
Abstract
Salinity is the second most important abiotic stress after drought that hampers rice production, especially in south and Southeast Asia. Breeding approach supplemented with molecular markers-assisted selection is the most promising approach in terms of efficiency to increase the productivity under salt-affected soils. Thirty-day-old rice seedlings of 300 \(\hbox {F}_{5:6 }\) recombinant-inbred lines derived from a cross between the salt sensitive, IR29 (indica), and a salt tolerant, Hasawi (aus), were used to identify quantitative trait loci (QTLs) linked to salinity tolerance. One hundred and ninety four polymorphic SNP markers were used to construct a genetic linkage map involving 142 selected RILs that covered 1441.96 cM genome with an average distance of 7.88 cM between loci. Twenty new QTLs (LOD > 3) were identified on chromosomes 1, 2, 4, 6, 8, 9 and 12 using composite interval mapping with \(R^{2}\) as high as >20% with LOD value of 7.21. Many earlier studies reported big qSaltol for seedling stage salinity tolerance in rice is on short arm of chromosome 1 but none of the QTL in our study was on qSaltol or nearby position, therefore, Hasawi conferred salinity tolerance in RILs due to novel QTLs. It is suggested to fine map the novel QTLs so that the level of salinity tolerance could be further enhanced by pyramiding of the different QTLs in one genetic background through marker-assisted selection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rice (Oryza sativa L.) is a major source of food and energy for more than 2.7 billion people on a daily basis and is planted on about one-tenth of the earth’s arable land (GRISP 2013). Besides its economic significance, it is rich in genetic diversity in the form of thousands of landraces and progenitor species. Various stresses limits its productivity worldwide, among which abiotic stress alone contributes to more than 50% of the total yield losses (Munns and Tester 2008). The soil salinity is one of the most serious biophysical constraints of rice production in many rice-producing areas of the world (Munns and Tester 2008) with over 800 million hectares of total salt-affected soil. In Africa, of the total arable land of 1899 million ha, 73 million are salt affected (Ahmed et al. 2010; Yadav et al. 2011).
Breeding for salt tolerance offers more promising, energy efficient, economical, and socially acceptable approach to overcome problems related to the salt-stress (Ray and Islam 2008). Tolerance to salinity is a complex trait both genetically and physiologically and controlled by polygenes (Baby et al. 2010); and to develop new varieties with a high level of salinity tolerance, it requires an understanding of the genetic control underlying salt-tolerance mechanisms. Identification of the robust quantitative trait locus (QTLs) for salinity tolerance with closely linked flanking markers would be ideal to supplement theconventional breeding programme which heavily relies on phenotypic evaluation (Sabouri and Sabouri 2008; Thomson 2009). A major QTL for salt tolerance named Saltol was mapped using IR29/Pokkali recombinant-inbred lines (RIL) mapping population on short arm of chromosome 1 at 14.7 cM (Gregorio 1997; Singh et al. 2007; Thomson et al. 2010) but deploying this QTL alone in various mega rice varieties was not providing enough tolerance as none of the adapted variety with qSaltol find adoption in salt-affected fields. Therefore, some novel source of tolerance and identification of associated QTL for pyramiding would be an appropriate approach to breed rice varieties with high level of salinity tolerance (Rahman et al. 2016). This study is aimed to identify the large-effect novel QTL for salinity tolerance using new source of tolerance. Hasawi, a highly tolerant rice variety, has seldom been investigated for QTLs for seedling stage is used as novel source of salt tolerance (Bimpong et al. 2014a, b, 2016).
Materials and methods
Materials
A set of 300 \(\hbox {F}_{5}\) RILs from International Rice Research Institute (IRRI) was used for genotyping in this study. Each RIL derived from seeds of a single \(\hbox {F}_{4}\) plant from a cross between IR29 (salt sensitive) and Hasawi (salt tolerant) were advanced to constitute the phenotyping population (\(\hbox {F}_{5:6}\)) where \(\hbox {F}_{5}\) plants were genotyped while \(\hbox {F}_{6}\) seeds phenotyped for seedling stage salinity tolerance.
Phenotyping: evaluation of \(F_{ 5:6}\) RILs for salt tolerance
Screening of Hasawi, IR29 and the 300 \(\hbox {F}_{5:6}\) RILs for salinity tolerance was carried out in hydroponic system following IRRI standard protocol (Gregorio et al. 1997). Seeds were heat-treated for four days in a convection oven set at \(48{^{\circ }}\hbox {C}\) to break seed dormancy, and were then placed in Petri dishes with two layers of paper towels, moistened with distilled water during 48 h for uniform germination. The germinated seeds were sown one seed per hole on styrofoam sheet with 96 holes, attached to a nylon net bottom, and the sheet was floated on modified Yoshida nutrient solution (Singh et al. 2010). Seedlings were salinized after five days using \(6 \hbox { dSm}^{-1}\) salt (NaCl) concentration (equivalent about to 60 mM NaCl). This concentration was increased to \(12 \hbox { dSm}^{-1}\, (\sim 120\,\,\hbox {mM}\)) after two days of \(6 \hbox { dSm}^{-1}\) treatment to reduce the immediate shock. Each genotype represented by five seedlings per row of styrofoam and replicated thrice in the experiment. This experiment was conducted in a house covered with plastic only on top with a minimum temperature of 24 and maximum of \(37{^{\circ }}\hbox {C}\). The minimum relative humidity was 51% and maximum 84% with natural daylight of about 14 h. The pH of the solution was adjusted and maintained at 5.0 to 5.1 everyday with acid (1 N HCl) or base (1 N NaOH). The nutrient solution was renewed once every week to limit the effect of algae and replenish the nutrients. Initial standard evaluation system (SES) scoring (SESI) and final scoring (SESF) were recorded 12 and 25 days after the imposition of salinity stress respectively. The IRRI modified SES (IRRI 2007) was used (table 1).
In addition to salt injury scores, the length of roots and shoots (RL, SL) from each 30 days old \(\hbox {F}_{6}\) RILs were measured from three plants to get the average length (in cm). Similarly, roots and shoot fresh weight (RFW, SFW) from each 30 days old \(\hbox {F}_{6}\) RILs were weighed from three plant replicates using a top loading electronic balance and the average measurements (in g) was recorded. The roots and shoot dry weight (RDW, SDW) from each 30 days old \(\hbox {F}_{6}\) RILs were dried for five days in an oven set at \(75{^{\circ }}\hbox {C}\). Upon drying, the root and shoot dry weights were weighed using a top loading electronic balance and the average measurements (in g) was recorded.
Genotyping
DNA extraction, quantification and quality control
Selective genotyping was followed (Lander and Botstein 1989) to find out marker-trait association to identify the QTLs linked with desired traits (Lin and Ritland 1996). A total of 142 RILs were selected for genotyping of 300 RILs phenotyped which comprises of seven and 34 from both tolerant and sensitive extremes and 101 as random from intermediate RILs for SES (score 5–7). Genomic DNA was isolated from young leaves using the cetyl trimethyl ammonium bromide (CTAB) minipreparation method (Murray and Thompson 1980) in Genotyping Service Laboratory (GSL), IRRI, Philippines. The DNA quality was tested by staining DNA with \(\hbox {SYBR}^{\textregistered }\) Safe DNA gel staining solution after electrophoresis in 1% agarose gel at 150 V for 45 min in \(0.5\times \) TBE buffer and the image was visualized with gel documentation system (Alphaimager HP, California, USA). DNA concentration and purity were also determined by measuring the absorbance of diluted DNA solution at 260 nm and 280 nm through spectrophotometer (NanoDrop8000, Thermo Scientific, Waltham, USA).
Scoring of SNPs and analysis of polymorphism
A chip (\({ indica} \times { indica}\)) comprised of 384 SNP markers spread throughout 12 chromosomes of rice genome was used for parental polymorphic survey between two parents (Hasawi and IR29). For each oligo pool all (OPA) run, a plate of 96 samples with \(5 \,\mu \hbox {L}\) of unamplified genomic DNA normalized to \(50\, \hbox {ng} \mu \hbox {L}^{-1}\) concentration was genotyped using the “\(\hbox {GoldenGate}^{\mathrm{TM}}\) Genotyping Assay for \(\hbox {VeraCode}^{\mathrm{TM}}\) Manual Protocol” following the manufacturer’s instructions (https://www.illumina.com/documents/products/technotes/technote_veracode_goldengate_genotyping.pdf). The analysis employed the VC0011439-OPA set of 384-SNP markers designed to be informative across indica and aus germplasm (Thomson 2014; Thomson et al. 2012) and was run at the Genotyping Services Lab at IRRI (http://gsl.irri.org). Raw hybridization intensity data processing was performed using the genotyping module in the BeadStudio package (Illumina, San Diego, USA), followed by allele calling using Alchemy software (Wright et al. 2010). SNP call with the same genotype as Hasawi were scored as ‘1’, while as IR29 were scored as ‘2’, heterozygous SNP calls were scored as ‘3’, and missing SNPs were scored ‘0’. The marker position in bp was converted to cM using the equation: 1 cM \(=\) 250 kb (Tanksley et al. 1989; Famoso et al. 2011). Graphical genotyping of both IR29 and Hasawi were performed using Flapjack (http://bioinf.scri.ac.uk/flapjack) software developed by Scottish Crop Research Institute to check polymorphisms. The calls for Hasawi and IR29 were checked carefully to find polymorphism (Milne et al. 2010).
SNPs linkage map and QTL analysis
Three hundred eighty four SNPs were used for parental polymorphism survey. The polymorphic markers were selected for QTL analysis and construction of linkage map. QGene software ver. 4.3.1 (Joehanes and Nelson 2008) was used to construct the genetic linkage map by using Kosambi functions and linkage evaluation of \(P=0.001\) based on genotypic and phenotypic data of \(\hbox {F}_{5:6}\) RILs. Windows QTL Cartographer ver. 2.5 (Wang et al. 2011) software was used to construct the genetic linkage map (\(P =\) 0.001) based on genotypic and phenotypic data of \(\hbox {F}_{5:6}\) RILs. The threshold of logarithm of odds (LOD) score for the test of independence of marker pairs was set at \({>}\)3.0 (Collard et al. 2005). Composite interval mapping (CIM) was performed to examine the association between phenotypic data and marker genotype. The proportion of the total phenotypic variance explained (\(R^{2}\)) by each QTL was calculated.
Results
Polymorphism between parents
One hundred and ninety four SNPs of total 384 (50.52%) showed polymorphism between Hasawi and IR29 during genetic survey. No heterozygote alleles were found. The SNP genotyping data was used to perform linkage analysis with 194 markers which were distributed throughout the rice genome and covered a total length of 1441.96 cM. The average interval size between markers was 7.88 cM. Highest marker distribution was found on chromosome 1 (28 markers over a total number of 194), with an average interval of 6.38 cM and the highest average interval was in chromosome 10 (11.74 cM). Figure 1 shows different alleles across chromosomes contrasting the two parents for each chromosome. The physical position for the polymorphic markers is also given in base pair (bp).

Allele calls due to the presence of SNPs showing polymorphism between two parents used to develop recombinant inbred lines. The physical position of the polymorphic SNP is given in base pair (bp); A, adenine; T, thymine; G, guanine; C, cytosine; Chr1 to Chr12, chromosomes 1 to 12.

Morphological parameters
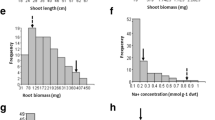
All the 300 RILs in \(\hbox {F}_{6}\) generation were phenotyped after 12 and 25 days of salinity treatment for SES and other parameters. The morphological differences for different traits observed within RILs derived from IR29/Hasawi cross are illustrated in figure 2. All the traits were not normally distributed, many were found skewed but the performance of parents were quite contrasting.

Frequency distribution of phenotypic traits after 12 days for initial SES and 25 days of salt stress at \(\hbox {EC } 12 \hbox { dSm}^{-1}\) for the 300 recombinant inbred lines (\(\hbox {F}_{6})\) derived from Hasawi and IR29.
Correlation between evaluated traits and growth reduction
The correlation coefficients between traits are provided in table 2. Under salinized conditions, highly significant (\(P=0.01\)) and positive correlations were found between the RL and SL, RL and RFW, RL and RDW, RL and SDW. Between initial salt injury scores (SESI) and final salt injury scores (SESF), a highly significant correlation was noticed but both showed inverse and highly significant correlation with other parameters. The SESF showed highly significant negative correlation with RL (\(r = -0.581^{**}\)), SL (\(r = -0.537^{**}\)), RFW (\(r = -0.708^{**}\)), SFW (\(r = -0.782^{**}\)), RDW (\(r= -0.601^{**}\)), and with SDW (\(r = -0.703^{**}\)). Generally as the tolerance score (SESF) increases (higher the score, more sensitive genotypes are), the percentage reduction of RL and SL, RFW and SFW, RDW and SDW decreases over nonstress for all RILs. Strangely, the results showed a different trend especially for root. For the genotypes with low SESF score, the RL, RFW, and RDW were found better in saline conditions.
QTL identification
Twenty putative and significant QTLs were identified for the visual initial and final SES salinity tolerance scores (SESI, SESF), seedling root length (RL), shoot length (SL), root fresh and dry weight (RFW, RDW), and shoot fresh and dry weight (SFW, SDW). The three QTLs associated with initial SES (qSESI2.1, qSESI6.1 and qSESI12.1) were identified on chromosomes 2, 6 and 12; two QTLs associated with final SES (qSESF2.1 and qSESF12.1) were identified on chromosomes 2 and 12; two QTLs associated with RL (qRL6.1 and qRL12.1) were identified on chromosomes 6 and 12; four QTLs associated with SL (qSL1.1, qSL1.2, qSL6.1 and qSL12.1) were identified on chromosomes 1, 6 and 12, three QTLs associated with SFW (qSFW4.1, qSFW9.1 and qSFW12.1) were identified on chromosomes 4, 9 and 12; two QTLs associated with RFW (qRFW4.1 and qRFW9.1) were identified on chromosomes 4 and 9; three QTLs associated with SDW (qSDW2.1, qSDW8.1 and qSDW12.1) were identified on chromosomes 2, 8 and 12; and one QTL associated with RDW (qRDW4.1) was identified on chromosome 4 (table 3; figure 3). Based on the computed \(R^{2}\) values which ranged from 9.3 to 20.6, 16 QTLs over the 20 identified QTLs explained 10% or more of the phenotypic variance explained for the concerned trait and therefore were having relatively major effect. Also, the two parents had different additive effect on phenotypic traits. Indeed, the IR29 allele increased the SES (SESI and SESF) at all loci. The Hasawi allele increased the RL, SL, RFW, SFW, RDW and SDW at all loci (table 3).

Linkage map with positions of QTLs for salinity tolerance using 142 RILs, the cross IR29/Hasawi. The labels on the right of the chromosomes reveal marker names while the numbers on the left indicate SNP maker positions in cM.
Discussion
The parents of mapping populations must have sufficient variation for the traits of interest at both the DNA sequence and phenotypic level (Meksem and Kahl 2005; Semagn et al. 2006). The requisite significant differences were found between Hasawi and IR29 for the genetic constitution and morphological performance, which is clearly revealed from studied traits and their relative percentage reduction under salinity stress. This confirmed the high sensitivity of IR29 and high tolerance of Hasawi under salt stress.
There were 194 polymorphic markers (50.52%) observed between the parents in this study which is much higher than that noted in earlier studies. Ammar et al. (2007) reported 18.88% polymorphism, when mapping QTLs for salinity tolerance at seedling stage, while Islam et al. (2011) and Alam et al. (2011) 34.35% and 16.45% respectively. Therefore, 194 polymorphic markers were good enough to be used for making linkage map and QTL analysis as well.
Genetically and phenotypically, the results obtained during the present study showed significant difference between Hasawi and IR29. This confirms that the RILs mapping population derived from the cross between IR 29 \(\times \) Hasawi were suitable for mapping of the QTLs for salinity tolerance traits. These results fit to the suggestion of Flowers (2004) to use the variation which is already present in existing crops to enhance salinity tolerance.
We tried to cover whole genome evenly using 384 SNP chip. Some marker intervals were relatively large due to the lack of polymorphism. It is also important to note that the RILs used for this study already reached to 92.3% of homozygosity which corroborate well with the theoretical expectation of homozygosity in \(\hbox {F}_{5:6}\) RILs generation (92.25%) (Vinod 2006). Several QTL mapping studies have been undertaken for the seedling stage salinity tolerance using different kind of mapping populations from early segregating to the fixed lines (Gregorio 1997; Masood et al. 2004; Ming-zhe et al. 2005; Lee et al. 2006; Ammar et al. 2007; Haq 2009; Thomson et al. 2010; Islam et al. 2011; Kanjoo et al. 2011). These studies used various kinds of markers like restriction fragment length polymorphism (RFLP), amplified fragment length polymorphism (AFLP), expressed sequence tags (EST), sequenced-tagged site (STS), cleaved amplified polymorphic sequence (CAPS) and simple sequence repeat (SSR); hence difficult to compare the exact chromosomal locations of QTLs directly. However, recent studies have used SNPs to identify robust markers for QTLs responsible for seedling stage salinity tolerance (Bimpong et al. 2014a, b; Kumar et al. 2015). Saltol QTL region located between 14.7 and 18.6 cM on short arm of chromosome 1 (Singh et al. 2007; Thomson et al. 2010) is the most robust QTL so far reported for seedling stage salinity tolerance (Gregorio 1997; Niones 2004) but we did not find any QTL on short arm of chromosome 1 in this study. This could be probably because of different donor used in IR29/Hasawi population which is novel and also quite diverse in origin from Pokkali rice variety, a donor for Saltol. Of the 20 QTL identified in the present study, 16 were major QTL as more than 10% phenotypic variance was explained by them (Tuberosa et al. 2002; Singh 2012).
There are specific locations on different chromosomes which appear like QTL clusters. For example, five different QTLs for various traits were identified on short arm of chromosome 12. Other QTL clusters were also identified on chromosomes 2, 4 and 6. This could be either due to very close or linked loci or may be pleiotropic effect of some genes. This could be ascertained through fine mapping in further studies. Beside this, all the traits except RDW are governed by more than one QTL which are mostly contributed by Hasawi. Only SES traits are governed by both IR29 and Hasawi. It suggests that there could be epistatic interactions among the QTLs for a trait that are distributed across the genome; and also in some case, the QTLs for one trait, e.g. SL (qSL12.1) is associated with another trait like survival which is reflected by low SES scores (initial or final) through indirect path. The QTLs associated with the initial and final SES scores (SESI and SESF) for salinity tolerance were located on different chromosomes, namely chromosomes 2, 6 and 12, which means that expression of the seedling performance under stress is the overall sum effect of QTLs for this trait and there are number of genes which govern this trait. A major QTL ‘Saltol’ was mapped on chromosome 1 using a RIL population derived from IR29/Pokkali. This qSaltol was responsible for \(\hbox {Na}^{+}\) and \(\hbox {K}^{+}\) absorption as well as \(\hbox {Na}^{+}/\hbox {K}^{+}\) ratio and each accounted for more than 60% of the variation in this population (Gregorio 1997). Higher \(\hbox {Na}^{+}\) uptake has been considered as a negative trait as far as salinity tolerance is concerned in rice (Flowers 2004; Lee et al. 2006; Singh and Flowers 2010; Thomson et al. 2010). Some earlier studies also reported QTLs responsible for physiological parameters conferring seedling stage salinity tolerance in rice on different chromosomes (Koyama et al. 2001), and substantiated the independence of \(\hbox {Na}^{+}\) and \(\hbox {K}^{+}\) uptake as they were located on different linkage groups. However few studies suggested colocalization of QTLs for \(\hbox {Na}^{+}\), \(\hbox {K}^{+}\), and/or their ratios on same chromosome 1 (Gregorio 1997; Koyama et al. 2001). But the present study did not find any QTL for SES or any other traits on chromosome 1 except for SL (qSL1.1 and qSL1.2), thus indicating that tolerance conferred in RILs derived from cross with Hasawi is due to other then qSaltol. Thomson et al. (2010) identified two QTLs associated with final SES tolerance scores on chromosomes 4 and 9. Ammar et al. (2007) found six QTLs linked to salinity injury scores on chromosomes 1, 3, 4, and 5 and among the two QTLs identified on chromosome 1; one was located on short arm close to RM84 marker at 0.11 cM position while the other one was located on long arm at 14 cM from RM572. In addition to that, different location of the SES from the previous findings confirmed that qSESI2.1, qSESF2.1, qSESF12.1 and qSESI12.1 mapped in this study were novel QTLs and reported for the first time.
The QTLs associated with the RL were located on chromosomes 6 and 12, but previous works (Sabouri and Sabouri 2008; The 2010) identified QTLs which controlled RL at different chromosomes (on chromosomes 2, 7 and 9). This also confirms that qRL6.1 and qRL12.1 were also novel QTLs. Also, the results suggested that RL were controlled by different genes.
Several studies have identified the QTLs associated with the SL under salinity stress on chromosomes 1, 2, 3, 4, 7, 10 and 12 (Singh et al. 2007; Sabouri and Sabouri 2008; The 2010; Thomson et al. 2010; De Leon et al. 2016). These findings suggested that there were several genes controlling SL under salinity stress. In the current study, QTLs for the SL (qSL1.1, qSL1.2, qSL6.1, and qSL12.1) were located on chromosomes 1, 6 and 12 and their physical positions on chromosomes were at different positions compared with the QTLs identified for SL under salt stress in previous studies. There was only commonality of report for qSL on chromosome 12 but the peak marker position in the present study was 6.9 cM comparing to 93 cM reported be De Leon et al. (2016), hence all the QTLs associated with SL could be therefore novel QTLs. In addition, with regards to the contribution of 20.6% in total phenotypic variation explained by qSL1.1, this QTL had main effect on expression of salt tolerance for root length.
During previous studies, QTLs associated with RFW were identified on chromosomes 1, 3, 4, and 9 (Singh et al. 2007; The 2010). In this study, the QTLs were identified on chromosomes 4 and 9. These results confirmed that RFW was controlled by multiple genes. Comparing the above previous findings with the results of the present work, qRFW4.1 and qRFW9.1 were new QTLs associated with root fresh weight.
In this study, 16 QTLs (80%) of 20 QTLs, all the alleles showing positive effects on the vigour related traits derived from Hasawi. This led to think that the transgressive segregants obtained from cross between Hasawi and IR29 could be the result of accumulation of favourable genes for salinity tolerance and high vigour, which could be one of the reasons beside tolerance that make Hasawi as a novel donor.
The results of this study suggested that SFW was controlled by multiples genes/QTLs and the associated QTLs (qSFW4.1, qSFW9.1 and qSFW12.1) with SFW were located on chromosomes 4, 9 and 12. These QTLs could also be designated as novel as these are not reported earlier. QTLs associated with SDW were identified on chromosomes 2, 8 and 12. However, in previous studies QTLs associated with SFW were identified on chromosomes 1, 2, 3, 6, 7, 8, 9 and 11 (Masood et al. 2004; Singh et al. 2007; Sabouri and Sabouri 2008; Haq 2009; The 2010; Bimpong et al. 2014b). This confirmed that the qSDW12.1 of three is novel as not reported earlier.
Finally, contrary to the previous studies where numerous QTLs associated with RDW (Masood et al. 2004; Sabouri and Sabouri 2008; The 2010) were identified on different chromosomes (1, 3, 4, 5, 8, 9 and 11), during this study, a single QTL (qRDW4.1) was identified on chromosome 4 to be associated with RDW.
Overall 20 QTLs were identified of which 11 were linked either to SL or SFW and SDW. These findings are very encouraging and corroborate with other reports that the shoot growth is severely affected with the salt stress than roots (Läuchli and Grattan 2007), hence it has opened the doors to discover unknown candidate genes for shoot growth (Munns and Tester 2008). Interestingly, among all the 20 QTLs identified six were located on chromosome 12; five were located at 6.9 cM position, while only one was located at 84.9 cM. Two QTLs were located at same position (18.1 cM) on chromosome 4; two QTLs at same position (18.5 cM) on chromosome 6; and also two QTLs were located at same position (27.0 cM) on chromosome 9. The fact that these QTLs were located at the same position, suggest that one or a group of genes controls the group of traits. The results also suggest the probability of pleiotropic effects of genes. As the traits, namely SESI, SESF, RL, SL, RFW, SFW, RDW and SDW were significantly correlated, the QTL corresponding to these traits were also found colocalized. According to Veldboom et al. (1994) and Xiao et al. (1996), often the correlated traits have QTLs mapping to the same chromosomal location. For example, in this study, RL and SESF was highly correlated (\(r= -0.581^{**}\)), similarly with SDW and SFW (\(r= 0.891^{**}\)) also. Trait correlation may result from either pleiotropic effects of genes or from tight linkage of several genes controlling the traits. This suggests that QTL affecting RL (qRL12.1) is closely linked to the QTL (qSESF12.1) affecting SESF in that region of chromosome 12. Likewise, the QTLs affecting SDW (qSDW12.1) and SFW (qSFW12.1) are closely linked.
Indeed, there were many colocalized regions showing a tight linkage of several QTLs/genes controlling the traits. For instance, the QTL affecting RL (qRL12.1) is closely linked to the QTL (qSESF12.1) affecting SESF, qSDW12.1 for SDW and qSFW12.1 for SFW in the same region of chromosome 12. The identified QTLs in the colocalized region may not be controlled by the same set of genes but may be controlled through different set of closely linked genes. The flanking markers were at 2.9 and 12.8 cM distance which is too much to accommodate many hundreds of genes but more precise locations of each QTL could be ascertained if more number of polymorphic markers could be included within 2.9–12.8 cM region of the chromosome 12. Thus, the large gaps in the chromosomal regions without polymorphic markers could miss some of the probable QTLs for traits related to salt tolerance. Additional markers within gaps could certainly increase the precision of QTLs detection. Thomson et al. (2010) identified two QTLs on chromosome 12 affecting SESF and SESI when they were characterizing Saltol region. Haq (2009) found a QTL affecting SDW on chromosome 2 like in our study. The findings of our study were in agreement with the previous reports that salinity tolerance is quantitative traits and controlled by many genes/QTLs (Singh et al. 2007, 2010; Baby et al. 2010; De Leon et al. 2016). Further, our results indicated some of the QTLs mapped for the seedling stage salinity tolerance are similar to the previously reported locations by various researchers but most of the putative QTL for seedling salinity tolerance from our study are new that could be due to the use of novel source of salinity tolerance which has not been used so far in any salt-tolerance related studies. The novelty of QTL could also be attributed to the use of SNP markers in this study with novel source of salinity tolerance. The study also suggested that novel sources for salinity tolerance should be explored to identify novel QTLs.
Conclusion
The findings of the present study confirmed that the mapping population developed from cross between Hasawi and IR29 was suitable to map QTLs for salinity tolerance. Consequently, Hasawi, tolerant check and parent are really a novel source of salinity tolerance since most of the identified putative QTLs are novel and have not been reported so far and none of the QTL is similar or colocalized with earlier reported major Saltol QTL on short arm of chromosome 1. These 20 new putative QTLs were identified on chromosomes 1, 2, 4, 6, 8, 9, and 12 using phenotyping of 142 RILs selected based on extremes for SES and percentage of reduction of RDW and SDW. Some of the novel QTLs with easy phenotyping procedures like SESI and SESF on chromosome 12 (qSESI12.1 and qSESF12.1) could be used to pinpoint the fine mapping of the regions and to identify the closely linked flanking markers for pyramiding of the fine mapped robust QTLs from different sources to enhance the level of salt tolerance in one background.
References
Ahmed M. A., Islam M. M., Begum S. N., Halder J. and Ahmed M. H. 2010 Evaluation of \(\text{ F }_{3}\) rich lines of JANGLIBORO\(\times \) BRRI Dhan40 for salt tolerance using SSR markers. Int. J. Biores. 1, 1–5.
Alam R., Sazzadur Rahman M., Seraj Z. I., Thomson M. J., Ismail A. M., Tumimbang-Raiz E. and Gregorio G. B. 2011 Investigation of seedling-stage salinity tolerance QTLs using backcross lines derived from Oryza sativa L. Pokkali. Plant Breed. 130, 430–437.
Ammar M. H. M., Singh R. K., Singh A. K., Mohapatra T., Sharma T. R. and Singh N. K. 2007 Mapping QTLs for salinity tolerance at seedling stage in rice (Oryza sativa L.). Afr. Crop Sci. Conf. Proc. 1, 617–620.
Baby J., Jini D. and Sujatha S. 2010 Biological and physiological perspectives of specificity in abiotic salt stress response from various rice plants. Asian J. Agri. Sci. 2, 99–105.
Bimpong I. K., Manneh B., Diop B., Ghislain K., Sow A., Amoah N. K. A. et al. 2014a New quantitative trait loci for enhancing adaptation to salinity in rice from Hasawi, a Saudi landrace into three African cultivars at the reproductive stage. Euphytica 200, 45–60.
Bimpong I. K., Manneh B., Namaky R., Diaw F., Amoah N. K. A., Sanneh B. et al. 2014b Mapping QTLs related to salt tolerance in rice at the young seedling stage using 384-plex single nucleotide polymorphism SNP, marker sets. Mol. Plant Breed. 5, 47–63.
Bimpong I. K., Manneh B., Sock M., Diaw F., Amoah N. K. A., and Ismail A. M. 2016 Improving salt tolerance of lowland rice cultivar ‘Rassi’ through marker-aided backcross breeding in West Africa. Plant Sci. 242, 288–299.
Collard B. C. Y., Jahufer M. Z. Z., Brouwer J. B. and Pang E. C. K. 2005 An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: The basic concepts. Euphytica 142, 169–196.
De Leon T. B., Linscombe S. and Subudhi P. K. 2016 Molecular dissection of seedling salinity tolerance in rice (Oryza sativa L.) using a high-density GBS-based SNP linkage map. Rice 9, (doi:10.1186/s12284-016-0125-2).
Famoso A. N., Zhao K., Clark R. T., Tung C.-W., Wright M. H., Bustamante C. et al. 2011 Genetic architecture of aluminum tolerance in rice (Oryza sativa) determined through genome-wide association analysis and QTL mapping. PLoS Genet. 7, e1002221.
Flowers T. J. 2004 Improving crop salt tolerance. J. Exp. Bot. 55, 307–319.
Gregorio G. B. 1997 Tagging salinity tolerance genes in rice using amplified fragment length polymorphism (AFLP). Ph.D. thesis, University of Philippines, Los Baños, Laguna, Philippines.
Gregorio G. B., Senadhira D. and Mendoza R. D. 1997 Screening rice for salinity tolerance. Int. Rice Res. Inst. 3–19.
GRISP (Global Rice Science Partnership) 2013 Rice almanac, 4th edition, pp. 283. International Rice Research Institute, Los Baños, Philippines.
Haq T. U. 2009 Molecular mapping of \(\text{ Na }^{+}\) accumulation quantitative trait loci (QTLs) in rice (Oryza sativa L.) under salt stress. Ph.D. thesis, University of Agriculture, Fasialabad, Pakistan.
IRRI 2007 Standard evaluation system for rice (SES), 4th edition, pp. 52. Philippines International Rice Research Institute, Los Banos, Philippines.
Islam M. R., Salam M. A., Hassan L., Collard B. C. Y., Singh R. K. and Gregorio G. B. 2011 QTL mapping for salinity tolerance at seedling stage in rice. Emir. J. Food Agric. 23, 137–146.
Joehanes R. and Nelson J. C. 2008 QGene 4.0, an extensible Java QTL-analysis platform. Bioinformatics 24, 2788–2789.
Kanjoo V., Jearakongman S., Punyawaew K., Siangliw J. L., Siangliw M., Vanavichit A. and Toojinda T. 2011 Co-location of quantitative trait loci for drought and salinity tolerance in rice. Thai J. Genet. 4, 126–138.
Koyama M. L., Levesley A., Koebner R. M. D., Flowers T. J. and Yeo A. R. 2001 Quantitative trait loci for component physiological traits determining salt tolerance in rice. Plant Physiol. 125, 406–422.
Kumar V., Singh A., Amitha Mithra S. V., Krishnamurthy S. L., Parida S. K., Jain S. et al. 2015 Genome-wide association mapping of salinity tolerance in rice (Oryza sativa). DNA Res. 22, 133–145.
Lander E. S. and Botstein D. 1989 Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121, 185–199.
Läuchli A. and Grattan S. R. 2007 Plant growth and development under salinity stress. In Advances in molecular breeding toward drought and salt tolerant crops (ed. M. A. Jenks, P. M. Hasegawa and S. M. Jain), pp. 1–32. Springer, Netherlands, USA.
Lee S. Y., Ahn J. H., Cha Y. S., Yun D. W., Lee M. C., Ko J. C. et al. 2006 Mapping of quantitative trait loci for salt tolerance at the seedling stage in rice. Mol. Cells 21, 192–196.
Lin J. Z. and Ritland K. 1996 The effects of selective genotyping on estimates of proportion of recombination between linked quantitative trait loci. Theor. Appl. Genet. 93, 1261–11266.
Masood M. S., Seiji Y., Shinwari Z. K. and Anwar R. 2004 Mapping quantitative trait loci (QTLs) for salt tolerance in rice (Oryza sativa) using RFLPs. Pak. J. Bot. 36, 825–834.
Meksem K. and Kahl G. 2005 The handbook of plant genome mapping: genetic and physical mapping. Wiley-Vch Verlag GmbH, Weinheim, Germany.
Milne I., Shaw P., Stephen G., Bayer M., Cardle L., Thomas W. T. B. et al. 2010 Flapjack-graphical genotype visualization. Bioinformatics 26, 3133–3134.
Ming-zhe Y., Jian-fei W., Hong-you C., Hu-qu Z. and Hong-sheng Z. 2005 Inheritance and QTL mapping of salt tolerance in rice. Rice Sci. 2, 25–32.
Munns R. and Tester M. 2008 Mechanisms of salinity tolerance. Ann. Rev. Plant Biol. 59, 651–681.
Murray M. G. and Thompson W. F. 1980 Rapid isolation of high molecular weight plant DNA. Nucl. Acids Res. 8, 4321–4325.
Niones J. M. 2004 Fine mapping of the salinity tolerance gene on chromosome 1 of rice (Oryza sativa L.) using near isogenic lines. MS dissertation. College, Laguna, Philippines: MSc thesis, University of the Philippines, Los Baños, Laguna.
Rahman M. A., Thomson M. J., Alam M. S., De Ocampo M., Egdane J. and Ismail A. M. 2016 Exploring novel genetic sources of salinity tolerance in rice through molecular and physiological characterization. Ann. Bot. 117, 1083–1097.
Ray P. K. S. and Islam M. A. 2008 Genetic analysis of salinity tolerance in rice. Bangladesh J. Agril. Res. 33, 519–529.
Sabouri H. and Sabouri A. 2008 New evidence of QTLs attributed to salinity tolerance in rice. Afr. J. Biotechnol. 7, 4376–4383.
Semagn K., Bjørnstad Å. and Ndjiondjop M. N. 2006 Principles, requirements and prospects of genetic mapping in plants. Afr. J. Biotechnol. 5, 2569–2587.
Singh B. D. 2012 Plant breeding: principles and methods. Kalyani Publishers, New Delhi, India.
Singh R. K. and Flowers T. J. 2010 The physiology and molecular biology of the effects of salinity on rice. In Handbook of plant and crop stress (ed. M. Pessarakli), pp. 901–942. Taylor and Francis, Florida, USA.
Singh R. K., Gregorio G. B. and Jain R. K. 2007 QTL mapping for salinity tolerance in rice. Physiol. Mol. Biol. Plants 13, 87–99.
Singh R. K., Redoña E. and Refuerzo L. 2010 Varietal improvement for abiotic stress tolerance in crop plants: special reference to salinity in rice. In Abiotic stress adaptation in plants: physiological, molecular and genomic foundation (ed. A. Pareek, S. K. Sopory, H. J. Bohnert and Govindjee), pp. 387–415. Springer, Netherlands, Germany.
Tanksley S. D., Young N. D., Paterson A. H. and Bonierbale M. W. 1989 RFLP mapping in plant breeding: new tools for an old science. Biotechnology 7, 257–264.
The H. V. 2010 QTL mapping and marker-assisted backcrossing for developing salinity tolerant rice (Oryza sativa L.). M.Sc. thesis, University of Philippines, Los Baños, Laguna, Philippines.
Thomson M. J. 2009 Marker-assisted breeding for abiotic stress tolerance in rice: progress and future perspectives. International Symposium, Rice Research in the Era of Global Warming, pp. 43–55. Taiwan Agricultural Research Institute, Taichung, Taiwan.
Thomson M. J. 2014 High-throughput SNP genotyping to accelerate crop improvement. Plant Breed. Biotech. 2, 195–212.
Thomson M. J., De Ocampo M., Egdane J., Rahman M. A., Sajise A. G., Adorada D. L. et al. 2010 Characterizing the Saltol quantitative trait locus for salinity tolerance in rice. Rice 3, 148–160.
Thomson M. J., Zhao K., Wright M., McNally K. L., Rey J., Tung C.-W. et al. 2012 High-throughput single nucleotide polymorphism genotyping for breeding applications in rice using the BeadXpress platform. Mol. Breed. 29, 875–886.
Tuberosa R., Salvi S., Sanguineti M. C., Landi P., Maccaferri M. and Conti S. 2002 Mapping QTLs regulating morpho-physiological traits and yield: case studies, shortcomings and perspectives in drought-stressed maize. Ann. Bot. 89, 941–963.
Veldboom L. R., Lee M. and Woodman W. L. 1994 Molecular-marker facilitated studies in an elite maize population: I. Linkage analysis and determination of QTLs for morphological traits. Theor. Appl. Genet. 88, 7–16.
Vinod K. K. 2006 Genome mapping in plant population. In Proceedings of training programme on modern approaches in plant genetics resources-collection, conservation and utilization, pp. 402–414. Tamil Nadu Agricultural University, Coimbatore, India.
Wang S., Basten C. J. and Zeng Z. B. 2011 Windows QTL Cartographer 2.5. North Carolina State University, Raleigh, USA.
Wright M., Tung C. W., Zhao K., Reynolds A., McCouch S. R. and Bustamante C. D. 2010 ALCHEMY: a reliable method for automated SNP genotype calling for small batch sizes and highly homozygous populations. Bioinformatics 26, 2952–2960.
Xiao J., Li J., Yuan L. and Tanksley S. D. 1996 Identification of QTLs affecting traits of agronomic importance in a recombinant inbred population derived from a sub specific rice cross. Theor. Appl. Genet. 92, 230–244.
Yadav S., Irfan M., Ahmad A. and Hayat S. 2011 Causes of salinity and plant manifestations to salt stress: a review. J. Environ. Biol. 32, 667–685.
Acknowledgements
The authors thank Ms Marydee Arceta for technical assistance in the genotyping. Technical editing by Dr Bill Hardy is sincerely acknowledged. Help from head of the Plant Breeding, Genetics and Biotechnology Division is duly acknowledged for permission to conduct the research at IRRI. Authors also acknowledge the funding support of Japan Rice Breeding Project for MSc degree and to carry out the research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Corresponding editor: U. C. Lavania
Rights and permissions
About this article

Cite this article
Bizimana, J.B., Luzi-Kihupi, A., Murori, R.W. et al. Identification of quantitative trait loci for salinity tolerance in rice (Oryza sativa L.) using IR29/Hasawi mapping population. J Genet 96, 571–582 (2017). https://doi.org/10.1007/s12041-017-0803-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12041-017-0803-x