified version of the cetyltrimethylam-monium bromide (CTAB) method (Shan et al. 2008). RNA quality and quantity were checked by a Nanodrop ND-2000 Spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, USA) after contaminating DNA had been removed using a TURBO DNase (Ambion, Austin, TX, USA). The remaining mRNA was enriched by cellulose oligo (dT) magnetic beads (Thermo Scientific) according to the manufacturer’s instructions. The mRNA was then cut into short fragments (about 200 bp). Double-stranded cDNA was synthesized and purified for end repair and 3ʹ-end single nucleotide A (adenine) addition. Finally, sequencing adaptors were ligated to the fragments and then the fragments were enriched by PCR amplification. After quantitative and qualitative assays, the three biological replicates of each sample were pooled and the resultant cDNA library products were sequenced via the Illumina HiSeq™ 2000 platform. The control samples were designated G1 and the samples harvested after treatment were designated G2. The same sample tags were used in all subsequent assays and data analyses.
Sequencing assessment, alignment, and annotation analysis
To create the assembly library, raw reads were filtered to remove those containing adapters and reads with more than 5% unknown nucleotides. Low quality reads, where the percentage of low Q value (≤10) bases was more than 20%, were also removed. Clean reads were de novo assembled by the Trinity Program (Grabherr et al. 2011). To annotate the unigenes, we performed a Basic Local Alignment Search Tool (BLAST) comparison (Altschul et al. 1997) against the NCBI Nucleotide Acid database (NT) (http://www.ncbi.nlm.nih.gov/nucleotide), the Non-redundancy Protein database (NR) (http://www.ncbi.nlm.nih.gov/protein), the Swiss-Prot database (http://www.expasy.ch/sprot), the TrEMBL database (http://www.uniprot.org/uniprot), the Gene Ontology database (GO) (http://www.geneontology.org/), the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg/), and the Cluster of Orthologous Groups of proteins (COG) database (http://www.ncbi.nlm.nih.gov/COG) to identify the putative functions of the unigene sequences. The sequence direction of the unigenes was determined using the best alignment results. When a unigene could not be aligned, sequence direction was confirmed using the ESTscan program. GO annotation was analyzed by Blast2GO software and functional classification of the unigenes was performed using WEGO software. KEGG pathway annotations were assigned by Blastall software against the KEGG database. The sequence data from this study have been submitted to the NCBI Sequence Read Archive (SRA, http://www.ncbi.nlm.nih.gov/Traces/sra) with accession number SRP073078.
In-depth analysis of differential gene expression
The number of unique, mapped, clean reads for each gene was calculated and normalized to the RPKM value (reads per kilobase of transcript per million reads mapped). DESeq was used to compare unigene expressions between the control and treatment according to Wang et al. (2010). A false discovery rate (FDR) ≤0.001 and the absolute value of log2 ratio ≥1 or ≤−1 (two-fold) were used as the thresholds for significant differences in gene expression (Ziliotto et al. 2008). The fold changes reported in this manuscript are all log2 fold changes. Cluster analysis of the gene-expression patterns was carried out by Cluster software (Hoon et al. 2004) and Java Tree view software (Saldanha 2004). Then, the differentially expressed genes (DEGs) were subjected to GO and COG functional classification, and KEGG pathway-enrichment analysis.
Gene-expression validation
Total RNA from the control and freeze-treated samples was extracted using the same method as described for RNA sequencing. One milligram of RNA was used for cDNA synthesis by a PrimeScript 1st Strand cDNA Synthesis Kit (TaKaRa, Dalian, China) in accordance with the manufacturer’s instructions. Ten-fold diluted cDNA was used as the template for qRT-PCR analysis. Primers for the candidate genes and actin were designed using Primer 5.0 and are listed in supplementary Online Resource 1. The PCR mixture (10 µl total volume) contained 5.0 µl of SYBR® Premix Ex Taq™ II (TaKaRa, Dalian, China), 0.4 µl of each primer (10 µM), 1.0 µl of diluted cDNA, and 3.2 µl of RNase-free water. PCR analysis was performed on a LightCycler® 96 instrument (Roche Diagnostics, Mannheim, Germany). The PCR was initiated by 95 °C for 30 s, which was then followed by 40 cycles of 95 °C for 5 s and 60 °C for 20 s. The final step was a melting curve analysis. The actin gene was used as an internal control. Relative transcript levels were calculated relative to actin using the formula 2−ΔΔCt. All reactions were performed in triplicate.
Physiological analysis
Soluble sugar sucrose, glucose, fructose, and sorbitol were identified and quantified by HPLC according to Xu and Chen (2011). The superoxide radical (O2 .−) generation rate was measured by monitoring the amount of nitrite formed from hydroxylamine in the presence of O2 .− as described by Elstner and Heupel (1976). Hydrogen peroxide (H2O2) levels were determined according to Velikova et al. (2000) and the malondialdehyde (MDA) content was determined using the thiobarbituric acid reaction method (Dhindsa et al. 1981). Proline contents were measured by the sulfosalicylic acid–acid ninhydrin method (Bates et al. 1973). Electrolyte leakage (EL) was estimated according to Lutts et al. (1996) and the activities of the antioxidant enzymes: superoxide dismutase (SOD, EC 1.15.1.1), catalase (CAT, EC 1.11.1.6), and ascorbate peroxidase (APX, EC 1.11.1.11), were determined according to Xu et al. (2014). Soluble protein was monitored according to Bradford (1976).
Statistical analysis
The results were expressed as mean values ± SE. All analyses were performed using three biological replicates. SYSTAT Version 11.5 (SPSS, Chicago, IL, USA) was used to statistically analyze the data. Significant differences between the treated and control samples were determined using one-way analysis of variance at P ≤ 0.05.
Results
Sequencing and assembly
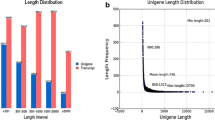
A summary of the Illumina sequencing and assembly is shown in Table 1. Approximately 74,023,888 and 83,601,630 raw reads were generated from the G1 and G2 libraries, respectively. After removing adaptor sequences, ambiguous nucleotides, and low-quality sequences, the clean reads in the two libraries were assembled into 63,608 and 63,423 unigenes, respectively, and average sequence lengths were 759.26 and 754.80 bp with a N50 length of 1,314 and 1,345 bp, respectively. The combined data in the G1 and G2 libraries were assembled into 87,379 unigenes, with a mean length of 709.98 bp and a N50 length of 1,200 bp.
To analyze the similarities and differences among the freezing-responsive transcriptomes, a hierarchical clustering was prepared to represent the transcripts of all the differentially expressed probes for the control and treatment replicates. The results showed significant gene expression profile differences between the two libraries (Fig. 1). Among the DEGs, 1,883 genes were up-regulated and 1,009 genes were down-regulated when the treatment/control samples were compared. More genes were expressed in the control fruitlets (27,250) than the treated fruitlets (26,589) (Table 2).

Hierarchical cluster analysis of all DEGs in the control (G1) and freeze treated (G2) loquat fruitlets. These include 2,892 DEGs before and after the freezing treatment. The median ratio (stressed/control) was log2 transformed and subjected to linkage hierarchical clustering. G1-1, G1-2, and G1-3 represent the control library. G2-1, G2-2, and G2-3 represent the treatment library
DEG annotation and functional classification
Estimating the number of genes and the level of transcript coverage is difficult because of the lack of genetic or genomic information on loquat. Functional annotations for the derived unigenes were assigned using BLAST comparisons against the public available databases such as NT, NR, Swiss-Prot, Tr-EMBL, GO, KEGG, and COG to identify the putative functions of the unigene sequences. Of the 2,892 DEGs from the G1 and G2 libraries, a total of 2,435 unigenes (84.2%) matched the NR database with an E value of 1E-5, and 1,866 unigenes (64.5%) matched genes in the NT database with an E value of 1E-10. BLASTx against the Swiss-Prot and Tr-EMBL database indicated that 2,007 (64.5%) and 2,241 (77.5%) unigenes were annotated, respectively, with an E-value of 1E-5 (Table 3).
All unigenes that matched Swiss-Prot database entries were further classified by GO terms according to the number of matched entries. A total of 1,980 unigenes (68.5%) had GO terms (Table 3) that could be classified into 63 functional groups (Fig. 2) in three main categories. These were cellular component, molecular function, and biological process. Cellular process, cell part, metabolic process, cell, organelle, binding, response to stimulus, catalytic activity, and biological regulation were the most dominant terms in the three categories. Very few genes were clustered into carbohydrate utilization, chemoattractant activity, phenotypic switching, carbon utilization, nutrient reservoir activity, protein tag, receptor regulator activity, nitrogen utilization, or metallochaperone activity.

Gene ontology classification of the assembled DEGs. a Cellular component; b molecular function; c biological process
In addition to the GO analysis, COG analysis was used to further evaluate the functions of our assembled unigenes (Fig. 3). In total, 956 unigenes (33.1%) were matched to COG terms (Table 3), and grouped into 25 COG categories. The ‘general function prediction only’ (33.7%) cluster represented the largest group, followed by ‘carbohydrate transport and metabolism’ (16.3%) and ‘amino acid transport and metabolism’ (14.5%). Few DEGs were assigned to ‘extracellular structures’ and ‘nuclear structure’.

Histogram of the clusters of orthologous groups (COG) classifications. A total of 956 DEGs were aligned to the COG database in order to predict and classify their possible functions
A KEGG analysis was also conducted to determine whether the freezing stress responsive genes were involved in specific pathways. A total of 574 unigenes (19.8%) (Table 3) were mapped to 102 KEGG pathways (Online Resource 2). Around 404 DEGs were assigned to metabolic pathways (Fig. 4). The top ten significant pathways included glycolysis/gluconeogenesis (ko00010, 36 unigenes); methane metabolism (ko00680, 27 unigenes); oxidative phosphorylation (ko00190, 22 unigenes); peroxisome (ko04146, 22 unigenes); pyruvate metabolism (ko00620, 19 unigenes); plant hormone signal transduction (ko04075, 19 unigenes); tryptophan metabolism (ko00380, 17 unigenes); alanine, aspartate, and glutamate metabolism (ko00250, 15 unigenes); arginine and proline metabolism (ko00330, 14 unigenes); and glycerophospholipid metabolism (ko00564, 14 unigenes).

Pathway assignment based on the Kyoto Encyclopedia of Genes and Genomes (KEGG)
The results showed that glycolysis/gluconeogenesis was the most significantly regulated pathway. A detailed view of the glycolysis/gluconeogenesis pathway is shown in Fig. 5. Ten genes, encoding key enzymes related to glycolysis/gluconeogenesis were significantly up-regulated. These were glucose-6-phosphate 1-epimerase (EC: 5.1.3.15, G2_Unigene_BMK.39779), N-acylglucosamine-6-phosphate2-epimerase (EC: 5.1.3.9, G2_Unigene_BMK.43313), fructose-bisphosphatase (EC: 3.1.3.11, G2_Unigene_BMK.18065), glyceraldehyde-3-phosphate dehydrogenase (EC: 1.2.1.12, G2_Unigene_BMK.39349), phosphoglycerate kinase (EC: 2.7.2.3), phosphopyruvate hydratase (EC: 4.2.1.11), pyruvate kinase (EC: 2.7.1.40, G2_Unigene_BMK.3052), pyruvate dehydrogenase (EC: 1.2.4.1, G2_Unigene_BMK.44631), aldehyde dehydrogenase (EC: 1.2.1.3, G2_Unigene_BMK.22352, G2_Unigene_BMK.56625, G2_Unigene_BMK.62115) and alcohol dehydrogenase (EC: 1.1.1.1, G2_Unigene_BMK.33253, G2_Unigene_BMK.46086, G2_Unigene_BMK.48258, G2_Unigene_BMK.48708, G2_Unigene_BMK.54783, and G2_Unigene_BMK.54784).

Regulatory changes in the glycolysis/gluconeogenesis pathway. Colored rectangles represent the loquat genes detected in the RNA-seq data. Red up-regulated; green down-regulated; and blue both up and down-regulated
Freezing stress regulation of transcription factors
Among the 2,892 DEGs, 66 up-regulated genes and 27 down-regulated ones were identified as putative transcription factors (TFs) (Fig. 6; Online Resource 3). According to the classification used to describe the 81 gene families in the Plant Transcription Factor Database (PlantTFDB, http://plntfdb.bio.uni-potsdam.de/v3.0/index.php?sp_id=ATH), 87 freezing-responsive TFs fell into 19 families (Fig. 6), while six TFs (G1_Unigene_BMK.1772, G2_Unigene_BMK.59122, G2_Unigene_BMK.50114, G1_Unigene_BMK.24034, G2_Unigene_BMK.628, and G2_Unigene_BMK.48919) were not classified. In our study, AP2-EREBP, NAC, and WRKY were the three major TF families. They contained 23, 11, and seven genes out of the 93 freezing-responsive TFs, respectively. The TFs in the ABISVP1, ARF, AUS/IAA, C2C2-Dof, C3H, HSF, and Trihelix families were all up-regulated, whereas the TFs in bHLH, C2C2-GATA, and GRAS were all down-regulated.

Distribution of differentially expressed transcription factors. The histograms showing the number of up- or down-regulated transcription factors
Validation of DEGs by qRT-PCR
Sixteen differentially expressed unigenes were selected for qRT-PCR analysis to determine their relative expression in the control and freezing treated loquat fruitlets, and to validate the transcriptome sequencing results. These candidates included nine functional genes and seven regulatory genes. In the functional gene categories, four cold response or cold acclimation related genes (G2_Unigene_BMK.46416, Indole-3-acetic acid-induced protein ARG2; G2_Unigene_BMK.35582, LEA14; G2_Unigene_BMK.38922, Cold-regulated 413 plasma membrane protein 2-like; and G1_Unigene_BMK.11095, Ferric reduction oxidase 6-like), two glycolysis related genes (G1_Unigene_BMK.6877, Fructose-bisphosphate aldolase; G2_Unigene_BMK.51393, Pyruvate kinase) and three oxidation–reduction process related genes (G2_Unigene_BMK.48798, SOD; G2_Unigene_BMK.56198, CAT; G2_Unigene_BMK.42434, POD) were tested. The regulatory gene categories analyzed consisted of seven transcription factors. These were AP2 (G2_Unigene_BMK.50090), DREB1 (G2_Unigene_BMK.53067), bZIP (G2_Unigene_BMK.61588), C2H2L (G2_Unigene_BMK.49546), HSF (G2_Unigene_BMK.40543), NAC (G2_Unigene_BMK.61591) and WRKY (G1_Unigene_BMK.17796). The expression profiles of the selected genes after exposure to freezing stress were determined using replicates of the qRT-PCR measurements and then compared with the results from an RNA-seq assay (Fig. 7). The qRT-PCR results showed that 14 of the 16 selected genes had significantly higher expressions in the freezing-treated samples than in the non-treated ones. All expression comparisons of qRT-PCR were generally in good agreement with the transcriptome sequencing analysis.

qRT-PCR analysis of 16 selected DEG genes, including G2_Unigene_BMK.46416 (a), G2_Unigene_BMK.35582 (b), G2_Unigene_BMK.38922 (c), G1_Unigene_BMK.11095 (d), G1_Unigene_BMK.6877 (e), G2_Unigene_BMK.51393 (f), G2_Unigene_BMK.48798 (g), G2_Unigene_BMK.56198 (h), G2_Unigene_BMK.42434 (i), G2_Unigene_BMK.50090 (j), G2_Unigene_BMK.53067 (k), G2_Unigene_BMK.61588 (l), G2_Unigene_BMK.49546 (m), G2_Unigene_BMK.40543 (n), G2_Unigene_BMK.61591 (o) and G1_Unigene_BMK.17796 (p). Error bars indicate standard errors of the means (n = 3)
Physiological changes to loquat fruitlets in response to freezing stress
Clear physiological changes were observed after freezing stress in loquat fruitlets. Changes to soluble sugar concentration, O2 .− generation rate, H2O2 content, MDA content, proline content, electrolyte leakage, SOD activity, CAT activity, and APX activity occurred (Table 4). The accumulation of soluble sugars could help maintain membrane stability. In this study, sucrose, glucose, fructose, sorbitol, and total sugar increased by 45.8, 4.1, 11.3, 68.8, and 36.0%, respectively, after freezing stress. The O2 .− generation rate and H2O2 level increased by 108.2 and 66.0%, respectively, in the freeze-treated fruitlets compared to the control. MDA is usually used to measure membrane system injuries and cellular metabolism deterioration. In this study, the MDA content increased by almost two-fold after the freezing treatment, the proline content increased by about 45.1% in the treated fruitlets compared to the control, and electrolyte leakage increased by about 47.5% after the freezing treatment. The antioxidant enzymes: SOD, CAT, and APX, are very important reactive oxygen species (ROS) suppressors, and their activities in the freeze-treated fruitlets were almost 20, 48, and 52%, respectively, higher than in the control.
Discussion
Extremely complex mechanisms are involved in abiotic stress defense in plants. Special receptors on the cell membrane detect the stress signals and then transmit them to secondary messengers. Consequently, a series of down-stream physiological activations and numerous stress-responsive gene expressions are triggered. This leads to protective responses by the whole plant (Lei et al. 2014). However, the molecular and genetic responses to freezing stress in loquat fruitlets have not been completely described. Transcriptome analysis is a sensitive and comprehensive technology that is used to detect relative response mechanisms during abiotic stress defense. In this study, the DEG expression profiles were analyzed using the transcriptome sequencing data from non-treated and freeze-treated loquat fruitlets, and several important freezing response metabolism pathways were detected.
Carbohydrate changes are of particular importance because of their direct relationship with physiological processes, such as photosynthesis, translocation, and respiration (Kerepesi and Galiba 2000). Carbohydrate metabolism, a fundamental metabolic process in living organisms, was the most sensitive to freezing stress in our study. Most of the DEGs related to carbohydrate metabolism were maped to the ‘glycolysis/gluconeogenesis’, ‘pyruvate metabolism’, ‘starch and sucrose metabolism’, ‘pentose and glucuronate interconversions’ and the ‘fructose and mannose metabolism’ pathways (Online Resource 4). Glycolysis/gluconeogenesis is the predominant pathway fueling plant respiration and the free energy released in this process is used to form the high-energy ATP and NADH compounds (Berg et al. 2010). Such a significant activation of the glycolysis/gluconeogenesis pathway suggests increased production of available energy. In wheat (Hamilton et al. 2001) and rice (Wang et al. 2014), glycolysis/gluconeogenesis related genes were induced at the transcriptional level to resist Al stress. Collectively, our RNA-seq data and KEGG analysis suggest that the glycolysis/gluconeogenesis pathway also plays an important role in protecting plants from freezing stress.
Many studies have suggested that sucrose, a soluble carbohydrate, has a potential role in adaptation to unfavorable environments (McKersis and Leshem 1994; Williams et al. 1992). The RNA-seq data showed that 12 regulated genes were involved in the ‘starch and sucrose metabolism’ pathway (Online Resource 4). The enzymes α-amylase, β-amylase, and isoamylase degrade starch to soluble sugar, which leads to increases in maltose, glucose, fructose, and sucrose levels (Wang et al. 2010). In this study, β-amylase (G2_Unigene_BMK.56377) and sucrose phosphate phosphatase (G2_Unigene_BMK.55707) were up-regulated by 1.5- and 1.7-fold after stress, respectively. In addition to the effects of freezing on β-amylase and sucrose phosphate phosphatase transcript levels, we found that freezing treatment increased the sucrose, sorbitol, and total sugar levels in loquat fruitlets (Table 4). The accumulation of sucrose also contributes to cold tolerance in Lilium lancifolium (Wang et al. 2014). Sorbitol is a major photosynthesis product and accounts for about 60–80% of the C translocated in the Rosaceae (Webb and Burley 1962). It also has an important role in osmotic adjustment during dehydration stress (Wang and Stutte 1992). Our results suggest that sorbitol is also involved in osmotic adjustment in loquat, but this needs further study.
Amino acid metabolic pathways are involved in synthesizing proteins, hormones, and enzymes, as well as degrading amino acids into metabolic intermediates of glucose, which can then be used in the citric acid cycle (Klowden 2007). Amino acids are also thought to mediate cold and desiccation resistance. In mycelia, amino acids biosynthesis increased after cold stress (Liu et al. 2017). It has also been reported that various of other amino acids, such as alanine, asparagine, asparate, glutamin and glutamate, over-accumulated in Arabidopsis (Klotke et al. 2004) and ryegrass (Bocian et al. 2015) after cold treatment. Several amino acid metabolic pathways were enriched after freezing stress, such as tryptophan metabolism, alanine, aspartate and glutamate metabolism, and arginine and proline metabolism (Fig. 4). The accumulation of proline, which is one of the most effective organic osmolytes, is usually associated with whole plant tolerance to various environmental stresses (An et al. 2012). In this study, the proline content also significantly increased in the treated fruitlets (Table 4). Furthermore, according to the metabolic pathway enrichment analysis, ‘arginine and proline metabolism’ included 14 DEGs that regulate and relieve the osmotic stress caused by freezing-induced dehydration (Online Resource 4). Proline levels are usually determined by the balance between the biosynthesis and catabolic pathways, which were controlled by the delta-1-pyrroline-5-carboxylate synthetase (P5CS) and proline dehydrogenase (ProDH) genes, respectively (Sharma et al. 2011). In this study, up-regulation of loquat homologous gene P5CS (G2_Unigene_BMK.60996) and and un-regulated ProDH (G1_Unigene_BMK.36875) might be correlated with the increased proline accumulation (Table 4).
The energy metabolism balance between assimilation and dissimilation is fundamental to the maintenance of cell life (Huner et al. 1998; Yinger et al. 1981). In the loquat fruitlets, of the 404 DEGs, 76 unigenes in the KEGG category ‘energy metabolism’ were significantly affected by freezing stress, and were mainly involved in six assimilation pathways: ‘methane metabolism (KO 00680)’, ‘nitrogen metabolism (KO 00910)’, ‘sulfur metabolism (KO 00920)’, ‘photosynthesis-antenna proteins (KO 00196)’, ‘photosynthesis (KO 00195)’, and ‘carbon fixation in photosynthetic organisms (KO 00710)’. There was one involved in dissimilation, which was ‘oxidative phosphorylation (KO 00190)’ (Online Resource 4). In ‘energy metabolism’, most of the DEGs were involved in ‘photosynthesis’, ‘photosynthesis-antenna proteins’, ‘carbon fixation in photosynthetic organisms’, and ‘sulfur metabolism’. There were more inhibited genes in the fruitlets after freezing stress. However, most of the DEGs involved in the ‘methane metabolic’, ‘nitrogen metabolic’, and ‘oxidative phosphorylation’ pathways showed increased transcript abundance after treatment (Fig. 4). Genes encoding F-type ATPase subunit A (G2_Unigene_BMK_20723), subunit E (G2_Unigene_BMK_35062), and subunit G (G2_Unigene_BMK_46437) notably increased in the treated samples. The transcript abundance of cytochrome c oxidase subunit 1 (COX1 G2_Unigene_BMK_32369), subunit 4 (COX4, G2_Unigene_BMK_44663), and subunit 17 (COX17 G2_Unigene_BMK_44269) increased significantly, whereas subunit 2 (COX2, G2_Unigene_BMK_27350) significantly decreased (Online Resource 4). These results suggested that the fruitlets were promoting dissimilation and that this could lead to increased energy production when defending against freezing stress.
Membrane systems are the primary site of freezing injury in plants, and are usually damaged by freeze-induced cellular dehydration (Kawamura and Uemura 2003). In this study, the membranes were clearly destroyed because increased MDA and electrolyte leakage were detected (Table 4). However, plants can increase cellular metabolic activity, and concentrations of unsaturated fatty acids and phospholipids to cope with low temperature stress (Moellering et al. 2010). Correspondingly, we identified a total of 60 genes (12.4% of DEGs) involved in ‘lipid metabolism’. High unsaturated fatty acid levels can improve low temperature tolerance and prevent cell damage (Degenkolbe et al. 2012). Thirty-seven genes involved in this process showed significant up-regulation of their transcripts after freezing stress. For example, four FAD3 (chloroplast omega-3 fatty acid desaturase) genes (G1_Unigene_BMK.19740, G2_Unigene_BMK.39043, G2_Unigene_BMK.56186, and G2_Unigene_BMK.40932), which contribute to freeze tolerance by altering the lipid composition, were up-regulated by 1.10–4.07-fold. The accumulation of certain proteins also contributes to membrane stabilization. Late embryogenesis abundant (LEA) protein acts as an osmotic regulator (Nylander et al. 2001), an antioxidant (Hara et al. 2003), and as a membrane structure stabilizer (Danyluk et al. 1998) during low-temperature stress. The dehydrins, which belong to the LEA IIs, have highly positive correlations with cold-stress tolerance (Kosová et al. 2011; Daviket al. 2013). In this study, several dehydrin genes were significantly up-regulated in the freezing treatment samples (Online Resource 5). In addition, the over-accumulation of sucrose and some other simple sugars also plays an important role in maintaining membrane stability (Table 4). The results showed that the loquat membrane protection system network was very complicated.
Peroxisome is also an important pathway in freeze stress resistance. Lipid peroxidation in plant membranes is primarily due to free-radical attack (Thompson et al. 1987). In this study, ROS rapidly increased after freezing stress, and the activity of antioxidant enzymes dramatically increased (Table 4). Twenty-two DEGs involved in this process showed significantly different expressions between the treated and control samples. Among the DEGs, three SOD genes (G1_Unigene_BMK.10607, and G1_Unigene_BMK.821, and G2_Unigene_BMK.52099) and 13 CAT genes (G1_Unigene_BMK.12498, G1_Unigene_BMK.12688, G1_Unigene_BMK.14342, G1_Unigene_BMK.16824, G1_Unigene_BMK.4309, G1_Unigene_BMK.52747, G1_Unigene_BMK.8150, G2_Unigene_BMK.23983, G2_Unigene_BMK.23984, G2_Unigene_BMK.26517, G2_Unigene_BMK.26518, G2_Unigene_BMK.56198, and G2_Unigene_BMK.9001) were significantly up-regulated (Online Resource 4). This indicated that the high expression levels of antioxidant genes may be an important functional trait in the freeze tolerance mechanism found in loquat fruitlets.
Transcription factors are involved in plant development and environmental stress resistance. AP2-EREBP, NAC, and WRKY are well known TF families that are involved in the low-temperature stress response process, and have been studied in numerous species (Zou and Yu 2010; Liu et al. 1998; Nakashima et al. 2012; Kovi et al. 2016). In our study, 23 genes belonging to the AP2-EREBP family were freezing-regulated (Fig. 6), which indicated that the AP2-EREBP family may play an important role in the loquat fruitlet freezing response. The AP2-EREBP family can be subdivided into four subfamilies, which are DREB/CBF, AP2, ERF, and RAP (Gilmour et al. 1998). However, we did not identify any CBFs that responded to freezing stress. The ICE-CBF cold response pathway is prominent in cold acclimation and has been well studied in Arabidopsis (Thomashow 1999; Chinnusamy et al. 2007). It had been reported that the DREB/CBF subfamily is involved in the early stages of the cold response (Cook et al. 2004). In tea plants, it has been suggested that DREB/CBF TFs play a vital role in the chilling, but not the freezing stress response (Zheng et al. 2015). Zou and Yu (2010) suggested that the CBFs were not up-regulated in response to cold stress because components of the calcium signaling cascade had been down-regulated. These results indicated that the AP2-EREBP family was critical to low-temperature resistance, and that different subfamilies might be involved in different low-temperature stress stage responses. Most of the AP2 and ERF members were up-regulated after freezing stress, which suggested that AP2 and ERF TFs had key roles in freezing stress resistance. The RAP member (G1_Unigene_BMK.11038), which is seldom investigated in cold response studies, was up-regulated over one-fold after freezing treatment, which indicated that it also had a potential role in the freezing response.
The plant-specific NAC transcription factor family has been implicated in many biotic and abiotic stress responses (Nakashima et al. 2012). It is notable that nine out of 11 NAC family members were up-regulated in response to freezing stress (Fig. 6). In rice, OsNAC5, OsNAC6, OsNAC045, and OSNAP have been characterized as being responsive to cold stress and lead to enhanced cold tolerance in transgenic rice (Chen et al. 2014; Nakashima et al. 2007; Takasaki et al. 2010; Zheng et al. 2009). In cotton, GhNAC8 and GhNAC11 are induced by cold temperatures and may regulate cotton development under abiotic stress (Huang et al. 2013). In Medicago truncatula, overexpression of the Medicago falcata NAC transcription factor MfNAC3 enhances cold tolerance (Qu et al. 2016), and the WRKY transcription factor family has complicated roles in many plant-environment interactions (Bakshi and Oelmüller 2014). Our RNA-seq data also revealed that the WRKY transcription factor family was freezing-regulated (Fig. 6). Three out of four members were up-regulated, and one member was down-regulated. In cotton, it has been reported that GbWRKY1 is a negative regulator of the jasmonic acid-mediated defense response and plant resistance to pathogens (Li et al. 2014). In Arabidopsis, loss of AtWRKY1 function could enhance stomatal movement and improve plant drought tolerance (Qiao et al. 2016). The WRKY1 gene was also found to be down-regulated in our study, which may indicate that it has a potential role in loquat freezing resistance. Furthermore, many other transcription factor families were also identified, such as ABISVP1, ARF, AUS/IAA, bHLH, C2C2-Dof, C2C2-GATA, C3H, HSF, GRAS, and Trihelix. Some of their homologous genes in other plant species have not yet been reported to respond to freezing stress, which suggests that these genes might be specific to loquat and should be targets for further functional characterization.
In conclusion, this study has provided the first large-scale transcriptome data set for loquat fruitlets when they respond to freezing stress. More than 157.63 million high quality reads were generated and assembled into 87,379 unigenes in loquat fruitlets. Furthermore, 2892 DEGs were identified under freezing stress, classified, and their functions and metabolic pathways shown for the first time. The higher expression levels of genes involved in carbohydrate transport and metabolism, amino acid metabolism, energy metabolism, and lipid metabolism in loquat fruitlets responding to freezing stress could play important roles in the freeze tolerance mechanism.
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
An D, Yang J, Zhang P (2012) Transcriptome profiling of low temperaturetreated cassava apical shoots showed dynamic responses of tropical plant to cold stress. BMC Genom 13:64
Bakshi M, Oelmüller R (2014) WRKY transcription factors. Plant Signal Behav 9:e27700
Bates LS, Waldren RP, Teare ID (1973) Rapid determination of free proline for water-stress studies. Plant Soil 39:205–207
Berg JM, Tymoczko JL, Stryer L (2010) Glycolysis and gluconeogenesis. Biochemistry, 7th edn. WH Freeman, New York, pp 433–469
Bocian A, Zwierzykowski Z, Rapacz M, Koczyk G, Ciesiołka D, Kosmala A (2015) Metabolite profiling during cold acclimation of Lolium perenne genotypes distinct in the level of frost tolerance. J Appl Genet 56:439–449
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem 72:248–254
Cai C, Li X, Chen KS (2006) Acetylsalicylic acid alleviates chilling injury of postharvest loquat (Eriobotrya japonica Lindl.) fruit. Eur Food Res Technol 223:533–539
Chen X, Wang Y, Lv B, Li J, Luo L, Lu S, Zhang X, Ma H, Ming F (2014) The NAC family transcription factor OsNAP confers abiotic stress response through the ABA pathway. Plant Cell Physiol 55:604–619
Chinnusamy V, Zhu J, Zhu JK (2007) Cold stress regulation of gene expression in plants. Trends Plant Sci 12:444–451
Cook D, Fowler S, Fiehn O, Thomashow MF (2004) A prominent role for the CBF cold response pathway in configuring the low-temperature metabolome of Arabidopsis. Proc Natl Acad Sci 101:15243–15248
Danyluk J, Perron A, Houde M, Limin A, Fowler B, Benhamou N, Sarham F (1998) Accumulation of an acidic dehydrin in the vicinity of the plasma membrane during cold acclimation of wheat. Plant Cell 10:623–638
Davik J, Koehler G, From B, Torp T, Rohloff J, Eidem P, Wilson RC, Sønsteby A, Randall SK, Alsheikh M (2013) Dehydrin, alcohol dehydrogenase, and central metabolite levels are associated with cold tolerance in diploid strawberry (Fragaria spp.). Planta 237:267–277
Degenkolbe T, Giavalisco P, Zuther E, Seiwert B, Hincha DK, Willmitzer L (2012) Differential remodeling of the lipidome during cold acclimation in natural accessions of Arabidopsis thaliana. Plant J 32:972–982
Dhindsa RS, Plumb-Dhindsa P, Thorpe TA (1981) Leaf senescence: correlated with increased levels of membrane-permeability and lipid peroxidation, and decreased levels of superoxide dismutase and catalase. J Exp Bot 32:93–101
Die J, Rowland L (2014) Elucidating cold acclimation pathway in blueberry by transcriptome profiling. Environ Exp Bot 106:87–98
Ding CK, Chachin K, Ueda Y, Imahori Y, Wang CY (2002) Modified atmosphere packaging maintains postharvest quality of loquat fruit. Postharvest Biol Technol 24:341–348
Elstner EF, Heupel A (1976) Inhibition of nitrite formation from hydroxylammonium chloride: a simple assay for superoxide dismutase. Anal Biochem 70:616–620
Gilmour SJ, Zarka DG, Stockinger EJ, Salazar MP, Houghton JM, Thomashow MF (1998) Low temperature regulation of the Arabidopsis CBF family of AP2 transcriptional activators as an early step in cold-induced COR gene expression. Plant J 16:433–442
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotech 29:644–652
Hamilton CA, Good AG, Taylor GJ (2001) Induction of vacuolar ATPase and mitochondrial ATP synthase by aluminum in an aluminum-resistant cultivar of wheat. Plant Physiol 125:2068–2077
Hara M, Terashima S, Fukaya T, Kuboi T (2003) Enhancement of cold tolerance and inhibition of lipid peroxidation by citrus dehydrin in transgenic tobacco. Planta 217:290–298
Hare PD, Cress WA, Staden JV (1998) Dissecting the roles of osmolyte accumulation during stress. Plant Cell Environ 21:535–553
Hoon MJ, Imoto S, Nolan J, Miyano S (2004) Open source clustering software. Bioinformatics 20:1453–1454
Huang GQ, Li W, Zhou W, Zhang JM, Li DD, Gong SY, Li XB (2013) Seven cotton genes encoding putative NAC domain proteins are preferentially expressed in roots and in responses to abiotic stress during root development. Plant Growth Regul 71:101–112
Huner N, Öquist G, Sarhan F (1998) Energy balance and acclimation to light and cold. Trends Plant Sci 3:224–230
Kawamura Y, Uemura M (2003) Mass spectrometric approach for identifying putative plasma membrane proteins of Arabidopsis leaves associated with cold acclimation. Plant J 36:141–154
Kerepesi I, Galiba G (2000) Osmotic and salt stress-induced alteration in soluble carbohydrate content in wheat seedlings. Crop Sci 40:482–487
Klotke J, Kopka J, Gatzke N, Heyer A (2004) Impact of soluble sugar concentrations on the acquisition of freezing tolerance in accessions of Arabidopsis thaliana with contrasting cold adaptation-evidence for a role of raffinose in cold acclimation. Plant Cell Environ 27:1395–1404
Klowden MJ (2007) Physiological systems in insects. Academic Press, Burlington
Kosová K, Vítámvás P, Prášil IT (2011) Expression of dehydrins in wheat and barley under different temperatures. Plant Sci 180:46–52
Koster KL, Lynch DV (1992) Solute accumulation and compartmentation during the cold acclimation of Puma rye. Plant Physiol 98:108–113
Kovi MR, Ergon A, Rognli OA (2016) Freezing tolerance revisited-effects of variable temperatures on gene regulation in temperate grasses and legumes. Curr Opin Plant Biol 33:140–146
Lei X, Xiao Y, Xia W, Mason AS, Yang Y, Ma Z, Peng M (2014) RNA-seqanalysis of oil palm under cold stress reveals a different C-repeat bindingfactor (CBF) mediated gene expression pattern in Elaeis guineensis compared to other species. PloS One 9:e114482
Li C, He X, Luo X, Xu L, Liu L, Min L, Jin L, Zhu L, Zhang X (2014) Cotton WRKY1 mediates the plant defense-to-development transition during infection of cotton by Verticillium dahliae by activating JASMONATE ZIMDOMAIN1 expression. Plant Physiol 166:2179–2194
Lin S, Sharpe RH, Janick J (1999) Loquat: botany and horticulture. Hortic Rev 23:233–276
Liu Q, Kasuga M, Sakuma Y, Abe H, Miura S, Yamaguchi-Shinozaki K, Shinozaki K (1998) Two transcription factors, DREB1 and DREB2, with an EREBP/AP2 DNA binding domain separate two cellular signal transduction pathways in drought- and low-temperature-responsive gene expression, respectively, in Arabidopsis. Plant Cell 10:1391–1406
Liu JY, Men JL, Chang MC, Feng CP, Yuan LG (2017) iTRAQ-based quantitative proteome revealed metabolic changes of Flammulina velutipes mycelia in response to cold stress. J Proteom 156:75–84
Lutts S, Kinet J, Bouharmont J (1996) NaCl-induced senescence in leaves of rice (Oryza sativa L.) cultivars differing in salinity resistance. Ann Bot 78:389–398
McKersie BD, Leshem YY (1994) Stress and stress coping in cultivated plants. Kluwer Academic Publishers, London
Moellering ER, Muthan B, Benning C (2010) Freezing tolerance in plants requires lipid remodeling at the outer chloroplast membrane. Science 330:226–228
Nakashima K, Tran LS, Van Nguyen D, Fujita M, Maruyama K, Todaka D, Ito Y, Hayashi N, Shinozaki K, Yamaguchi-Shinozaki K (2007) Functional analysis of a NAC-type transcription factor OsNAC6 involved in abiotic and biotic stress-responsive gene expression in rice. Plant J 51:617–630
Nakashima K, Takasaki H, Mizoi J, Shinozaki K, Yamaguchi-Shinozaki (2012) NAC transcription factors in plant abiotic stress responses. BBA Gene Regul Mech 1819:97–103
Nylander M, Svensson J, Palva ET, Welin BV (2001) Stress-induced accumulation and tissue-specific localization of dehydrins in Arabidopsis thaliana. Plant Mol Biol 45:263–279
Peleg Z, Blumwald E (2011) Hormone balance and abiotic stress tolerance in crop plants. Curr Opin Plant Biol 14:290–295
Qiao Z, Li CL, Zhang W (2016) WRKY1 regulates stomatal movement in drought-stressed Arabidopsis thaliana. Plant Mol Biol 91:53–65
Qu Y, Duan M, Zhang Z, Dong J, Wang T (2016) Overexpression of the Medicago falcata NAC transcription factor MfNAC3 enhances cold tolerance in Medicago truncatula. Environ Exp Bot 129:67–76
Saldanha AJ (2004) Java Treeview—extensible visualization of microarray data. Bioinformatics 20:3246–3248
Sham A, Al-Azzawi A, Al-Ameri S, Al-Mahmoud B, Awwad F, Al-Rawashdeh A, Iratni R, AbuQamar S (2014) Transcriptome analysis reveals genes commonly induced by Botrytis cinerea infection, cold, drought and oxidative stresses in Arabidopsis. PLoS One 9:e113718
Shan LL, Li X, Wang P, Cai C, Zhang B, Sun CD, Zhang WS, Xu CJ, Ferguson I, Chen KS (2008) Characterization of cDNAs associated with lignification and their expression profiles in loquat fruit with different lignin accumulation. Planta 227:1243–1254
Sharma S, Villamor JG, Verslues PE (2011) Essential role of tissue-specific proline synthesis and catabolism in growth and redox balance at low water potential. Plant Physiol 157:292–304
Takasaki H, Maruyama K, Kidokoro S, Ito Y, Fujita Y, Shinozaki K, Yamaguchi-Shinozaki K, Nakashima K (2010) The abiotic stress-responsive NAC-type transcription factor OsNAC5 regulates stress-inducible genes and stress tolerance in rice. Mol Genet Genom 284:173–183
Tao DL, Oquist G, Wingsle G (1998) Active oxygen scavengers during cold acclimation of scots pine seedlings in relation to freezing tolerance. Cryobiology 37:38–45
Thomashow MF (1998) Role of cold-responsive genes in plant freezing tolerance. Plant Physiol 118:1–8
Thomashow MF (1999) Plant cold acclimation: freezing tolerance genes and regulatory mechanisms. Annu Rev Plant Biol 50:571–599
Thompson JE, Legge RL, Barber RF (1987) The role of free radicals in senescence and wounding. New Phytol 105:317–344
Velikova V, Yordanov I, Edreva A (2000) Oxidative stress and some antioxidant systems in acid rain-treated bean plants: protective role of exogenous polyamines. Plant Sci 151:59–66
Wang Z, Stutte GW (1992) The role of carbohydrates in active osmotic adjustment in apple under water stress. J Am Soc Hortic Sci 117:816–823
Wang L, Feng Z, Wang X, Wang X, Zhang X (2010) DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26:136–138
Wang ZQ, Xu XY, Gong QQ, Xie C, Fan W, Yang JL, Lin QS, Zheng SJ (2014) Root proteome of rice studied by iTRAQ provides integrated insight into aluminum stress tolerance mechanisms in plants. J Proteom 98:189–205
Webb KL, Burley JWA (1962) Sorbitol translocation in apple. Science 137:766
Williams JHH, Williams AL, Pollock CJ, Farrar JF (1992) Regulation of leaf metabolism by sucrose. Sov Plant Physiol 39:443–446
Wisniewski M, Bassett C, Arora R (2004) Distribution and partial characterization of seasonally expressed proteins in different aged shoots and roots of ‘Loring’ peach (Prunus persica). Tree Physiol 24:339–345
Xu HX, Chen JW (2011) Commercial quality, major bioactive compound content and antioxidant capacity of 12 cultivars of loquat (Eriobotrys japonica Lindl.) fruits. J Sci Food Agric 91:1057–1063
Xu HX, Yang Y, Xie L, Li XY, Feng C, Chen JW, Xu CJ (2014) Involvement of multiple types of dehydrins in the freezing response in loquat (Eriobotrya japonica). PLoS One 9:e87575
Yinger JM (1981) Toward a theory of assimilation and dissimilation. Ethn Rac Stud 4:249–264
Zheng X, Chen B, Lu G, Han B (2009) Overexpression of a NAC transcription factor enhances rice drought and salt tolerance. Biochem Biophys Res Commun 379:985–999
Zheng C, Zhao L, Wang Y, Shen J, Zhang Y, Jia S, Li Y, Ding Z (2015) Integrated RNA-Seq and sRNASeq analysis identifies chilling and freezing responsive key molecular players and pathways in tea plant (Camellia sinensis). PLoS One 10:e0125031
Zhu H, Yu X, Xu T, Wang T, Du L, Ren G, Dong K (2015) Transcriptome profiling of cold acclimation in bermudagrass (Cynodon dactylon). Sci Hortic 194:230–236
Ziliotto F, Begheldo M, Rasori A, Bonghi C, Tonutti P (2008) Transcriptome profiling of ripening nectarine (Prunus persica L. Batsch) fruit treated with 1-MCP. J Exp Bot 59:2781–2791
Zou C, Yu J (2010) Analysis of the cold-responsive transcriptome in the mature pollen of Arabidopsis. Plant Biol 53:400–416
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 31101530); Key Project for New Variety Breeding in Agriculture of Zhejiang Province (Grant No. 2016C02052-3), the Local Science and Technology Cooperation Project of Zhejiang Academy of Agricultural Sciences (Grant No. LS20140003), and the Program for Young Science Technology Talent Cultivation of Zhejiang Academy of Agricultural Sciences.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xu, Hx., Li, Xy. & Chen, Jw. Comparative transcriptome profiling of freezing stress responses in loquat (Eriobotrya japonica) fruitlets. J Plant Res 130, 893–907 (2017). https://doi.org/10.1007/s10265-017-0942-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10265-017-0942-4