
Abstract
Advances in DNA sequencing provide tools for efficient large-scale discovery of markers for use in plants. Discovery options include large-scale amplicon sequencing, transcriptome sequencing, gene-enriched genome sequencing and whole genome sequencing. Examples of each of these approaches and their potential to generate molecular markers for specific applications have been described. Sequencing the whole genome of parents identifies all the polymorphisms available for analysis in their progeny. Sequencing PCR amplicons of sets of candidate genes from DNA bulks can be used to define the available variation in these genes that might be exploited in a population or germplasm collection. Sequencing of the transcriptomes of genotypes varying for the trait of interest may identify genes with patterns of expression that could explain the phenotypic variation. Sequencing genomic DNA enriched for genes by hybridization with probes for all or some of the known genes simplifies sequencing and analysis of differences in gene sequences between large numbers of genotypes and genes especially when working with complex genomes. Examples of application of the above-mentioned techniques have been described.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Molecular genetic markers have found wide application in plants (Henry 1997). Molecular markers are used increasingly in plant breeding to guide selection and plant identification and play an important role in determining plant identity and or purity in forensic and industrial situations. Protection of intellectual property rights such as plant variety rights is strongly supported by the use of molecular markers, while research on population genetics, conservation genetics and evolutionary genetics of plant population also makes extensive use of molecular genetic markers (Henry 2005).
DNA-based molecular markers have largely replaced earlier biochemical markers as techniques for DNA analysis in plants have improved (Henry 2001, 2008). Methods for both the discovery and analysis of molecular markers have been improved (Henry and Edwards 2009) with advances in high-throughput analysis of genetic polymorphisms focusing most recently on single nucleotide polymorphisms (SNP) (Kharabian-Masouleh et al. 2009; Varshney et al. 2010). For some time the application of high-throughput SNP analysis technology was limited by the availability of plant genome DNA sequence information, and screening populations for DNA marker discovery employed a range of techniques to deal with the lack of direct sequence information (Cross et al. 2008). Recently dramatic advances in second- and third-generation DNA sequencing technology have removed this barrier (Thudi et al. 2012).
2 Advances in DNA sequencing technologies
2.1 Sequencing strategies
Plant genome sequencing has progressed rapidly since the first genome (Arabidopsis thaliana) was completed in 2000 (Arabidopsis Genome Initiative 2000). The 389 Mb rice genome was completed in 2004 (IRGSP International Rice Genome Sequencing Project 2005). These projects used the ‘traditional’ Sanger sequencing approach. A well-proven method for large genome sequencing is known as BAC-by-BAC (bacterial artificial chromosome) sequencing and involves the production of an overlapping tiling path of large genomic fragments (around 120,000 bases) maintained within BACs. An alternative cheaper, high-throughput approach is the whole genome shotgun (WGS) method, where the entire genome is fragmented into many smaller pieces that are individually sequenced, and computational algorithms are applied to assemble the complete genome sequence. Second-generation sequencing (SGS) methods allow huge increases in data output at much lower costs; however, the read quality is more variable. This variability is mainly overcome by increased depth of coverage, which is especially important during SNP marker discovery (Deschamps and Campbell 2010, Varshney et al. 2010).
The assembly of large plant genome sequences is very problematic with short-read technologies due to sequence repeats. Various experimental designs that can facilitate sequence assembly include longer reads, paired-end reads, mate-pair libraries (large insert libraries), RNA-Seq data, reduced representation libraries, optical mapping and genetic mapping (Birney 2011). Paired-end or mate-pair protocols help to overcome some of these limitations of short reads by providing information about relative location and orientation of a pair of reads. In paired-end sequencing, the actual ends of rather short DNA molecules (<1 kb) are determined, while mate-pair sequencing requires the preparation of special libraries. In these protocols, the ends of longer, size-selected molecules (e.g. 8, 12 or 20 kb) are connected with an internal adapter sequence in a circularization reaction (figure 1) (Kircher and Kelso 2010).

Sequencing library preparations and de novo assembly of whole genome sequence requires a range of insert lengths using second-generation short read technologies (Glenn 2011).
Sample preparations often involve methods to select targeted regions in multiple individuals or samples instead of sequencing one sample to excessive depth in order to reduce costs and take advantage of the large output capacities in a single sequencing process. These methods also called ‘genome partitioning’, ‘enrichment’ or ‘genomic capture’ are being utilized. For example, megabase-size candidate genomic regions, identified through linkage studies, can be sequenced to find the exact genes and mutations underlying the trait. This is facilitated by techniques such as hybridization to preselected probes, mRNA extraction or attachment of barcode (sometimes called ‘index’) sequences to the library molecules. Samples are separated later (computationally) based on their individual barcode sequence. Commercial kits presently contain from 12 to 384 unique indexes (Edwards and Henry 2011). The complexity of polyploid genome analysis has been reduced through chromosome sorting preparations (Imelfort et al. 2009a).
2.2 Sequencing Technologies
For decades, DNA sequencing relied on the Sanger di-deoxy method with gradual improvements in the technology. Beginning in the mid 2000s a series of new techniques were developed and continuously improved. These techniques have allowed SGS to generate sequence data in very large volumes much more rapidly and cost-effectively (Varshney et al. 2010). The major technologies are detailed in table 1 (figure 2). SGS technologies have delivered large volumes of data, but the lowest cost options have been very short sequence reads. Third-generation technologies based upon single molecular analysis are currently being developed commercially. These technologies promise a number of different advances, lower cost per analysis, longer reads and direct RNA sequencing and direct analysis of methylation. A range of different applications for SGS have been developed and applied to marker discovery in plants (table 2).

Early whole genome assembly used overlapping BACs (bacterial artificial chromosomes) with original Sanger sequencing technology (Kircher 2010).
SGS methods follow a similar pattern for library preparation that can be summarized in three major steps: (1) random shearing of DNA, either via nebulization or sonication; (2) ligation of universal adapters at both ends of the sheared DNA fragments and (3) immobilization and amplification of the adapter-flanked fragments to generate clustered amplicons to serve as templates for the sequencing reactions (Shendure and Ji 2008). These platforms parallelize from thousands to millions of sequencing reactions. Through alternating cycles of base incorporation and image capture, short DNA sequences ranging in size from 25 to 500 bases are generated (table 2) (Deschamps and Campbell 2010).
The dominant SGS sequencing platforms are the Roche Genome Sequencer FLX System, ABI SOLiD and the Illumina platforms. The 454 Genome Sequencer was initially introduced in 2005 by 454 Life Sciences and subsequently acquired by Roche Diagnostics and renamed the Roche Genome Sequencer FLX System. This platform utilizes pyrosequencing technology parallelized on separate beads in a picotiter plate. In pyrosequencing, one pyrophosphate per nucleotide is released and converted to ATP by an ATP sulphurylase. The ATP drives the light reaction of luciferases present (Kircher and Kelso 2010). The 454 sequencing at present produces the longest reads but the total sequence output per run is low (Brautigam and Gowik 2010; Magi et al. 2010). Illumina (former Solexa) sequencing introduced in early 2007 is the most widely used SGS technology. This method uses a solid-phase bridge PCR, and a ‘sequencing by synthesis’ approach, with fluorescent dye-labelled reversible terminator nucleotides allowing each cycle of the sequencing reaction to occur simultaneously in the presence of the four nucleotides that are distinguished by four fluorescent dyes. The ABI SOLiD (Sequencing by Oligo Ligation and Detection) sequencer was introduced in 2005 by Agencourt Personal Genomics. This method employs sequential ligation producing short reads (up to 50 bases). The system features a two-base encoding mechanism that interrogates each base twice providing a form of built-in error detection (Brautigam and Gowik 2010).
The current SGS methods all have a requirement for numerous and slow wash–scan cycles. As each base is added, the population of molecules loses synchronicity (called dephasing), and this causes an increase in noise and sequencing errors as the read extends, effectively limiting the read length. In common with Sanger sequencing, the error rate also increases with sequence length due to a reduction in enzyme efficiency or loss of enzymes. In addition, to obtain sufficient light signal intensity for detection, the sequencing-by-synthesis reaction step requires PCR amplifications, which can introduce errors in the template sequence as well as amplification bias. Finally, the generation of huge amounts of sequence data has created challenges for storage and bioinformatics operations such as sequence annotation tasks (figure 3) (Imelfort et al. 2009a; Schadt et al. 2010).

High-throughput assays using molecular markers facilitate plant identification and forensics and can guide early selection of a trait in a breeding program. Statistical analyses relate markers to physical characteristics. Innumerable marker-trait associations can be developed though marker effects can be inconsistent.
A recent survey of public and private sequencing laboratories in the US and Europe estimated an increase in utilization of SGS technologies from ~37% of to 56% within 2 years. Interest for third-generation platforms is expected to replace ~47% of all SGS activity in the next 3 years (Peterson et al. 2010). Third-generation DNA-sequencing technologies (table 1) are mostly distinguished by direct inspection of single molecules with methods that do not require the repetitive wash and scan steps during DNA synthesis, synchronization of multiple reactions or problems associated with PCR amplifications or phasing (Thudi et al. 2012). Several technologies are currently under development utilizing diverse approaches such as captured polymerases, nanopores, electronic detection and fluorescence resonance energy transfer (FRET) detection and transmission electron microscopy (TEM). Read lengths, time to results and cost are expected to greatly improve (Edwards and Henry 2011; Schadt et al. 2010).
3 Applications to plants
3.1 Amplicon sequencing
In plants, polymerase chain reaction (PCR) allows sequences of interest to be amplified and isolated from the genome, transcriptome or even organelles. Even with the limited capacity of first-generation (Sanger) sequencing, amplicons generated by PCR provided the basis for large scale analysis of allelic variations (SNPs and InDels) in genes, gene families and specific sequence regions of interest for population studies and marker development.
Advances in SGS have resulted in the continued application of amplicon sequencing in plant genetics (Bundock et al. 2009; Kulheim et al. 2009; Kharabian-Masouleh et al. 2011; Malory et al. 2011; Sexton et al. 2011). Because of the massive capacity of SGS, amplicons are now sequenced at coverage levels significantly higher than Sanger sequencing (deep sequencing). Deep sequencing utilizes the high sequencing capacity of SGS and ensures that base calling is internally validated, effectively allowing SNPs to be defined from sequencing errors (Bundock et al. 2009; Cronn et al. 2008). PCR amplification is used to isolate the sequence(s) of interest using individuals or pooled of DNA from the population of interest as a template. Amplicons from multiple genes or sequence regions can be combined (multiplexed or pooled) and sequenced together, so long as the genes are of sufficient difference to allow sequences to be assembled later by alignment of the sequence reads generated with the reference DNA sequences. Early application SGS to two different polyploid sugarcane genotypes identified 1632 and 1013 SNPs for the Q165 (242 amplicons) and IJ76–514 (247 amplicons) varieties, respectively (Bundock et al. 2009). Cronn et al. (2008) used multiplex PCRs to amplify 35 amplicons representing ~120 kb of chloroplast sequence in individual genotypes representing seven Pinus and one Picea species. Following amplification genotypes were barcoded with 3 bp indexing tags and pooled prior to SGS. This experiment provided the basis for phylogenetic studies between many Pinus species (Parks et al. 2009; Whittall et al. 2010).
SGS can be effectively integrated with long-range PCR (LR-PCR) of pooled DNA samples. This maximizes the efficiency of sequencing and reduces the cost of amplification, oligonucleotides and labour (Out et al. 2009). Utilizing a LR-PCR approach, targeted deep sequencing can be employed to analyse regions surrounding candidate genes involved in various aspects of physiology, stress tolerance, quality, disease, etc. (Varley and Mitra 2008). This technology has the power to detect new mutations and discover rare alleles as a fragment of 10 kb may be read 100,000 times or more (Druley et al. 2009; Out et al. 2009; Thomas et al. 2006).
Pooling populations of individuals prior to sequencing, either at the tissue or DNA level, is another way to utilize the high sequencing capacity of SGS. By ensuring equal concentrations of DNA are pooled, accurate allele frequencies can be estimated for pooled populations (Out et al. 2009; Futschik and Schlotterer 2010; Sexton et al. 2011).Therefore, sequence polymorphisms can be identified between sequence reads bases of relative frequency (Kulheim et al. 2009; Kharabian-Masouleh et al. 2011; Malory et al. 2011; Sexton et al. 2011). In pooling experiments, genotype (individual) identities are confounded, and so it is important that pool size is not so high that target allele frequencies and expected error frequencies do not converge. For example, if 50 diploid genotypes are pooled, a rare allele in one haplotype is expected to occur at a frequency of 1%; if the pooling depth increases further, there is a risk that errors (e.g. Taq, sequencing, etc.) could be mistakenly confounded with SNP calls. Because of this issue it is not uncommon for researchers to only confirm SNPs at conservative allele frequencies of 5 to 10% (Novaes et al. 2008; Kulheim et al. 2009).
For several plant species trans-specific primers have been used to amplify orthologous genes in distantly related species not previously characterized. In conjunction with pooling and SGS-based amplicon sequencing, large-scale SNP discovery in candidate genes has been achieved for populations in species previously uncharacterized (Kulheim et al. 2009; Malory et al. 2011; Sexton et al. 2011). Using this approach with multiple species in parallel, researchers working with species in the diverse Eucalyptus genus have utilized comparative genomics for the discovery of shared (trans-specific) SNPs which may be of biological and commercial significance (Kulheim et al. 2009; Sexton et al. 2011). Subsequent association studies have validated SNPs identified with this method and also revealed several significant correlations with specific phenotypes (Kulheim et al. 2011; Sexton et al. 2012).
A similar pooling and amplicaon-based SGS approach was used to characterize genetic variation in starch biosynthesis genes, where the DNA of 233 rice genotypes was pooled and LR-PCR of 17 genes amplified 116,403 bp of genomic sequence. Subsequent SGS generated 60.9 million reads corresponding to the coverage between 12,000- and 38,000-fold for each of the genes, within the sequence reads 501 SNP and 113 InDels were identified (Kharabian-Masouleh et al. 2011). These SNPs have been related to starch properties explaining much of the variation in starch properties in cultivated rice (Kharabian-Masouleh et al. 2011 unpublished). Analysis of some of these genes in wild rice relatives is helping define the changes that have been introduced by human selection for desirable grain qualities.
A total of 399 SNPs for the targeted loci had already been reported at OryzaSNP@MSU database ( http://oryzasnp.plantbiology.msu.edu/ ) domain for 20 rice cultivars. As expected, the total number of detected polymorphisms by Illumina sequencing was significantly higher than that reported in the rice public domain (OryzaSNP). Furthermore, discovery of InDels was another outcome of this approach as there were no InDels reported for these genes (Kharabian-Masouleh et al. 2011). A similar but more limited analysis of starch biosynthesis genes in sorghum germplasm using more laborious and expensive conventional sequencing has been used to explain variation in starch properties in sorghum (Hill et al. 2011).
3.2 Transcriptome sequencing
Transcriptome sequencing can not only define the sequences of all transcripts de novo, revealing previously undiscovered genes and splice variants, but can also indicate the relative abundance of the transcripts. This contrasts with traditional microarray analysis, which requires all genes of interest be known and represented on the microarray and does not return sequence/genotype data for a particular individual. Comparison of the transcriptome sequences of different tissues can be used follow changes in gene expression through development, in different tissues or in different genotypes to understand genetic differences that are due to differential gene expression (Gillies et al. 2012).
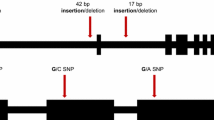
Genetic variation within commercial crop varieties is not usually well characterized or quantified. It follows then that the effect of intra-varietal genetic variation on crop performance under stress is also poorly understood, which may put production at risk from changing climate and rapidly evolving pests and diseases. Transcriptome sequencing allows genome-wide analysis of large, complex plant genomes and the potential to identify biologically significant SNPs. The genetic variation between and within barley varieties was defined by deep sequencing and assembled into unigenes the transcriptomes of two barley varieties Baudin and Gairdner (Pattemore et al. unpublished). A large number of SNP were identified, with more than 200,000 SNP between reads for variety Baudin and reference EST sequences, and more than 300,000 SNP between Baudin reads and de novo assembled reads from the variety Gairdner. Putatively significant SNPs (SNP allele frequency > 0.1) represented 9.65% and 14.64% of Baudin and Gairdner genetic variation. Background genetic diversity (SNP allele frequency ≤ 0.1) accounted for 90.23% and 85.52% of genetic variation in Baudin and Gairdner, respectively. The SNP dataset was further refined to produce a set of very high-quality SNPs for varietal genotyping. Although SNP variation within varieties has not been widely examined in other species, analyses of SNP between varieties have been undertaken to facilitate varietal distinction in many plant species like wheat (Trebbi et al. 2011), rice (Gopala Krishnan et al. 2012), maize (Barbazuk et al. 2007), chickpea (Hiremath et al. 2011), pigeonpea (Dubey et al. 2011), soybean (Wu et al. 2010) and oilseed rape (Trick et al. 2009) (see figure 4 for examples of SNP discovery strategies). Genome-wide marker sets, such as these developed from transcriptome sequencing, may become a valuable and complementary addition to the plant breeding toolbox uniquely aiding both selection and management of diversity within varieties.

(A) SNP discovery scheme (magnifying glass)Illustrating common risk of ascertainment bias through limitation of sample numbers causing selection of non-representative or uninformative SNPs, and SNPs that cannot distinguish the two populations (Garvin et al. 2010). (B) Common strategy for marker–trait association analyses. Bioinformatics analyses compare sequence data and identify phenotype association. Marker Assisted Selection is dependent on efficient and effective phenotypic evaluation programmes (Xu et al. 2008).
3.3 Whole genome sequencing
Recent major improvements in whole genome shotgun sequencing has drastically reduced the cost of whole genome sequencing, providing low cost coverage of any genome, generating a sequence resource for molecular marker and gene discovery, comparative genomics and genome assembly. Draft genome sequences have been assembled based on second-generation sequencing technology in several crop species such as castor (Chan et al. 2010), pigeonpea (Dubey et al. 2011), cocoa (Argout et al. 2010). Assembly of reference genome sequences for complex plant genomes remains difficult (Souza et al. 2011). Coupled with the developments in bioinformatics to assemble short reads, next-generation sequencing technologies enables the discovery of millions of DNA polymorphisms such as SNPs and Indels by comparing the whole genome sequences of several individuals within a species. SNPs and InDels are becoming the preferred markers in molecular breeding due to multiple advantages such as high frequency, stability, high-throughput capability and cost-effectiveness over other DNA markers (Henry and Edwards 2009). An approach based on annotation has been developed for discovery of genome-wide SNPs without a reference genome in Aegilops tauschii, a diploid progenitor of the hexaploid wheat (Yan et al. 2011). More recently, high-quality reference of autotetraploid potato has been achieved by whole genome sequencing of homozygous doubled monoploid derived from conventional tissue culture and has been compared with its heterozygous diploid breeding line for SNP discovery and evolution of potato (TPGSC 2011).
Whole genome sequencing accelerates the rate of gene and molecular marker discovery. For instance, large-scale SNP markers have been developed in a wide array of crops such as rice (Yamamoto et al. 2010; Arai-Kichise et al. 2011; Gopala Krishnan, et al. 2011), wheat (Akhunov et al. 2009), maize (Yan et al. 2010), soybean (Kim et al. 2010) and grapes (Myles et al. 2010). Whole genome re-sequencing has been utilized in rice for identifying SNPs, taking advantage of the high-quality reference genome sequences of the japonica cultivar Nipponbare (IRGSP 2005). SNP resources based on differences between a few cultivars pose limitations in its utility as only a subset of them may be applicable to other combinations of genotypes. Therefore, emphasis has been placed on enlarging the SNP resources through re-sequencing of diverse Oryza accessions (Tung et al. 2010). Through whole genome re-sequencing using sequencing by synthesis, large numbers of SNPs have been discovered in japonica cultivars by comparison of Nipponbare with japonica cultivars Koshihikari (Yamamoto et al. 2010) and Omachi (Arai-Kichise et al. 2011). More recently, whole genome re-sequencing of six elite indica rice parental inbreds (three CMS and three restorer lines) have been carried out resulting in discovery of 2,819,086 non-redundant DNA polymorphisms including 2,495,052 SNPs, 160,478 insertions and 163,556 deletions across the rice genome, providing an average of 6.8 SNPs/kb across the genome, which is significantly higher than the previously reported SNP rates in rice (Gopala Krishnan et al. 2011).
Whole genome re-sequencing of the A genome wild relatives from Australia, O. rufipogon and O. meridionalis, has aided discovery of a large number of SNPs with a mean SNP rate of 6.71 and 6.33 SNPs/kb across the rice genome. A comparison of the SNP distribution across the rice genome between these wild relatives based on whole genome re-sequencing reveals that the elite cultivars have lost huge variability due to selection and crop improvement (Gopala Krishnan et al. unpublished). Relative to the Nipponbare reference sequence, the mean number of non-synonymous SNPs (nsSNPs) discovered per kb in 30,294 genes across the genome in the wild rice accessions is more than twice (0.89 and 0.87 nsSNPs/kb in O. rufipogon and O. meridionalis, respectively) compared to cultivated rice (0.30 nsSNPs/kb).
High levels of homology and gene synteny based on whole genome sequencing allows knowledge of gene function to be applied across species by comparison to high-quality reference genome sequences. Whole genome re-sequencing has been employed to study the patterns of evolution during the process of domestication and crop improvement in rice (He et al. 2011), maize (Gore et al. 2009) and soybean (Kim et al. 2010); discovering novel alleles for crtRB1 gene in maize (Buckler et al. 2010); for genome-wide association studies of 14 different traits in rice and southern leaf blight of maize ; and for studying the genome-wide variations between parental lines of hybrids in rice (Krishnan et al. 2011) and maize (Lai et al. 2010).
3.4 Gene-enriched genome sequencing
Sequencing gene-rich fractions rather than whole genomes is much more efficient for the discovery of polymorphisms within genes. Polymorphisms detected within and around genes (as opposed to outside genes) are much more likely to be phenotypically relevant (i.e. to influence traits of interest) or to remain in linkage disequilibrium with causal mutations in the same gene through generations and across populations. The technique of genome enrichment can be used to define allelic differences between genotypes across the whole genome. Probes that are designed to hybridize with each of the genes in the genome can be used to isolate a genomic fraction enriched for complementary gene sequences for sequencing. The development of this technique has been driven particularly by the desire to discover rare sequence variants from the exome of the human genome (Frazer et al. 2009). Extremely promising results were achieved using probe sequences that are synthesized on microarray slides – the microarray probes capture the genomic DNA, which is then eluted from the microarray and sequenced (Albert et al. 2007; Okou et al. 2007). Using this approach, Hodges et al. (2007) designed a number of arrays to capture the entire human exome. A technical improvement on array hybridization is to carry out the hybridization in solution with free oligonucleotides using the microarray as a convenient platform only for oligonucleotide synthesis (Gnirke et al. 2009). Enrichment would appear to be a highly desirable approach for the discovery of large numbers of polymorphisms in plants although few reports using plant genomes have been published. A two-stage capture protocol was tested on maize targeting a chromosomal region and 43 dispersed maize genes (Fu et al. 2010). The first stage involved subtracting repeats, and in the second stage the targets were captured for sequencing. The solution hybridization-based approach has been used to enrich the sugarcane genome using probes mainly designed from the Sorghum genome sequence (Bundock et al. 2012) with hundreds of thousands of SNPs discovered within the polyploid sugarcane genome for 13000–16000 target genes.
3.5 Organelle sequencing
Historically, organellar sequences have been an important source of plant molecular markers. Early approaches to whole plastome sequencing involved either prior isolation of organellar DNA or PCR amplification. These approaches have been used in conjunction with second-generation sequencing to generate plastid genome sequences, producing large numbers of SNP and SSR markers for phylogenetic, barcoding and intraspecific studies (e.g. Cronn et al. 2008; Atherton et al. 2010; Doorduin et al. 2011; Zhang et al. 2011). Chloroplast (cp) isolation was reportedly ineffective as sequence reads from cp extracts contained over 80% non-cp DNA, whereas sequenced PCR amplicons had up to 24% non-cp DNA but required the use of universal primers.
Organelle sequencing is a routine outcome of sequencing DNA from plants because of the high number of copies of these genomes in the cell relative to the nuclear genome (figure 5). The chloroplast or mitochondrial sequences in the sample will usually greatly outnumber the nuclear sequences and, by assessing relative coverage, are easily distinguished from any organellar-like sequences resulting from genetic transfer to the nuclear genome. Sequencing of a total DNA sample can be used to readily determine the sequence of the chloroplast without prior purification or amplification of plastid DNA (Nock et al. 2011; Straub et al. 2011). This approach has been applied in phylogenetic analysis of plant species (Waters et al. 2012).

Relative coverage of the 12 rice chromosomes, mitochondrial and chloroplast genomes. From reference assembly of resequenced Oryza sativa japonica to the reference genome sequence (single lane of 35 bp paired-end reads; Illumina GAII).
In comparison to animals, the mitochondrial genome of plants is characterized by structural complexity and highly variable size, ranging from 200 to 2400 kb (Ward et al. 1981). Second-generation sequencing of plant mitochondrial genomes has been used to identify markers involved in important crop breeding traits such as cytoplasmic male sterility (Fuji et al. 2010). It also reveals the prevalence of recombination and DNA transfer to the mitochondria from the chloroplast and nucleus and by horizontal gene transfer (e.g. Rodríguez-Moreno et al. 2011; Alverson et al. 2011). Large-scale sequencing of plant organelles will accelerate the discovery of markers for use in plant identification, population and phylogenetic studies; and improve understanding of DNA exchange among the organellar and nuclear genomes within the plant.
3.6 Epigenetic markers
Epigenomics refers to the large-scale study of epigenetic features on the genome, which include covalent modifications of histone tails (acetylation, methylation, phosphorylation and ubiquitination), DNA methylation and small RNA machinery (Rival et al. 2010). Bisulphite sequencing involves the pre-treatment of DNA with sodium bisulphite, which causes deamination of cytosine to uracil, unless this reaction is blocked by methylation at the 5-carbon position. Third-generation sequencing technologies are capable of detecting sites of methylation directly. The delineation of regional DNA methylation patterns, and broader DNA methylation profiles, has important implications for understanding why certain regions of the genome can be expressed in specific developmental contexts and how epigenetic changes might enable aberrant expression patterns and disease (Laird 2010). Epigenetic phenomena have economic relevance in the case of somaclonal variation: a genetic and phenotypic variation among clonally propagated plants from a single donor genotype (Rival et al. 2010). Identification of heritable, epigenetic markers linked to important traits have potential in marker-assisted breeding.
3.7 Genetic markers from high-throughput sequencing
Genetic markers have played a major role in our understanding of heritable traits. As the cost of genome sequencing continues to decrease, it will become routine to re-sequence the genome of individual plants in place of the targeted genotyping with current SNP platforms (Imelfort et al. 2009b). MAS allows the breeder to achieve early selection of a trait in a breeding program. However, the vast majority of SNP are not readily associated with agronomic traits (Edwards and Batley 2010). While a few simple traits have been well characterized at the plant genome level, there are many other traits that are poorly understood. This is particularly true for complex traits that are controlled by interacting gene networks. The functions of many of the genes identified by genome sequencing remain unknown and the genetic control of the majority of agronomic traits is yet to be determined (Edwards and Batley 2010). Improvement of complex polygenic traits may be possible in the future by combining the application of newer technologies based on high-throughput genotyping associated with newer marker systems (e.g. SNP), and new selection strategies such as AB-QTL, Mapping-As-You-Go, Marker-Assisted Recurrent Selection and Genome-Wide Selection (Gupta et al. 2010).
Genetic mapping places molecular genetic markers in linkage groups based on their co-segregation in a population. The development of these markers also allows the integration of genetic and physical maps. The use of common molecular genetic markers across related species permits the comparison of linkage maps. Knowledge of the gene underlying a particular trait enables the transfer of the trait between cultivars and even species using genetic modification. Furthermore, the integration of molecular marker data with genomics, proteomics and phenomics data allows researchers to link sequenced genome data with observed traits (Edwards and Batley 2010). Advanced genome sequencing technologies generate large quantities of molecular marker genotyping data, which favours Association Studies over traditional QTL mapping (Edwards and Batley 2010).
4 Developments in plant molecular genetics driven by advances in sequencing technology
Molecular marker technology in plants is no longer limited by the capacity to discovery polymorphisms. Whole genome sequencing has been shown to now be a viable strategy for discovery of gene variants in the complex genomes of distant wild relatives of crops (Malory et al. 2011). Tools for polymorphism analysis are probably again the limiting factor. Genotyping by sequencing is an option that will possibly overcome this limitation. Ultimately all genotyping will be based upon determining the complete DNA sequence of every sample. Technological developments continue to make this a more realistic option. Species with simple genomes or for which a high-quality reference genome exists are most amenable to this approach using existing technology. These tools will deliver large numbers of genetic markers for variety identification and marker-assisted selection (Pattemore et al. 2010; Sexton 2010a and 2010b). Sequencing of plant populations will increasingly allow analysis of population and evolutionary genetics at the whole genome level (Fitzgerald et al. 2011; Shapter et al. 2012). These developments will deliver new insights into plant biology and support conservation of biodiversity, and plant improvement to deliver food and energy security (Henry 2010).
References
Albert TJ, Molla MN, Muzny DM, Nazareth L, Wheeler D, Song XZ, Richmond TA, Middle CM, Rodesch MJ, Packard CJ, Weinstock GM and Gibbs RA 2007 Direct selection of human genomic loci by microarray hybridization. Nat. Method. 4 903–905
Akhunov E, Nicolet C and Dvorak J 2009 Single nucleotide polymorphism genotyping in polyploid wheat with the Illumina Goldengate assay. Theor. Appl. Genetics 119 507–517
Alverson AJ, Rice DW, Dickinson S, Barry K and Palmer JD 2011 Origins and recombination of the bacterial-sized mitochondrial genome of cucumber. Plant Cell 23 2499–2513
Arai-Kichise Y, Shiwa Y, Nagasaki H, Ebana K, Yoshikawa H, Yano M and Wakasa K 2011 Discovery of genome-wide DNA polymorphisms in a landrace cultivar of japonica rice by whole-genome sequencing. Plant Cell Physiol. 52 274–282
Argout X, Salse J, Aury J-M, Guiltinan MJ, Droc G, Gouzy J, Allegre M, Chaparro C, et al. 2010 The genome of Thebroma cacao. Nat. Genet. 43 101–108
Atherton RA, McComish BJ, Shepherd LD, Berry LA, Albert NW and Lockhart PJ 2010 Whole genome sequencing of enriched chloroplast DNA using the Illumina GAII platform. Plant Methods 6 22
Barbazuk WB, Emrich SJ, Chen LL and Schnable PS 2007 SNP discovery via 454 transcriptome sequencing. Plant J. 51 910–918
Birney E 2011 Assemblies: the good, the bad, the ugly. Nat. Method. 8 59–60
Brautigam A and Gowik U 2010 What can next generation sequencing do for you? Next generation sequencing as a valuable tool in plant research. Plant Biol. 12 831–841
Buckler ES, Warburton ML and Rocheford T 2010 Rare genetic variation at Zea mays crtRB1 increases beta-carotene in maize grain. Nat. Genet. 42 322–327
Bundock PC, Eliott F, Ablett G, Benson AD, Casu R, Aitken K and Henry RJ 2009 Targeted SNP discovery in sugarcane using 454 sequencing. Plant Biotechnol. J. 7 347–354
Bundock PC, Casu R and Henry RJ 2012 Enrichment of genomic DNA for polymorphism detection in a non-model highly polyploidy crop plant. Plant Biotechnol. J. 10 657-667
Chan AP, Crabtree J, Zhao Q, Lorenzi H, Orvis J, Puiu D, Melake-Berhan A, Jones KM, et al. 2010 Draft genome sequence of the oilseed species Ricinus communis. Nat. Biotechnol. 28 951–956
Cronn R, Liston A, Parks M, Gernandt DS, Shen R and Mockler T 2008 Multiplex sequencing of plant chloroplast genomes using Solexa sequencing-by-synthesis technology. Nucleic Acids Res. 36 e122
Cross M, Waters D, Lee LS and Henry RJ 2008 Endonucleolytic Mutation Analysis by Internal Labeling (EMAIL). Electrophoresis 29 1291–1301
Deschamps S and Campbell MA 2010 Utilization of next-generation sequencing platforms in plant genomics and genetic variant discovery. Mol. Breed. 25553–570
Doorduin L, Gravendeel B, Lammers Y, Ariyurek Y, Chin-A-Woeng T and Vrieling K 2011 The complete chloroplast genome of 17 individuals of pest species Jacobaea vulgaris: SNPs, microsatellites and barcoding markers for population and phylogenetic studies. DNA Res. 18 93–105
Druley TE, Vallania FLM, Wegner DJ, Varley KE, Knowles OL, Bonds JA, Robison SW, Doniger SW, Hamvas A and Cole FS 2009 Quantification of rare allelic variants from pooled genomic DNA. Nat. Method 6 263–265
Dubey A, Farmer A, Schlueter J, Cannon SB, Abernathy B, Tuteja R, Woodward J, Shah T, et al. 2011 Defining the transcriptome assembly and its use for genome dynamics and transcriptome profiling studies in pigeonpea (Cajanus cajan L.) DNA Res. 18 153–164
Edwards D and Batley J 2010 Plant genome sequencing: applications for crop improvement. Plant Biotechnol. J. 8 2–9
Edwards M and Henry R 2011 DNA sequencing methods contributing to new directions in cereal research. J. Cereal Sci. 54 395–400
Fitzgerald TL, Shapter FM, McDonald S, Waters DLE, Chivers IH, Drenth A, Nevo E and Henry RJ 2011 Genome diversity in wild grasses under environmental stress. Proc. Natl. Acad. Sci. USA 108 21139–21144
Frazer KA, Murray SS, Schork NJ and Topol EJ 2009 Human genetic variation and its contribution to complex traits. Nat. Rev. Genet. 10 241–251
Fu Y, Springer NM, Gerhardt DJ, Ying K, Yeh CT, Wu W, Swanson-Wagner R, D’Ascenzo M, et al. 2010 Repeat subtraction-mediated sequence capture from a complex genome. Plant J. 62 898–909
Fuji S, Kazama T, Yamada M and Toryama K 2010 Discovery of global genomic re-organization based on comparison of two newly sequenced rice mitochondrial genomes with cytoplasmic male sterility-related genes. BMC Genomics 11 209
Futschik A and Schlotterer C 2010 The next generation of molecular markers from massively parallel sequencing of pooled DNA samples. Genetics 186 207–218
Garvin MR, Saitoh K and Gharrett AJ 2010 Application of single nucleotide polymorphisms to non-model species: a technical review. Mol. Ecol. Resour. 10 915–934
Gillies S, Furtado A and Henry RJ 2012 Gene expression in the developing aleurone and starchy endosperm of wheat. Plant Biotechnol. J. 10 668-679
Glenn TC 2011 Field guide to next-generation DNA sequencers. Mol. Ecol. Resour. 11 759–769
Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, Fennell T, Giannoukos G, et al. 2009 Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 27 182–189
Gopala Krishnan S, Waters DLE, Katiyar SK, Sadananda AR, Satyadev V and Henry R 2011 Genome-wide DNA polymorphisms in elite indica rice inbreds discovered by whole-genome sequencing. Plant Biotechnol. J. 10 623-634
Gore MA, Chia JM, Elshire RJ, Sun Q, Ersoz ES, Hurwitz BL, Peiffer JA, McMullen MD, et al. 2009 A first generation haplotype map of maize. Science 326 1115–1117
Gupta PK, Langridge P and Mir RR 2010 Marker-assisted wheat breeding: present status and future possibilities. Mol. Breed. 26 145–161
He Z, Zhai W, Wen H, Tang T, Wang Y, Lu X, Greenberg AJ, Hudson RR, Wu CI and Shi S 2011 Two evolutionary histories in the genome of rice: the roles of domestication genes. PLoS Genet. 7 e1002100
Henry RJ and Edwards K 2009 New tools for single nucleotide polymorphism (SNP) discovery and analysis accelerating plant biotechnology. Plant Biotechnol. J. 7 311
Henry RJ 1997 Practical applications of plant molecular biology (London: Chapman and Hall)
Henry RJ 2001 Plant genotyping: The DNA fingerprinting of plants (Oxon: CABI Publishing)
Henry RJ 2005 Plant diversity and evolution: Genotypic and phenotypic variation in higher plants (Oxon: CABI Publishing)
Henry RJ 2008 Plant genotyping II: SNP technology (Wallingford: CABI Publishing)
Henry RJ 2010 Plant resources for food fuel and conservation (London: Earthscan)
Hill H, Lee LS and Henry RJ 2011 Variation in sorghum starch synthesis genes associated with differences in starch phenotype. Food Chem. doi:10.1016/j.foodchem.2011.08.057
Hiremath PJ, Farmer A, Cannon SB, Woodward J, Kudapa H, Tuteja R, Kumar A, BhanuPrakash A, et al. 2011 Large-scale transcriptome analysis of chickpea (Cicer arietinum L.) an orphan legume crop of the semi-arid tropics of Asia and Africa. Plant Biotechnol. J. 9 922–931
Hodges E, Xuan Z, Balija V, Kramer M, Molla MN, Smith SW, Middle CM, Rodesch MJ, Albert TJ, Hannon GJ and McCombie WR 2007 Genome-wide in situ exon capture for selective resequencing. Nat. Genet. 39 1522–1527
Imelfort M, Batley J, Grimmond S and Edwards D 2009a Genome sequencing approaches and successes; in Methods in molecular biology, plant genomics (eds) D Somers, P Langridge and Gustafson JP (Humana Press) pp 345–258
Imelfort M, Duran C, Batley J and Edwards D 2009b Discovering genetic polymorphisms in next-generation sequencing data. Plant Biotechnol. J. 7 312–317
IRGSP (International Rice Genome Sequencing Project) 2005 The map based sequence of the rice genome. Nature 436 793–800
Kharabian-Masouleh A, Waters D, Reinke R and Henry R 2009 A high-throughput assay for rapid and simultaneous analysis of perfect markers for important quality and agronomic traits in rice using multiplexed MALDI-TOF mass spectrometry. Plant Biotechnol. J. 7 355–363
Kharabian-Masouleh A, Waters DLE, Reinke RF and Henry RJ 2011 Discovery of polymorphisms in starch related genes in rice germplasm by amplification of pooled DNA and deeply parallel sequencing. Plant Biotechnol. J. 9 1074–1085
Kim MY, Lee S, Van K, Kim TH, Jeong SC, Cho IY, Kim DS, Lee YS, et al. 2010 Whole-genome sequencing and intensive analysis of the undomesticated soybean (Glycine soja Sieb. and Zucc.) genome. Proc. Natl. Acad. Sci. USA 107 22032–22037
Kircher M and Kelso J 2010 High-throughput DNA sequencing - concepts and limitations. Bioessays 32 524–536
Kulheim C., Yeou SH, Maintz J, Foley W and Moran G 2009 Comparative SNP diversity among four Eucalyptus species for genes from secondary metabolite biosynthetic pathways. BMC Genomics 10 452
Kulheim C,Yeoh, SH, Wallis IR, S Laffan S, Moran GF, et al. 2011 The molecular basis of quantitative variation in foliar secondary metabolites in Eucalyptus globulus. New Phytol. 191 1041–1053
Lai J, Li R, Xu, X, Jin W, Xu M, Zhao H, Xiang Z, Song W, et al. 2010. Genome-wide patterns of genetic variation among elite maize inbred lines. Nat. Genet. 42 1027–1030
Laird PW 2010 Principles and challenges of genome-wide DNA methylation analysis. Nat. Rev. Genet. 11 191–203
Magi A, Benelli M, Gozzini A, Girolami F, Torricelli F and Brandi ML 2010 Bioinformatics for next generation sequencing data. Genes 1 294–307
Malory S, Shapter FM, Elphinstone MS, Chivers IH and Henry RJ 2011 Characterizing homologues of crop domestication genes in poorly described wild relatives by high-throughput sequencing of whole genomes Plant Biotechnol. J. 9 1131–1140
McMullen MD, Kresovich S, Villeda HS, Bradbury P, Li H, Sun Q, Flint-Garcia S, Thornsberry J, et al. 2009 Genetic properties of the maize nested association mapping population. Science 325 737–740
Myles S, Chia JM, Hurwitz B, Simon C, Zhong GY, Buckler E and Ware D 2010 Rapid genomic characterization of the genus Vitis. PLoS ONE 5 e8219
Nock C, Waters DLE, Edwards MA, Bowen S, Rice N, Cordeiro GM and Henry RJ 2011 Chloroplast genome sequence from total DNA for plant identification. Plant Biotechnol. J. 9 328–333
Novaes E, Drost D, Farmerie W, Pappas G, Grattapaglia D, et al. 2008 High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genomics 9 312
Okou DT, Steinberg KM, Middle C, Cutler DJ, Albert TJ and Zwick ME 2007 Microarray-based genomic selection for high-throughput resequencing. Nat. Method. 4 907–909
Out AA, van Minderhout I, Goeman JJ, Ariyurek Y, Ossowski S, Schneeberger K, Weigel D, van Galen M, Taschner PEM and Tops CMJ 2009 Deep sequencing to reveal new variants in pooled DNA samples. Hum. Mutat. 30 1703–1712
Parks M, Cronn R and Liston A 2009 Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 7 84
Pattemore JA, Rice N, Marshall DF, Waugh R and Henry RJ 2010 Cereal Variety Identification using MALDI-TOF mass spectrometry SNP genotyping. J. Cereal Sci. 52 356–361
Peterson TW, Nam SJ and Darby A 2010 Next-gen sequencing survey; in North America equity research (New York: JP Morgan Chase & Co.)
Rival A, Beule T, Bertossi FA, Tregear J and Jaligot E 2010 Plant epigenetics: From genomes to epigenomes. Notulae Botanicae Horti Agrobotanici Cluj-Napoca 38 9–15
Rodríguez-Moreno L, González VM, Benjak A, Martí MC, Puigfomènech P, Aranda MA and Garcia-Mas J 2011 Determination of the melon chloroplast and mitochondrial genome sequences reveals that the largest reported mitochondrial genome in plants contains a significant amount of DNA having a nuclear origin. BMC Genomics 12 424
Schadt EE, Turner S and Kasarskis A 2010 A window into third-generation sequencing. Hum. Mol. Genet. 19 R227–R240
Sexton T, Henry R, McManus LJ, Bowen S and Shepherd M 2010a Capture of assay template by multiplex PCR of long amplicons for genotyping SNPs and InDels with MALDI-TOF mass spectrometry. Mol. Breed. 25 471–480
Sexton TR, Henry RJ, McManus LJ, Henson M, Thomas DS and Shepherd M 2010b Genetic association studies in Eucalyptus pilularis Smith (blackbutt). Aust. Forest. J. 73 254–258.
Sexton T, Henry R, Harwood C, Thomas D, L. McManus L, et al. 2011 SNP discovery and association mapping in Eucalyptus pilularis (blackbutt). BMC Proc. 5 O9
Sexton TR, Henry RJ, Harwood CE, Thomas DS, McManus LJ, Raymond C, Henson M and Shepherd M 2012 Pectin methylesterase genes influence solid wood properties of Eucalyptus pilularis. Plant Physiol. 158 531–541
Shendure J and Ji HL 2008 Next-generation DNA sequencing. Nat. Biotechnol. 26 1135–1145
Shapter FM, Fitzgerald TL, Waters DLE, McDonald S, Chivers IH and Henry RJ 2012 Analysis of adaptive ribosomal gene diversity in wild plant populations from contrasting climatic environments. Plant Signal. Behav. 7 1-3 accepted 8 February 2012
Shendure J and Ji HL 2008 Next-generation DNA sequencing. Nat. Biotechnol. 26 1135–1145
Souza GM, Berges H, Bocs S, Casu R, D’Hont A, Ferreira JE, Henry R, Ming R, et al. 2011 The sugarcane genome challenges: Strategies for sequencing a highly complex genome. Trop. Plant Biol. 4 145–156
Straub SCK, Fishbein M, Livshultz T, Foster Z, Parks M, Weitemier K, Cronn RC and Liston A 2011 Building a model: Developing genomic resources for common milkweed (Ascleplas syriaca) with low coverage genome sequencing. BMC Genomics 12 211
Thomas RK, Nickerson E, Simons JF, Jänne PA, Tengs T, Yuza Y, Garraway LA, LaFramboise T, Lee JC and Shah K 2006 Sensitive mutation detection in heterogeneous cancer specimens by massively parallel picoliter reactor sequencing. Nat. Med. 12 852–855
Thudi M, Li Y, Jackson SA, May GD and Varshney RK 2012 Current state-of-the-art sequencing technologies for plant genomics research. Brief. Functional Genomics 11 3–11
Trick M, Long Y, Meng J and Bancroft I 2009 Single nucleotide polymorphism (SNP) discovery in the polyploidy Brassica napus using Solexa transcriptome sequencing. Plant Biotechnol. J. 7334–346
Trebbi D, maccaferri M, de Heer P, Sorensen A, Giuliani S, Sanguineti MC, Massi A, van der Vossen EAG and Tuberosa R 2011 High-throughput SNP discovery and genotyping in durum wheat (Triticum durum Desf.). Theor. Appl. Genet. 123 555–569
TPGSC (The Potato Genome Sequencing Consortium) 2011 Genome sequence and analysis of the tuber crop potato. Nature 475 189–195
Tung C, Zhao K, Wright LM, Ali ML, Jung J, Kimball J, Tyagi W, Thomson MJ, et al. 2010 Development of a research platform for dissecting phenotye-genotype associations in rice (Oryza spp.). Rice 3 205–217
Varley KE and Mitra RD 2008 Nested Patch PCR enables highly multiplexed mutation discovery in candidate genes. Genome Res. 18 1844–1850
Varshney RV, Glaszmann J-C, Leung H and Ribaul J-M 2010 More genomic resources for less studies crops. Trends Biotechnol. 28 452–460
Ward BL, Anderson RS and Bendich AJ 1981 The mitochondrial genome is large and variable in a family of plants (Cucurbitaceae). Cell 25 793–803
Waters DLE, Nock CJ, Ishikawa R, Rice N and Henry RJ 2012 Chloroplast genome sequence confirms distinctness of Australian and Asian wild rice. Ecol. Evol. 2 211–217
Whittall JB, Syring J, Parks M, Buenrostro J, Dick C, et al. 2010 Finding a (pine) needle in a haystack: chloroplast genome sequence divergence in rare and widespread pines. Mol. Ecol. 19 100–114
Wu X, Ren C, Joshi T, Vuong T, Xu D and Nguyen HT 2010 SNP discovery by high-throughput sequencing in soybean. BMC Genomics 11 469
Xu YB, Crouch JH and Jonathan H 2008 Marker-assisted selection in plant breeding: From publications to practice. Crop Sci. 48 391–407
Yamamoto T, Nagasaki H, Yonemaru J, Ebana K, Nakajima M, Shibaya T and Yano M 2010 Fine definition of the pedigree haplotypes of closely related rice cultivars by means of genome-wide discovery of single-nucleotide polymorphisms. BMC Genomics 11 267
Yan JB, Yang XH, Shah T, Sanchez-Villeda H, Li JS, Warburton M, Zhou Y, Crouch JH and Xu YB 2010 High-throughput SNP genotyping with the GoldenGate assay in maize. Mol. Breed. 25 441–451
Yan J, Kandianis CB, Harjes CE, Bai L, Kim EH, Yang X, Skinner DJ, Fu Z, et al. 2011 Annotation-based genome-wide SNP discovery in the large and complex Aegilops tauschii genome using next-generation sequencing without a reference genome sequence. BMC Genomics 12 59
Zhang Y, Ma P and Li D 2011 High-throughput sequencing of six bamboo chloroplast genomes: phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PLoS ONE 6 e20596
Acknowledgements
Financial support from the Australian Research Council, Grains Research and Development Corporation, Sugar Research and Development Corporation, and Rural Industries Research and Development Corporation is acknowledged. GKS visit to SCPS was sponsored by the BOYSCAST fellowship of Department of Science and Technology, India.
Author information
Authors and Affiliations
Corresponding author
Additional information
[Henry RJ, Edwards M, Waters DLE, Gopala Krishnan S, Bundock P, Sexton TR, Masouleh AK, Nock CJ and Pattemore J 2012 Application of large-scale sequencing to marker discovery in plants. J. Biosci. 37 1–13] DOI 10.1007/s12038-012-9253-z
Rights and permissions
About this article
Cite this article
Henry, R.J., Edwards, M., Waters, D.L.E. et al. Application of large-scale sequencing to marker discovery in plants. J Biosci 37, 829–841 (2012). https://doi.org/10.1007/s12038-012-9253-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12038-012-9253-z