
Abstract
Mechanisms of anti-oxidant action of 6-gingerol as a hydroxyl radical scavenger have been investigated using the transition state theory within the framework of density functional theory. Hydrogen abstraction by a hydroxyl radical from the different sites of 6-gingerol and addition of the former to the different sites of the latter were studied. Electron transfer from 6-gingerol to a hydroxyl radical was also studied. Solvent effect in aqueous media was treated using the integral equation formalism of the polarizable continuum model (IEF-PCM). Reaction rate constants in aqueous media were generally found to be larger than those in gas phase. The tunneling contributions to rate constants were found to be appreciable. Our results show that 6-gingerol is an excellent anti-oxidant and would scavenge hydroxyl radicals efficiently.

Hydrogen abstraction, radical adduct formation and single electron transfer as three mechanisms of antioxidant action of 6-gingerol as a hydroxyl radical scavenger have been investigated using transition state theory within the framework of density functional theory. 6-Gingerol is shown to be an excellent hydroxyl radical scavenger.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The hydroxyl radical (OH·) is highly reactive and plays prominent roles in several reactions leading to modifications of DNA bases, sugars, proteins and lipids, etc. present in the human body and other biological systems.[1‐4]It is generated due to both enzymatic and non-enzymatic reactions and by radiolysis of water.[5‐7]Modifications of bio-molecules can be hazardous to normal cell functioning and can cause several non-neurodegenerative and neuro-degenerative diseases e.g., inflammation, mutation, aging, cancer, cardiovascular diseases, the Alzheimer’s and Parkinson’s diseases.[8‐13]Formation of hydroxyl radicals can take place in biological media during cell metabolism as well as due to reactions with external agents. Though free radicals are also in some sense useful as they can prevent some harmful effects of bacteria,[14]they need to be scavenged as they damage biomolecules involved in life processes. Free radicals are eliminated or deactivated by different mechanisms e.g., by hydrogen abstraction, addition and electron transfer reactions.[15‐17]If the concentration of antioxidants becomes inadequate to scavenge the toxic free radicals, it can lead to a state of disease. In such a situation, human diet needs to be supplemented with exogenous anti-oxidants most of which come from plants.[15,17]Human beings have depended on herbal medicines to treat several diseases from the ancient times.[18] As several modern drugs are isolated from medicinal and herbal sources, these sources fulfill the medicinal needs to a large extent for a large part of the world’s population.[18] Ginger or Zingiber officinalis has been used as a medicinal source in the Indian ayurvedic and Chinese systems of medicine. Ginger has a strong anti-oxidant property as a radical scavenger.[19] Out of about 400 different compounds identified in fresh ginger, large amounts of the phenolic compounds gingerol, shogaol and paradol are found in it. The pharmacological activity of ginger is mainly due to gingerol and shogaol.[19] Gingerols form a group of compounds for which nomenclature depends on the position of the keto group and length of an unbranched carbon chain attached to the phenolic ring. Bhattarai et al.,[20] have studied the pH and temperature dependence of 6-gingerol and concluded that dehydration of gingerols produces the corresponding shogaols. For example, after dehydration, 6-gingerol gets converted to 6-shogaol.[20] 6-Gingerol is known to have anti-oxidant, anti-inflammatory, anti-cancer, anti-diabetic and other valuable pharmaceutical properties.[21‐33]
Due to the numerous useful pharmaceutical properties of 6-ginegerol mentioned above, a study of its properties is important. We have investigated here the anti-oxidant property of 6-gingerol, particularly as a hydroxyl radical scavenger using a theoretical approach.
2 Computational details
Geometries of 6-gingerol, its reactant complexes with an OH radical, transition states and reaction products involved in hydrogen abstraction, radical addition and electron transfer were optimized using the B3LYP[34,35] and M06-2X[36,37]functionals of density functional theory (DFT) along with the 6-31G(d,p) basis set. These calculations were usually followed by single point energy calculations using the same two density functionals but along with the 6-311 ++G(d,p) basis set in gas phase as well as aqueous media.[36,37]However, for all the systems involved in electron transfer, final results were obtained by full geometry optimization in aqueous media at both the B3LYP/6-311 ++G(d,p) and M06- 2X/6-311 ++G(d,p) levels of theory. The B3LYP and M06-2X functionals were chosen for the present study as these have been shown to be reliable to predict molecular properties, the latter functional being particularly suitable to study reaction kinetics.[34‐37]Rate constants were calculated employing the transition state theory.[38]Wigner transmission coefficients giving tunneling contributions to rate constants were also evaluated. As the study has more relevance in aqueous phase than in gas-phase, we would preferably consider rate constants for hydrogen abstraction in aqueous media. The integral equation formalism of the polarizable continuum model (IEF-PCM) was used to treat solvation in aqueous media.[39,40]All the molecules, complexes and transition states involved in hydrogen abstraction and addition reactions were solvated in aqueous media at the level of single point energy calculations. Gibbs barrier energies for hydrogen abstraction and radical addition reactions were calculated separately for each site of reaction with respect to the corresponding reactant complexes (RCs) while released energies were calculated with respect to the corresponding transition states (TSs). Reaction kinetics for various sites is affected to different extents by stabilities of RCs which are usually different. In these calculations, thermal energy corrections obtained at the B3LYP/6-31G(d,p) and M06-2X/6-31G(d,p) levels in gas phase were also considered to be valid for the corresponding single point energy calculations including those in aqueous medium. Values of the spin operator < S2 > for all the optimized geometries involving a hydroxyl radical was found to be close to 0.76 showing that spin contamination was negligible. Vibrational frequency analysis was carried out and necessary thermal energy corrections to total energy made in order to obtain Gibbs barrier energies at 298.15 K in gas phase. Genuineness of transition states was ensured by a visual inspection of vibrational modes corresponding to the imaginary frequencies. Thus, for all the transition states, vibrational motions corresponding to the imaginary frequencies were found to connect the reactants and products convincingly. The Windows version of the Gaussian G09 suite of programs (G09W) was used for all the calculations while the Gauss View 5.0 program was used for visualization of molecular structures and vibrational modes.[41,42]
3 Results and discussion
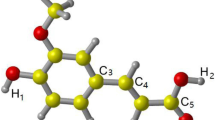
The optimized structure of 6-gingerol at the M06-2X/6-31G(d,p) level is shown in Figure 1 where the adopted atomic numbering scheme is also shown. 6-Gingerol has a methoxy group and a hydroxyl group attached to the C2 and C3 positions of its ring respectively (Figure 1). A long unbranched, 10 carbon atom chain including a keto group and a hydroxyl group bonded to its C9 and C11 positions, respectively, is attached to the C6 position of the ring. Gibbs barrier and released energies having different superscripts and subscripts presented in Tables 1–3 would correspond to different reactions as follows (n =variable integer): ΔG n: hydrogen abstraction from the carbon site Cn, \({\Delta } \textit {G}^{\prime \prime }_{\mathrm {n}}\): addition of OH· at the carbon site Cn and \({\Delta } \textit {G}_{\mathrm {n}^{\prime }}\): abstraction of the hydrogen atom of the OH group attached to the carbon site Cn.

Optimized geometry and atomic numbering scheme for 6-gingerol obtained at the M06-2X/6-31G(d,p) level of theory in gas phase.
3.1 Hydrogen abstraction reactions
There are 26 hydrogen atoms in 6-gingerol each of which can be abstracted by a hydroxyl radical. However, out of the two hydrogen atoms attached to each of the sites C7, C8, C10, C12-C15, and also out of the three hydrogen atoms of each of the two methyl groups, abstraction of only one was considered, since their abstraction barrier energies are not expected to be significantly different. Abstraction of a hydrogen atom Hn from a carbon site Cn of 6-gingerol (6-G(Hn)) by a hydroxyl radical may be represented by the following general scheme.
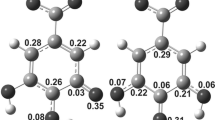
where 6-G(Hn)- OH⋅ is the reactant complex (RC) formed from the free reactants 6-G(Hn) and OH·, \({\Delta } \textit {G}_{\mathrm {n}}^{\mathrm {b}}\) is the Gibb’s barrier energy corresponding to the transition state [TS]n and \({\Delta } \textit {G}_{\mathrm {n}}^{\mathrm {r}}\) is the Gibbs released energy following the reaction. A 6-gingerol radical (6-G·) along with a water molecule (Hn-OH) constitute the product complex (PC). The calculated Gibbs barrier and released energies for abstraction of hydrogen atoms from the various ring sites of 6-gingerol by a hydroxyl radical are given in Table 1 while the corresponding barrier energies for hydrogen atom abstraction from the different chain sites are given in Table 2. The barrier and released energies involved in reactions at the different sites are appreciably different (Tables 1, 2). These energies would mainly depend on reorganization of electron density due to the reactions which would depend on valencies of the reacting centers and local reaction environments. Smaller barrier energies would reveal that the reactions under consideration would occur more easily while larger released energies would imply thermodynamically more stable products and vice versa. Negative barrier energies would imply that the reactions under consideration are barrierless. The structures of the different TSs, some important interatomic distances at these TSs and the imaginary vibrational frequencies corresponding to the hydrogen abstraction reactions for which barrier energies are presented in Tables 1 and 2 are shown in Figures 2 and 3. Optimized geometries of the reactant and product complexes corresponding to each transition state are shown in Figures S1–S4 (Supporting Information). There was a single common reactant complex for hydrogen abstraction from the sites C13 and C14 of the side chain. Both the forward and reverse barrier energies are informative as while the former shows the ease with which the reaction would occur, the latter reveals how stable would be the product formed consequent to the reaction. The reverse barrier energies would be obtained by reversing the sign of the released energies given in Tables 1 and 2.

Optimized transition state structures involved in hydrogen abstraction by an OH radical from the ring sites of 6-gingerol and the OCH3 and OH groups attached to the same along with the corresponding imaginary vibrational frequencies (cm−1) and some important bond lengths (Å) obtained at the M06-2X/6-31G(d,p) level of theory in gas phase.

Optimized transition state structures involved in hydrogen abstraction by an OH radical from the side chain of 6-gingerol along with the corresponding imaginary vibrational frequencies (cm−1) and some important bond lengths (Å) obtained at the M06-2X/6-31G(d,p) level of theory in gas phase.
Hereafter, the forward barrier energies would simply be referred to as barrier energies. The barrier heights for hydrogen abstraction from the phenolic OH group (O3 ′ site) (Figure 1) obtained at the B3LYP/6-31G(d,p) and B3LYP/6-311 ++G(d,p) levels of theory in gas phase as well as aqueous media are small and negative. However, the corresponding barrier energies obtained at the M06-2X/6-31G(d,p) and M06-2X/6-311 ++G(d,p) levels of theory in both gas phase and aqueous media are small and positive. The C2 ′ site (Figure 1) is the next site from where hydrogen abstraction would occur efficiently. In this case, the calculated barrier energies using the B3LYP functional and both the basis sets are smaller than those obtained using the M06-2X functional and the corresponding basis set in gas phase as well as aqueous media. The barrier energies, excepting those corresponding to the C2 ′ and O3 ′ sites, in both gas phase and aqueous media obtained using the B3LYP functional lie in the range ∼3-7.5 kcal/mol while those obtained using the M06-2X functional lie in the range ∼5-9 kcal/mol.
The released energies for hydrogen abstraction reactions from the C2 ′ and O3 ′ sites (Figure 1) are much larger than the corresponding barrier energies. In other words, these reactions are highly exothermic. However, the differences between the barrier and released barrier energies for the hydrogen abstraction reactions at the other sites are much smaller than those at the C2 ′ and O3 ′ sites (Table 1). It shows that the reactions at the other sites than the C2 ′ and O3 ′ would be mildly exothermic, and would not lead to as stable products as those involving the C2 ′ and O3 ′ sites.
The Gibbs barrier and released energies involved in hydrogen abstraction reactions by a hydroxyl radical from the different sites of the side chain of 6-gingerol obtained using the B3LYP and M06-2X functional s along with the 6-31G(d,p) and 6-311 ++G(d,p) basis sets in gas phase and aqueous media are presented in Table 2. It is noted that the Gibbs barrier energies obtained at the B3LYP/6-311 ++G(d,p) level are mostly small, positive or negative in both gas phase and aqueous media. However, at the M06-2X/6-311 ++G(d,p) level of theory, for hydrogen abstraction from five sites of the side chain, the Gibbs barrier energies are small and negative in both gas phase and aqueous media while for the corresponding reactions from the other sites, the barrier energies are positive in both the media, the maximum value in aqueous media being ∼6 kcal/mol (Table 2). The Gibbs released energies are much larger in magnitude than the corresponding Gibbs barrier energies in all the cases (Table 2). Therefore, all the reactions would be appreciably exothermic.
Zingerone and 6-gingerol are both found in ginger and act as anti-oxidants. Both the molecules have a phenolic ring, each having methoxy and hydroxyl groups attached to its C2 and C3 positions, respectively.[43] Further, both the molecules consist of a long unbranched carbon chain having 5 and 10 carbon atoms attached to the C6 site of the ring. There is no hydroxyl group attached to the side chain of zingerone while such a group is present in 6-ginerol attached to the C11 site of the corresponding chain.[43] The number of sites in zingerone that contribute to hydrogen abstraction by an OH radical where the calculated barrier energies at the B3LYP/6-31G(d,p) level are 2, 3 and 4 kcal/mol or less are 5, 6 and 7 while the corresponding numbers in 6-gingerol are 12, 12 and 13, respectively. Thus, for operation through the hydrogen abstraction mechanism, 6-gingerol appears to be a better OH radical scavenger than zingerone.[43]
We could not perform the CCSD(T)/6-31G(d,p) level of calculation for the reaction channels associated with the 6-gingerol molecule. This is because of our limited computational resource. Nevertheless, in order to test relative accuracies available with the B3LYP and M06-2X functionals, we performed single point CCSD(T)/6-31G(d,p) calculations for the CH3 COCH 2CH(OH)CH3 segment of the side chain obtained by replacing C7 and C13 atoms by a H atom each in gas phase. Further, abstraction of only two hydrogen atoms i.e., H11 and H11 ′ (Figure 1) by an OH radical was considered in these calculations. In these calculations, the geometries optimized at both the B3LYP/6-31G(d,p) and M06-2X/ 6-31G(d,p) levels were used. Gibbs barrier and released energies were calculated at the B3LYP/6-31G(d,p) and M06-2X/6-31G(d,p) levels employing the necessary thermal energy corrections obtained after vibrational frequency analyses. However, vibrational frequency analysis was not performed at the CCSD(T)/6-31G(d,p) level but barrier and released energies were calculated at this level with and without consideration of the corresponding thermal energy corrections obtained at the B3LYP/6-31G(d,p) and M06-2X/6-31G(d,p) levels. The barrier and released energies obtained for abstraction of the two hydrogen atoms are given in Table S1 (Supporting Information). The results presented in this table reveal that the barrier energies obtained at the M06-2X/6-31G(d,p) level are closer to those obtained at the CCSD(T)/6-31G(d,p) level than those obtained at the B3LYP/6-31G(d,p) level.
It is seen from Table S1 (Supporting Information) that the barrier energies for hydrogen abstraction from the CH and OH sites of the above mentioned segment of the side chain of 6-gingerol obtained at the CCSD(T)/6-31G(d,p) level of theory are appreciably larger than those obtained at the M06-2X/6-31G(d,p) level. Considering both the hydrogen abstraction and addition reactions involving an OH radical and different sites of 6-gingerol (Tables 2, 3), we find that many barrier energies predicted at M06-2X/6-31G(d,p) level are small positive or negative which indicate that these reactions would be nearly barrierless or barrierless. If we assume similar relative accuracies in barrier energies obtained at the CCSD(T)/6-31G(d,p) and M06-2X/6-31G(d,p) levels of theories for the various reactions involving the different sites of 6-gingerol (Tables 2, 3) as that found for hydrogen abstraction from the CH and OH sites of the selected segment of the side chain of the molecule, the barrier energies would be expected to be significantly increased in a CCSD(T)/6-31G(d,p) calculation. In that situation, several nearly barrierless or barrierless reactions mentioned above would be expected to be associated with small positive barriers. It shows that, broadly speaking, our conclusions based on the M06-2X/6-31G(d,p) level calculations are valid.
3.2 Radical addition reactions
Addition reactions between a hydroxyl radical and a site Cn of 6-gingerol can be expressed by the following general scheme.
Where, Cn-OH· is the RC between 6-gingerol and OH·, the latter being positioned near the Cn site of the former. After the necessary Gibb’s energy to overcome the barrier energy \(({\Delta } \mathrm {G}^{\prime \prime \;\;\mathrm {b}}_{\;\;\mathrm {n}})\) has become available, the transition state [TS]n is formed from the RC. Subsequently, after releasing the Gibbs energy \(({\Delta } \mathrm {G}^{\prime \prime \;\; \mathrm {r}}_{\;\;\mathrm {n}})\), the product complex i.e., the adduct (CnOH⋅) is formed from the transition state. The calculated Gibb’s barrier \(({\Delta } \mathrm {G}^{\prime \prime \;\;\mathrm {b}}_{\;\;\mathrm {n}})\) and released \(({\Delta } \mathrm {G}^{\prime \prime \;\;\mathrm {r}}_{\;\;\mathrm {n}})\) energies at 298.15 K for addition at the different carbon sites of the ring of 6-gingerol are presented in Table 3.
The optimized geometries of the transition states along with the barrier and released energies, certain inter-atomic distances and the imaginary vibrational frequencies at the TSs involved in the addition reactions are shown in Figure 4 while the geometries of the corresponding reactant and product complexes are shown in Figures S5 and S6 (Supporting Information). Though there was a common reactant complex for the hydroxyl radical addition reactions at the C2 and C3 sites, the corresponding barrier energies are noticeably different (1.86 and −0.6 kcal/mol in aqueous media at the M06-2X/6-311 ++G(d,p) level) (Table 3). Thus, reaction kinetics at two sites can be appreciably different even if the stabilities of the corresponding RCs are similar or the same.

Optimized transition state structures involved in the formation of adducts of an OH radical at the different sites of the ring of 6-gingerol along with the corresponding imaginary vibrational frequencies (cm−1) and some important bond lengths (Å) obtained at the M06-2X/6-31G(d,p) level of theory in gas phase.
At the M06-2X/6-31G(d,p) level, in the different reactant complexes, the interatomic distances between the corresponding carbon atom and the oxygen atom of the hydroxyl radical lie in the range 2.3-2.4 Å while at the different transition states, the corresponding interatomic distances lie in the range 2.0 – 2.07 Å (Figure 4). Thus, we find that there is substantial decrease of interatomic distance between the corresponding carbon atom and the oxygen atom of the hydroxyl radical in going from RCs to TSs.
At the M06-2X/6-311 ++G(d,p) level of theory, the calculated Gibbs barrier energies \(({\Delta } \mathrm {G}^{\prime \prime \;\;\mathrm {b}}_{\;\;\mathrm {n}})\) for addition reactions at the different carbon sites of the ring of 6-gingerol in aqueous media lie in the order C3 < C4 < C6 < C2 < C5 < C1, the smallest and largest barrier energies being −0.6 and 3.53 kcal/mol (Table 3). Thus, the addition reaction would occur most and least efficiently at the ring position C3 and C1, respectively. The results obtained using the B3LYP functional are significantly different from the obtained at the M06-2X functional (Table 3). In this situation, we would consider the results obtained using the M06-2X functional as more reliable in view of the previous reports.[36,37]The released energy at any of the reaction steps is appreciably larger in magnitude than the corresponding barrier energy (Table 3). Thus, all these reaction steps are appreciably exothermic (Table 3).
A graphical comparison of Gibbs barrier energies obtained at the M06-2X/6-311 ++G(d.p) level of theory in aqueous media involved in hydrogen abstraction by a hydroxyl radical from the different sites of the ring of 6-gingerol or from the groups attached to the same is made in Figure 5a while those involved in hydrogen abstraction by a hydroxyl radical from the various sites of the side chain of 6-gingerol in aqueous media is made in Figure 5b. Figure 5a reveals that hydrogen abstraction by a hydroxyl radical from the C1, C4 and C5 sites of the ring of 6-gingerol would be much less likely than from the C2 ′ and O3 ′ sites while Figure 5b shows that the reaction would take place most rapidly at the O11 ′, C12, C14, C15 and C16 sites of the side chain of 6-gingerol. Further, Figure 5b shows that the reaction would occur much less efficiently at the C8 and C10 sites while it would occur with intermediate efficiencies at the remaining sites. A graphical comparison of Gibbs barrier energies in aqueous media involved in the addition of a hydroxyl radical at the different sites of the ring of 6-gingerol obtained at the M06-2X/6-311 ++G(d,p) level of theory is made in Figure 5c. This figure clearly reveals that the addition reactions would occur most rapidly at the ring site C3.

Graphical comparison of Gibbs barrier energies (kcal/mol) obtained at the M06-2X/6-311 ++G(d,p) levels of theory in aqueous media involved in, (a) hydrogen abstraction by a hydroxyl radical from the different sites of the ring of 6-gingerol or groups attached to the same, (b) hydrogen abstraction from different sites of the side chain of 6-gingerol, and (c) addition reactions at different sites of the ring of 6-gingerol.
3.3 Electron transfer reaction
The Marcus theory of electron transfer[44‐46]considers electron jump from an electron donor to an electron acceptor. The electron jump between the donor and acceptor involves the barrier energy \(({\Delta } \mathrm {G}^{\mathrm {b}}_{\text {ET}})\) which can be obtained in terms of the Gibbs free energy of reaction \(({\Delta } \mathrm {G}^{\mathrm {r}}_{\text {ET}})\) and reorganization energy (λ) as follows.[47,48]
where \({\Delta } \textit {G}^{\mathrm {r}}_{\text {ET}}\) is the difference between the sums of Gibbs free energies of products and reactants. The reorganization energy (λ) is the amount of energy involved in structural reorganization of the electron donor and the acceptor. If electron transfer occurs from a species A to another species B producing a cation of A and an anion of B, according to the Nelsen’s four point scheme,[47,48]the reorganization energy involved in this process would be the sum of Gibbs free energy differences between the vertical and relaxed cation of A and the vertical and relaxed anion of B. Thus, we can obtain the following equation
where the first term on the r.h.s. is the vertical Gibbs free energy difference between the cation, anion pair while the second term is the Gibbs free energy difference for the relaxed pair. The rate constant can be obtained using the equation given in Section 3.4.
Gibbs barrier energy (\({\Delta } \mathrm {G}^{\mathrm {b}}_{\text {ET}}\)) and reorganization energy (λ) for single electron transfer (SET) from 6-gingerol to an OH radical obtained by full geometry optimization at the B3LYP/6-31G(d,p), B3LYP/6-311 ++G(d,p), M06-2X/6-31G(d,p) and M06-2X/6-311 ++G(d,p) levels of theory in gas phase and aqueous media are presented in Table 4. It is noted that the Gibbs barrier energy \(({\Delta } \mathrm {G}^{\mathrm {b}}_{\text {ET}})\) is very sensitive to the level of theory used, the medium in which the electron transfer process takes place and whether geometrical relaxation in the medium under consideration is allowed or not. The reorganization energy (λ) is also sensitive to these factors but to much smaller extents. The Gibbs barrier energies in gas phase are very large due to which electron transfer cannot occur in this phase. The Gibbs barrier energies in aqueous media obtained at the B3LYP/6-311 ++G(d,p) and M06-2X/6-311 ++G(d,p) levels of theory are 2.45 and 6.85 kcal/mol. Due to a greater accuracy available with the M06-2X functional, as also discussed later, the latter value should be considered more reliable. Thus, we find that SET barrier energies in aqueous media become comparable to those of many hydrogen abstraction and addition reactions.
Another possible mechanism for electron transfer reaction is the proton coupled electron transfer (PCET) mechanism.[49,50]The calculated reaction barrier energy for PCET for the C3 site of the ring was calculated and found to be 0.28 kcal/mol in aqueous media at the M06-2X/6-311 ++G(d,p) level of theory. As this barrier energy is much smaller than the SET barrier energy (6.9 kcal/mol) (Table 4), the PCET mechanism would be much more favored than the SET mechanism. We chose the C3 site for this reaction since the hydrogen atom of the phenolic OH group attached to this site is expected to involve lowest barrier energy.
3.4 Rate constants
The rate constant k for a reaction is given by the following general expression according to the transition state theory.[38]
where Γ(T) is the quantum mechanical tunneling factor, kB is the Boltzmann constant, T is the temperature on the absolute scale (room temperature = 298.15 K), h is the Planck’s constant, R is the gas constant and ΔG b is the Gibbs barrier energy involved in the reaction under consideration. Γ(T) can be obtained as the Wigner transmission coefficient given by,[51,52]
where ω is the imaginary frequency (in cycles/sec.) at the transition state under consideration. The imaginary frequency can also be expressed in cm−1 and represented by ν. The calculated values of Γ(T) for the various imaginary frequencies ν (cm−1) at the different transition states are given in Tables S2–S4 (Supporting Information). It is noted that the calculated values of Γ(T) range between 1.01 and 4.24 (Tables S2–S4). Therefore, Γ(T) cannot be ignored and must be evaluated to obtain the overall rate constant k (equation 3) reliably. The calculated rate constants k including Wigner transmission coefficients Γ(T) for hydrogen abstraction by an OH radical from the different ring sites of 6-gingerol at the various levels of theory in gas phase and aqueous media are presented in Table 5 while those corresponding to hydrogen abstraction from the side chain and addition of an OH radical at the different sites of the ring of 6-gingerol are presented in Tables S5 and S6 (Supporting Information), respectively. As negative barrier energies imply barrierless reactions, barrier energies in such cases were taken to be zero for calculating rate constants. It is noted that rate constants for different steps of the above mentioned reactions in gas phase and aqueous media, broadly speaking, lie in the range 106 - 10 13 M−1s−1.
The rate constants including Wigner transmission coefficients for hydrogen abstraction by an OH radical from the various carbon sites of the side chain and those for addition of an OH radical at the different carbon sites of the ring of 6-gingerol obtained at the various levels of theory and presented in Tables S5 and S6 (Supporting Information) lie approximately in the range 10 8−1013 M−1s−1. The calculated values of the rate constant including Wigner transmission coefficient for hydrogen abstraction from the OH group of phenol in aqueous media were found to be 1.1 × 1010 and 3.7 × 1010 M−1s−1 at the M06-2X/6-31G(d,p) and M06-2X/6-311 ++G(d,p) levels of theory, respectively, while the corresponding experimental value[53] is 1.0 × 1010 M−1s−1. The hydrogen abstraction reaction under consideration was found at the B3LYP/6-31G(d,p) and B3LYP/6-311 ++G(d,p) levels to be barrierless, the corresponding rate constants including Wigner transmission coefficient being 6.21 × 1012 M−1s−1 each. Thus, the rate constants obtained using the M06-2X functional appear to be more reliable than those obtained using the B3LYP functional. We had arrived at the same conclusion earlier in sub-section 3.1 on the basis of comparison of barrier energies obtained using these functionals and the CCSD(T) method. Further, the rate constant for phenol calculated at the M06-2X/6-311 ++G(d,p) level is closest to the experimental value. Rate constants for electron transfer from 6-gingerol to the OH radical at the B3LYP/6-311 ++G(d,p) and M06-2X/6-311 ++G(d,p) levels of theory in aqueous media were found using the barrier energies discussed earlier to be 9.9 × 1010 and 5.9 × 107 M−1s−1, respectively. In view of the above discussion, the rate constant obtained at the M06-2X/6-311 ++G(d,p) level of theory would be expected to be more reliable than that obtained at the B3LYP/6-311 ++G(d,p) level. Thus, the electron transfer process would also contribute efficiently to scavenging of the OH radical, and, on the whole, 6-gingerol would act as a very efficient hydroxyl radical scavenger.
4 Conclusions
The present study leads us to the conclusion that 6-gingerol is an excellent hydroxyl radical scavenger and operates efficiently through hydrogen abstraction, radical addition and electron transfer mechanisms. Some of these reactions are barrierless or are associated with small or moderate barriers. The tunneling contributions to rate constants are usually significant and need to be evaluated. The different thermal reaction rate constants lie in the range 106 -10 13 M−1s−1.
References
Hussain S P, Hofseth L J and Harris C C 2003 Nat. Rev. Cancer 3 276
White B, Smyth M R, Stuart J D and Rusling J F 2003 J. Am. Chem. Soc. 125 6604
Jena N R and Mishra P C 2005 J. Phys. Chem. B 109 14205
Ramakrishnan N, Kalinich J F and McClain D E 1996 Biochem. Pharmacol. 51 1443
Kehrer J P 2000 Toxicology 149 43
Finkel T 1999 J. Leukocyte Biol. 65 337
Greenberg M M 2007 Org. Biomol. Chem. 5 18
Simons J 2006 Acc. Chem. Res. 39 772
Mishina Y, Duguid E M and He C 2006 Chem. Rev. 106 215
Wyatt M D and Pittman D L 2006 Chem. Res. Toxicol. 19 1580
Neeley W L and Essigmann J M 2006 Chem. Res. Toxicol. 19 491
Bignami M, O’Driscoll M, Aquilina G and Karran P 2000 Mutat. Res. 462 71
Loechler E L, Green C L and Essigmann J M 1984 Proc. Natl. Acad. Sci. U.S.A. 81 6271
Sarma A D, Mallick A R and Ghosh A K 2010 Int. J. Pharma Sci. Res. 1 185
Agnihotri N and Mishra P C 2009 J. Phys. Chem. B 113 12096
Yadav A and Mishra P C 2013 J. Mol. Model. 9 767
Tiwari M K and Mishra P C 2013 J. Mol. Model. 19 5445
Aoki K, Cortes A R, Ramirez M C, Gomez H M and Lopez M F J 2008 J. Ethnopharmacol. 116 96
Shukla Y and Singh M 2007 Food Chem. Toxicol. 45 683
Bhattarai S, Tran V H and Duke C C 2001 J. Pharm. Sci. 90 1658
Mariadoss A V, Kathiresan S, Muthusamy R and Kathiresan S 2013 Asian Pac. J. Cancer Prev. 14 3123
Suresh K, Manoharan S, Vijayaanand M A and Sugunadevi G 2010 Carcino. Pharmacol. 62 1178
Ghosh A K, Sarkar and Mahmud A K 2011 Int. J. Pharma Bio-Sci. 2 817
Dugasania S, Pichikac M R, Nadarajahc V D, Balijepalli M K, Satyanarayana T and Korlakuntab J N 2010 J. Ethnopharmacol. 127 515
Surh Y J 2002 Food Chem. Toxicol. 40 1091
Jung Park Y J, Wen J, Bang S, Park S W and Song S Y 2006 Yonsei Med. J. 47 688
Abdullah S, Abidin S A Z, Murad N A, Makpoll S, Ngah W Z W and Yusof Y A M 2010 Afr. J. Biochem. Res. 4 134
Hiserodt R D, Franzblau S G and Rosen R T 1998 J. Agric. Food Chem. 46 2504
Jolad S D, Lantz R C, Solyom A M, Chen G J, Bates R B and Timmermann B N 2004 Phytochemistry 65 1937
Navas-Acien A, Silbergeld E K, Pastor-Barriuso R and Guallar E 2008 JAMA 300 814
Longnecker M P and Daniels J L 2001 Environ. Health Perspect. 109 871
Chakraborty D, Mukherjee A, Sikdar S, Paul A, Ghosh S and Khuda-Bukhsh A R 2012 Toxicol. Lett. 210 34
Saha P, Das B and Chaudhuri K 2013 Antimicrob. Agents Chemother. 57 4373
Lee C, Yang W and Parr R G 1988 Phys. Rev. B Condens. Matter. 37 785
Becke A D 1993 J. Chem. Phys. 98 5648
Zhao Y and Truhlar D G 2006 J. Phys. Chem. A 110 5121
Zhao Y and Truhlar D G 2008 J. Chem. Theory Comput. 4 1849
Laidler K J 2004 In Chemical Kinetics 3rd Edn. (Delhi: Pearson Education (Singapore)). p 89
Miertus S, Scrocco E and Tomasi J 1981 Chem. Phys. 55 117
Miertus S and Tomasi J 1982 Chem. Phys. 65 239
Dennington R, Keith T and Millam J 2009 GaussView, Version 5. Semichem. Inc. Shawnee Mission, K S
Frisch M J, Trucks G W, Schlegel H B, Scuseria G E, Robb M A, Cheeseman J R, Scalmani G, Barone V, Mennucci B, Petersson G A, Nakatsuji H, Caricato M, Li X, Hratchian H P, Izmaylov A F, Bloino J, Zheng G, Sonnenberg J L, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery J A, Peralta J E, Ogliaro F, Bearpark M, Heyd J J, Brothers E, Kudin K N, Staroverov V N, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant J C, Iyengar S S, Tomasi J, Cossi M, Rega N, Millam M J, Klene M, Knox J E, Cross J B, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann R E, Yazyev O, Austin A J, Cammi R, Pomelli C, Ochterski J W, Martin R L, Morokuma K, Zakrzewski V G, Voth G A, Salvador P, Dannenberg J J, Dapprich S, Daniels A D, Farkas O, Foresman J B, Ortiz J V and Cioslowski J, (Gaussian 09, revision D.01, Gaussian Inc.: Wallingford, CT USA) (2009)
Prasad A K and Mishra P C 2014 J. Phys. Org. Chem. 27 18
Marcus R A 1964 Annu. Rev. Phys. Chem. 15 155
Marcus R A 1993 Rev. Mod. Phys. 65 599
Marcus R A 1997 Pure Appl. Chem. 69 13
Nelsen S F, Blackstock S C and Kim Y 1987 J. Am. Chem. Soc. 109 677
Nelsen S F, Weaver M N, Luo Y, Pladziewicz J R, Ausman L K, Jentzsch T L and O’Konek J J 2006 J. Phys. Chem. A 110 11665
Anglada J M 2004 J. Am. Chem. Soc. 126 9809
Litwinienko G and Ingold K U 2004 J. Org. Chem. 69 5888
Barreto P R P, Vilela A F A and Gargano R 2003 J. Mol. Struct. (THEOCHEM) 664 135
Ng M., Mok D K W, Lee E P F and Dyke J M 2013 J. Comput. Chem. 34 545
De A K, Chaudhuri B, Bhattacharjee S and Dutta B K 1999 J. Hazard. Mater. B 64 91
Acknowledgements
One of the authors (PCM) is thankful to the National Academy of Sciences (NASI) for awarding a Senior Scientist Fellowship and for financial support. MKT is thankful to the University Grants Commission (New Delhi) for a research fellowship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information (SI)
The electronic supporting information can be seen at www.ias.ac.in/chemsci.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
TIWARI, M.K., MISHRA, P.C. Anti-oxidant activity of 6-gingerol as a hydroxyl radical scavenger by hydrogen atom transfer, radical addition and electron transfer mechanisms. J Chem Sci 128, 1199–1210 (2016). https://doi.org/10.1007/s12039-016-1128-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12039-016-1128-7