
Abstract
Mis-priming associated with uncharacterised single nucleotide polymorphisms (SNPs) may lead to failure of PCR for genotyping. This is particularly troublesome in high-throughput SNP genotyping applications relying on multiplex PCR (2–40-plex) generating many short amplicons (80–120 bp) of similar size, an approach best suited for whole genome scans. However, if the target SNPs are clustered within a few target genes one option to ameliorate this is to increase the amplicon length, effectively reducing the potential for primer/template interactions and mis-priming. We tested this approach in a diverse population of 372 Eucalyptus pilularis individuals (π = 8.11 × 10−3, H e = 0.75) using a modified Sequenom iPLEX gold assay. Four candidate genes (MYB1, MYB2, CAD and CCR) were amplified in a single long range multiplex capture PCR generating 6 long amplicons ranging in size from 907 to 2,225 bp. This contrasts with the standard approach which would have required the amplification of 98 short amplicons in 4 multiplex reactions. These 6 long amplicons provided the assay template for 98 assays (87 SNP and 11 InDel) within the 4 candidate genes. Reaction results indicated that longer amplicons could provide a suitable template for genotyping assays, with 90.8% of assays functional and 84.3% of assays suitable for downstream analysis. Additional advantages of this approach were the capacity for troubleshooting using gel electrophoresis and savings of 94% in capture primer synthesis costs. This approach will have the greatest relevance for candidate gene approaches for association testing in uncharacterised populations of organisms with high sequence diversity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Association studies are becoming an important way of linking genetic variation in candidate genes with phenotypic variation in forest trees (Thumma et al. 2005; Gonzales-Martinez et al. 2007; Ingvarsson et al. 2008). Many forest trees pose a challenge for genotyping Single Nucleotide Polymorphisms (SNPs) and Insertions or Deletions (InDels) because of high heterozygosity and sequence polymorphism which relates to both recent domestication (Stoeckel et al. 2006) and an outcrossing breeding system (Ward et al. 2005). Estimates of SNP density and nucleotide diversity reported in Eucalyptus globulus of 18.49 SNPs per kb (Poke et al. 2003) π = 5 × 10−3 (Thavamanikumar et al., unpublished) and Eucalyptus pilularis 10.84 SNPs per kb π = 8.11 × 10−3 (Sexton et al., unpublished) are far higher than those reported for humans at 0.83 SNPs per kb (Zhao et al. 2003) π = 7.51 × 10−4 (Sachidanandam et al. 2001), the model organism for which most genotyping platforms are designed (Smith 1993). However, in forest trees a candidate gene approach is deemed appropriate for association studies rather than a genome-wide approach because of the large genome sizes, limited availability of genomic resources, and rapid decay of Linkage Disequilibrium (LD; Thumma et al. 2005; Gonzales-Martinez et al. 2007).
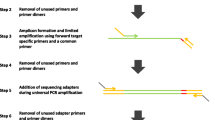
High throughput genotyping of SNPs and InDels is made possible on the Sequenom platform by a combination of assay design, capture PCR, liquid handling robotics, iPLEX gold chemistry, nano-dispensing and MALDI-TOF MS. This approach relies on the amplification of regions containing target SNPs in one round of multiplex capture PCR up to 36-plex. The standard approach uses one pair of capture primers per polymorphism which are used to amplify small amplicons, 80–120 bp by default, around the SNP or InDel. Unincorporated nucleotides and primers are then dephosphorylated by incubation with Shrimp Alkaline Phosphatase (SAP). In a subsequent nested iPLEX reaction, up to 40-plex, assay primers extend one base into the SNP and incorporate a ddNTP into the complimentary assay primer. This shift in molecular mass from unextended primer to extended primer is measured by Matrix Assisted Laser Desorption Ionization—Time of Flight Mass Spectrometry (MALDI-TOF MS). The difference in molecular mass of the incorporated ddNTP at the SNP site allows discrimination of the genotype (Ross et al. 1998; Oeth et al. 2008).
PCR amplification of the target still remains the bottleneck in most genotyping applications (Galvin 2002), including MALDI-TOF MS based genotyping (Pattemore et al. 2007). One potential limitation when transferring the standard assay design from the human model to forest trees is that assays may fail due to uncharacterised variation at primer sites across the population. In the standard approach the ability to evaluate the amplification of each capture primer pair individually is not possible because capture PCR is performed in a multiplex reaction of between 12 and 48plex (Ragoussis et al. 2006; Masouleh et al. 2009), generating similar sized short amplicons which can be impossible to resolve separately. One approach to overcome this would be to limit the number of capture primers by amplifying long PCR amplicons, with multiple SNPs and InDels assayed in each amplicon. Efficacy may be further increased by multiplexing the capture PCR, allowing higher equi-molar concentrations to be achieved than pooling, as well as reducing the total amount of genomic DNA required, which is often difficult to acquire from tree species in large quantities at sufficient purity (Tibbits et al. 2006).
By modifying the multiplex capture PCR to amplify long amplicons, we describe an alternate method to assay SNPs and InDels in candidate genes of the highly polymorphic and heterozygous tree species H e = 0.75 (Shepherd et al. 2009), Eucalyptus pilularis. Using our method a single multiplex capture PCR amplifying 6 long fragments representing four candidate genes provided the template to assay 87 SNPs and 11 InDels. This contrasts with a typical assay which would survey SNPs dispersed throughout the human (Buetow et al. 2001; Sladek et al. 2007) or rice genome (Masouleh et al. 2009). This approach optimises this technology for genotyping in populations of highly polymorphic species, typically undomesticated, where a candidate gene approach is optimal.
Materials and methods
DNA extraction from forest trees is often complicated because of high levels of polyphenolics, terpenes and polysaccharides in many leaf tissues (Shepherd et al. 2002; Tibbits et al. 2006). To minimise issues with DNA quality the Qiagen DNeasy 96 Plant Kit (Cat. No. 69181) was used for DNA extraction. The manufacture’s frozen tissue protocol was followed except that 1.5× volumes of AP1, AP2 and AP3 buffers were used. The AP1 lysis buffer was also modified by including 2% PVP-40 w/v (Stewart and Via 1993; Devey et al. 1996; Kim et al. 1997) and activated charcoal 2.5 mg/ml (Desai and Madamwar 2007). BSA 0.4% BSA was added to the AP2 precipitation buffer (Tibbits et al. 2006). Cell lysis was allowed to continue for 30 min at 65°C. Following the wash with AW buffer silica membranes were washed one additional time with 800 μl of 100% AR ethanol, then eluted in 2 × 50 μl of AE buffer (10 mM Tris–Cl, 0.5 mM EDTA; pH 9.0).
SNP discovery
Four genes, cinnamyl alcohol dehydrogenase 2 (CAD2), cinnamoyl coA reductase (CCR) and MYB transcription factors 1 and 2 (MYB1 and 2) were selected for this study based on their reported function Eucalyptus species (Grima-Pettenati et al. 1993; Goicoechea et al. 2005; Thumma et al. 2005; Legay et al. 2007). SNP Discovery was performed by cloning a minimum of 18 haplotypes for each gene into the Promega pGEMT easy vector (Cat. No. A1380). A total of 10,146 bp (comprising 3,839 bp of CAD2, 3,367 bp of CCR, 1,880 bp of MYB2 and 1,060 bp of MYB1 were sequenced on an ABI 3730 (Southern Cross Plant Genomics, Australia). Genotypes were aligned and polymorphisms were scored based on their representation in at least two genotypes. A total of 110 SNPs and 17 insertion-deletion mutations (InDels) were identified (Table 1) and represented the discovery of 10.84 novel SNPs per kb, 2.95 novel InDels per kb, or 12.81 novel polymorphisms per kb.
Capture PCR
Capture primers were designed using the PRIMO web interface (Li et al. 1997). Priming sites were checked manually for the presence of SNPs or InDels which would cause mis-priming. Once suitable combinations were identified a 10 base pair tag (ACGTTGGATG) was added to the 5′ end of each primer before synthesis (Appendix 1). This increased the molecular mass of each capture primer outside the range measured on the mass spectrometer.
A gradient PCR from 67 to 57°C was used to identify the optimal annealing temperature for all amplicons. Reaction components were then optimized with a fixed annealing temperature of 64°C which allowed amplification of all amplicons and provided minimal formation of non-specific amplicons (Fig. 1). The reaction component concentrations were optimized by titration and assessment of the reaction products by agarose gel electrophoresis. The following concentrations were found to be optimal for E. pilularis; 0.4 ng/μl of genomic DNA, 0.6 μg/μl of BSA, 1 mM total dNTP’s, 1× Platinum Taq buffer, 2.5 mM MgCl2, 0.1–2 μM primers and 0.2 U/μl Invitrogen Platinum Taq (Cat. No. 10966-034). The PCR products were visualised using agarose gel electrophoresis with 1.5% gels and stained with Ethidium Bromide (0.5 μg/ml).

Progression of multiplex optimization a annealing temperature gradient used to identify a fixed Ta from which PCR optimization was based b effect of BSA concentration on yield in 8 genotypes, with 0.6 ng/μl optimal c comparison of four DNA concentrations with 0.4 ng/μl optimal d individual amplicons separated e effect of polymerase on amplification efficiency using final primer concentrations
A multiplex PCR was used to amplify six amplicons representing four candidate genes (MYB1, MYB2, CCR and CAD) which formed the template for the iPLEX SNP assays. Reactions were performed in a 30 μl volume to provide sufficient volume to confirm the success on an agarose gel, as well as provide the template for four separate multiplex SNP assays.
Thermocycling conditions included a hot start step at 98°C for 1 min to activate the hot start polymerase, followed by a three step cycle of denaturing at 98°C for 8 s, annealing at 64°C for 12 s and extension at 72°C for 3 min, this cycle was repeated 45 times before a final extension at 72°C for 5 min.
iPLEX assay
Assay primers were designed using the Assay Design software 3.1. Parameters for capture primer design were relaxed to achieve the highest assay multiplex levels possible, as alternate capture primers designed in PRIMO were synthesized. A total of 127 polymorphisms were identified and presented for assay design software. Using relaxed capture primer design settings and default high plex assay design settings, 100 polymorphisms (89 SNPs and 11 InDels) were selected to be assayed in four separate multiplex reactions (1) 32-plex, (2) 29-plex, (3) 26-plex and (4) 13-plex (Fig. 2). Template for two assays in reaction #1 (32-plex) was not included so is termed a 30-plex reaction with two inbuilt negative controls. Primers were synthesised at a high concentration (400 μM) so that the reaction volume would not limit these high plex levels.

Distribution of quality calls for genotyping at differing multiplex levels
The thermocycling program for the iPLEX single base extension (SBE) assay consisted of one denaturing step at 94°C for 10 s followed by two steps of annealing at 52°C for 5 s, single base extension and simultaneous denaturing of the primer at 80°C. This cycling is repeated 120 times. This differed from the standard approach by; (1) increasing the denaturing time from 5 to 10 s, (2) incorporating a denaturing step after every second annealing extension cycle, opposed to every fifth and (3) an extra 45 cycles of annealing and extension. Complete reactions were then processed for genotyping on the Sequenom MassARRAY platform (Southern Cross Plant Genomics, Australia).
Results and discussion
Based on the high SNP density observed in Eucalyptus species, primers were designed within sequences conserved between genotypes. PRIMO software gave a more flexible alternative option to Assay Design 3.1 software for multiplex capture primer design allowing a choice of priming sites, amplicon number and amplicon length (Table 2).
By increasing the average length of amplicons in the capture PCR from 0.15 to 1.5 kb, more polymorphic sites were encompassed in each amplicon. This resulted in a significant reduction from 100 to 6 amplicons required for full coverage of the four candidate genes. This reduction in amplicons and capture primer numbers restricted opportunities for mis-priming and allowed a more detailed analysis of template/primer interactions. The choice of alternate priming sites designed with PRIMO software allowed flexibility of primer choice, and was successful in overcoming the problem of high SNP density. Since the six amplicons were amplified as a single low plex multiplex reaction, one reaction provided a template for all of the SNP and InDel assays within these candidate genes; opposed to four high plex multiplex reactions.
The range of amplicon lengths and low plex nature of this 6-plex multiplex capture PCR enabled trouble-shooting to be performed by agarose gel electrophoresis, available in any molecular laboratory. This would be extremely difficult for a standard high plex multiplex capture PCR with many short amplicons of a similar length. This approach also provided an economic advantage where less DNA template was required for full coverage of the genes, fewer primers were required (12 opposed to 200), and subsequently fewer bases are needed for primer synthesis (364 bp opposed to 6,000 bp).
The disadvantage of using alternate PRIMO software for the design of capture primers is that interactions between capture primers and assay primers could not be measured easily. One way of troubleshooting these interactions is to exclude all assays with calls in the minus template controls, where the assay primer has used the capture primer as template for the assay.
Optimisation of multiplex reactions
A gradient PCR from 67 to 57°C was used to identify the optimal annealing temperature for the multiplexed amplicons; 64°C was selected as the fixed annealing temperature allowing amplification of all primer pairs and minimal levels of non-specific amplification. Because longer amplicons were expected to be more difficult to amplify in multiplex at an equimolar concentration, preference was given to conditions which amplified longer amplicons at a high concentration (Fig. 1a). Pooling was not attempted because amplicons would possibly be diluted below the requirements for the iPLEX assay. Bovine Serum Albumin was assessed for its ability to standardise amplification efficiency of amplicons in multiplex (Henegariu et al. 1997). From this information 0.6 ug/μl of BSA was selected as optimal providing consistent amplification across all eight genotypes in the test panel (Fig. 1b). The optimal DNA concentration for multiplex PCR was identified by comparison of four PCRs containing DNA concentrations of 0.2, 0.3, 0.4 and 0.5 ng/μl. Based on these results a final DNA template concentration of 0.4 ng/μl was selected as the optimum for this population with consistent amplification of all primer pairs. These amplicons can be observed at the highest concentration (Fig. 1c). Normalization of amplicon concentrations was achieved by progressive adjustment of primer concentrations which ranged from 0.1 to 2 μm depending on the primer pair. Concentrations of individual amplicons in multiplex ranged between 0.01593 and 0.06057 pMol (Fig. 1d). Although equi-molar concentrations of each amplicon were not achieved, assays still returned results across all amplicons. This indicates that the assay was robust enough to cope with such variation in template concentrations.
The effect of doubling the final polymerase concentration was also assessed, 0.1 U/μl of Platinum Taq was found to give adequate yields but ultimately, 0.2 U/μl was selected for the final experiment because it gave higher yields of template providing a greater margin for error in the population of 372 individuals (Fig. 1e). Because longer amplicons were expected to be more difficult to amplify in multiplex at high enough concentrations, a long range proof reading polymerase was tested (Fig. 1a, Bio-Rad Iproof Cat# 1725302) which could be used at half the concentration of Platinum Taq polymerase (Fig. 1e) and amplified at a similar DNA concentration. Iproof polymerase was not used in the final experiment due to difficulty amplifying the short MYB1 amplicon. In hindsight lowering the fixed annealing temperature to 63°C may have solved this.
SNP assay synopsis
A total of 127 polymorphisms were identified and presented to the Assay Design 3.1 software. Assays were designed for 100 polymorphisms, the remaining 27 polymorphisms could not be assayed for a number of reasons including; (1) priming regions contained polymorphic sites, (2) primers bound non-specifically to other primers or template (Table 2). This is not expected to be a problem for association studies as neighbouring polymorphisms are likely to be LD with these sites. If necessary some of these 27 polymorphisms could be assayed using alternate techniques such as Cleaved Amplified Polymorphic Sequence (CAPS) markers (Konieczny and Ausubel 1993).
Because capture primers were designed using PRIMO software, as opposed to Assay Design 3.1, two of the assays would not have a template amplified in the capture PCR. From this point 98 assay primers had an equal opportunity to perform in the reactions. Of these 91% returned evidence that single base extension had worked (Table 3). Genotyping data in 5.6% of these working reactions was confounded by overlapping sodium or potassium peaks with primer extension products. Competition for time of flight between assays of low and high molecular mass caused a loss of signal for 7.9% of primer extension products. This was a particular problem for heterozygous alleles with high molecular mass assay primers, where the signal was split between the extension products and lost in the noise. Three SNP assays were recorded in the minus template negative controls indicating primer extension on the capture primers. This is thought to occur from the use of different software for the design of capture and assay primers, as possible interactions between these primer sets were not evaluated. Failed assays include those where only unextended primer (UEP) was observed. It is not clear why this occurred in 8.2% of total reactions. It is possible that tertiary structures of the longer amplicon may have been preventing access of assay primers to priming sites. One reaction failed because the signal of the UEP and extension products was completely lost in the noise, this could have been resolved by increasing the assay primer concentrations beyond those identified during normalization of UEP signal.
Of the 84.3% of reactions in the “total functional assays usable” category only 52 assays calls could be validated in the genotyping as real SNPs or InDels. The other 23 functional assays could not be validated (Table 3). A false InDel discovery rate of 57% and false SNP discovery rate of 28% can be calculated, with an average of 30.7% for SNPs and InDels together. These results indicate that the SNP discovery stage should be more stringent, and a more careful analysis of trace chromatograms can be employed to reduce erroneous identification of SNPs and InDels. It is believed that more InDels were misidentified because of gap placement settings of sequence analysis software.
Based on polymorphisms that were validated in the genotyping data, 5.3% of assays were missing. Heterozygosity of individual polymorphisms was 31% on average, with a maximum of 50%.
Assessment of call quality
Multiplex assays were then compared on the basis of total assays where Typer 4 software made a genotype call and those where no calls were made. Assays were then divided into four categories based on the relative significance a genotype would be called (Fig. 2). These four categories described in the software manual are as follows.
-
Conservative The most conservative assay call which makes no error on the training and test data, but has the most uncalled genotypes.
-
Aggressive The most aggressive assay call which makes the most errors (still less than 1%), but makes the largest number of calls.
-
Moderate The moderate set is a compromise between the conservative and aggressive extremes.
-
Failed Where no call was made, and only UEP was observed.
Analysis of this data for individual genotyping calls reveals a similar trend for the high 30- and 29-plex reactions. The 26-plex performed the worst with the lowest percentage of conservative calls (48%) and the highest rate of failed calls (32.3%). The call quality in the low 13-plex reaction had 10% more calls in the moderate category but, returned the lowest rate of failed calls (13.7%). From this data, call quality does not show a plex level trend, and the penalty for using higher plex levels appears to be negligible. However, this may be attributed to sequence-specific effects of individual assays in each multiplex. Examination of UEP levels between the different multiplex reactions reveals no trend towards plex level (Fig. 3), indicating that the level of UEP of assays is specific to the sequence and concentration of assay primers.

Distribution of unextended primer at the four multiplex levels
Conclusions
The method described adapts a system designed for SNP and InDel assay in genome wide scans and customises it for a candidate gene approach. This method has particular relevance to organisms with high sequence diversity. Sequences with a high SNP and InDel density were effectively genotyped by multiplexing long amplicons in the capture PCR. This provided a template for all SNP and InDel assays in four candidate genes. Because fewer amplicons were used, there was less opportunity for false priming. The difference in amplicon length allowed the multiplex to be separated by agarose by gel electrophoresis for troubleshooting, not possible with the standard approach. Because the amplicon number was reduced by 96%, and the number of multiplex reactions was reduced by 75%, far less time was spent designing and optimizing multiple multiplex PCRs. With the number of capture primers reduced by 94%, there were significant savings in primer syntheses. Capture PCRs were performed at a larger volume introducing a greater level of robustness for each reaction and reduced the overall quantity of genomic DNA template required. Long amplicons were expected to be more difficult to amplify in multiplex at equi-molar concentrations. BSA, DNA, polymerase and primer concentrations were optimized in order to normalize the equi-molar amplicons concentrations. A large range of amplicons concentrations was still observed in the optimised capture PCR. However, with 91% of assay primers extending and 84.3% of useful assays the method was robust enough to compensate for this variation. Increasing the amplicon length up to 12 kb might be possible. This would allow larger candidate genes to be studied.
References
Buetow KH, Edmonson M, MacDonald R, Clifford R, Yip P, Kelley J, Little DP, Strausberg R, Koester H, Cantor CR, Braun A (2001) High-throughput development and characterization of a genomewide collection of gene-based single nucleotide polymorphism markers by chip-based matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Proc Natl Acad Sci USA 98:581–584
Desai C, Madamwar D (2007) Extraction of inhibitor-free metagenomic DNA from polluted sediments, compatible with molecular diversity analysis using adsorption and ion-exchange treatments. Bioresour Technol 98:761–768
Devey M, Bell JC, Smith DN, Neale DB, Moran GF (1996) A genetic linkage map for Pinus radiata based on RFLP, RAPD, and microsatellite markers. Theor Appl Genet 92:673–679
Galvin P (2002) A nanobiotechnology roadmap for high-throughput single nucleotide polymorphism analysis. Psychiatr Genet 12:75–82
Goicoechea M, Lacombe E, Legay S, Mihaljevic S, Rech P, Jauneau A, Lapierre C, Pollet B, Verhaegen D, Chaubet-Gigot N, Grima-Pettenati J (2005) EgMYB2, a new transcriptional activator from Eucalyptus xylem, regulates secondary cell wall formation and lignin biosynthesis. Plant J 43:553–567
Gonzales-Martinez SC, Wheeler NC, Ersoz E, Nelson CD, Neale DN (2007) Association genetics in Pinus taeda L. I. Wood property traits. Genetics 175:399–409
Grima-Pettenati J, Feuillet C, Goffner D, Borderies G, Boudet AM (1993) Molecular cloning and expression of a Eucalyptus gunnii cDNA clone encoding cinnamyl alcohol dehydrogenase. Plant Mol Biol 21:1085–1095
Henegariu O, Heerema NA, Dlouhy SR, Vance GH, Vogt PH (1997) Multiplex PCR: critical parameters and step-by-step protocol. Biotechniques 23:504–511
Ingvarsson PK, Garcia MV, Luquez V, Hall D, Jansson S (2008) Nucleotide polymorphism and phenotypic associations within and around the phytochrome B2 locus in European aspen (Populus tremula, Salicaceae). Genetics 178:2217–2226
Kim CS, Lee CH, Shin JS, Chung YS, Hyung NI (1997) A simple and rapid method for isolation of high quality genomic DNA from fruit trees and conifers using PVP. Nucleic Acids Res 25:1085–1086
Konieczny A, Ausubel FM (1993) A procedure for mapping Arabidopsis mutations using co-dominant ecotype-specific PCR-based markers. Plant J 4:403–410
Legay S, Lacombe E, Goicoechea M, Brière C, Séguin A, Mackay J, Grima-Pettenati J (2007) Molecular characterization of EgMYB1, a putative transcriptional repressor of the lignin biosynthetic pathway. Plant Sci 173:542–549
Li P, Kupfer KC, Davies CJ, Burbee D, Evans GA, Garner HR (1997) PRIMO: a primer design program that applies base quality statistics for automated large-scale DNA sequencing. Genomics 40:476–485
Masouleh AK, Waters DL, Reinke RF, Henry RJ (2009) A high-throughput assay for rapid and simultaneous analysis of perfect markers for important quality and agronomic traits in rice using multiplexed MALDI-TOF mass spectrometry. Plant Biotechnol J 7:355–363
Oeth P, Beaulieu M, Park C, Kosman D, del Mistro G, van den Boom D, Jurinke C (2008) iPLEX gold assay for SNP genotyping biotechniques. Protocol Guide 2008:37
Pattemore JA, Trau M, Henry RJ (2007) Nanotechnology: the future of cost-effective plant genotyping. In: Henry RJ (ed) Plant genotyping II. CABI, Wallingford, pp 133–153
Poke FS, Vaillancourt RE, Elliott RC, Reid JB (2003) Sequence variation in two lignin biosynthesis genes, cinnamoyl CoA reductase (CCR) and cinnamyl alcohol dehydrogenase 2 (CAD2). Mol Breeding 12:107–118
Ragoussis J, Elvidge GP, Kaur K, Colella S (2006) Matrix-assisted laser desorption/ionisation, time-of-flight mass spectrometry in genomics research. PLoS genetics 2:e100
Ross P, Hall L, Smirnov I, Haff L (1998) High level multiplex genotyping by MALDI-TOF mass spectrometry. Nat Biotechnol 16:1347–1351
Sachidanandam R, Weissman D, Schmidt SC, Kakol JM, Stein LD, Marth G, Sherry S, Mullikin JC, Mortimore BJ, Willey DL, Hunt SE, Cole CG, Coggill PC, Rice CM, Ning Z, Rogers J, Bentley DR, Kwok PY, Mardis ER, Yeh RT, Schultz B, Cook L, Davenport R, Dante M, Fulton L, Hillier L, Waterston RH, McPherson JD, Gilman B, Schaffner S, Van Etten WJ, Reich D, Higgins J, Daly MJ, Blumenstiel B, Baldwin J, Stange-Thomann N, Zody MC, Linton L, Lander ES, Altshuler D (2001) A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409:928–933
Shepherd M, Cross M, Stokoe R, Scott L, Jones M (2002) High-throughput DNA extraction from forest trees. Plant Mol Biol Rep 20:425
Shepherd M, Sexton TR, Thomas D, Henson M (2009) Utility of symphyomyrt-derived microsatellite markers (Genus Eucalyptus Subgenus Symphomyrtus) in Eucalyptus pilularis (Subgenus Eucalyptus) (submitted)
Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, Boutin P, Vincent D, Belisle A, Hadjadj S, Balkau B, Heude B, Charpentier G, Hudson TJ, Montpetit A, Pshezhetsky AV, Prentki M, Posner BI, Balding DJ, Meyre D, Polychronakos C, Froguel P (2007) A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445:881–885
Smith LM (1993) The future of DNA sequencing. Science 262:530–532
Stewart NC, Via LE (1993) A rapid CTAB DNA isolation technique useful for RAPD fingerprinting and other PCR applications. Biotechniques 14:748–749
Stoeckel S, Grange J, Fernandez-Manjarres JF, Bilger I, Frascaria-Lacoste N, Mariette S (2006) Heterozygote excess in a self-incompatible and partially clonal forest tree species Prunus avium L. Mol Ecol 15:2109–2118
Thumma BR, Nolan MF, Evans R, Moran GF (2005) Polymorphisms in cinnamoyl CoA reductase (CCR) are associated with variation in microfibril angle in Eucalyptus spp. Genetics 171:1257–1265
Tibbits JFG, McManus LJ, Spokevicius AV, Bossinger G (2006) A rapid method for tissue collection and high throughput genomic DNA isolation from mature trees. Plant Mol Biol Rep 24:81–91
Ward M, Dick CW, Gribel R, Lowe AJ (2005) To self, or not to self…a review of outcrossing and pollen-mediated gene flow in neotropical trees. Heredity 95:246–254
Zhao Z, Fu YX, Hewett-Emmett D, Boerwinkle E (2003) Investigating single nucleotide polymorphism (SNP) density in the human genome and its implications for molecular evolution. Gene 312:207–213
Acknowledgments
This research was funded by the CRC for Forestry. The authors thank Forests NSW for providing leaf samples, as well as Chris Harwood, Peter Bundock, Julie Pattemore, Helen Hill, Dan Waters and Ardashir Kharabian Masouleh for many helpful comments and suggestions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sexton, T.R., Henry, R.J., McManus, L.J. et al. Capture of assay template by multiplex PCR of long amplicons for genotyping SNPs and InDels with MALDI-TOF mass spectrometry. Mol Breeding 25, 471–480 (2010). https://doi.org/10.1007/s11032-009-9345-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11032-009-9345-0