
Abstract
The aggregation of alpha-synuclein (α-Syn) plays a critical role in the development of Parkinson’s disease (PD) and other synucleinopathies. α-Syn, which is encoded by the SNCA gene, is a lysine-rich soluble amphipathic protein normally expressed in neurons. Located in the cytosolic domain, this protein has the ability to remodel itself in plasma membranes, where it assumes an alpha-helix conformation. However, the protein can also adopt another conformation rich in cross-beta sheets, undergoing mutations and post-translational modifications, then leading the protein to an unusual aggregation in the form of Lewy bodies (LB), which are cytoplasmic inclusions constituted predominantly by α-Syn. Pathogenic mechanisms affecting the structural and functional stability of α-Syn — such as endoplasmic reticulum stress, Golgi complex fragmentation, disfunctional protein degradation systems, aberrant interactions with mitochondrial membranes and nuclear DNA, altered cytoskeleton dynamics, disrupted neuronal plasmatic membrane, dysfunctional vesicular transport, and formation of extracellular toxic aggregates — contribute all to the pathogenic progression of PD and synucleinopathies. In this review, we describe the collective knowledge on this topic and provide an update on the critical role of α-Syn aggregates, both at the cellular and molecular levels, in the deregulation of organelles affecting the cellular homeostasis and leading to neuronal cell death in PD and other synucleinopathies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Signs and Clinical Symptoms of Parkinson’s Disease (PD)
Parkinson’s disease (PD) is the second most common neurodegenerative disorder worldwide, affecting 1–2% of the global population aged 65 years and older. PD is characterized by loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc), with a subsequent decrease in dopamine (DA) levels and alterations in motor system function, evidenced by tremor, muscular rigidity, bradykinesia, and postural instability [1]. Other non-motor symptoms of PD include hyposmia, autonomic alterations, urination dysfunction, rapid eye movement, sleep changes, dementia, and depression [2]. While 45% of patients with widespread cerebral Lewy pathology (LP) are diagnosed with dementia or motor symptoms [3], only 10% of patients with LP in the SNpc and/or basal forebrain are diagnosed with PD [4]. Moreover, neurodegeneration in the SNpc might precede LP [5]; therefore, caution should be taken in PD diagnosis.
Neuropathology of PD
Combined with the loss of mesencephalic nigrostriatal neurons, PD is histologically defined by the presence of Lewy bodies (LB) and Lewy neurites (LN) [6], both originally identified in the brains of PD patients by Henrich Lewy in 1912. For more than one century, Lewy’s disorders have been identified with the neuropathological characteristics of the postmortem PD brains [7]. Later, with the advent of more sophisticated histological techniques, α-Syn aggregates were discovered as a component of LB [8]. α-Syn aggregates were also identified in the central and peripheral nervous systems (CNS and PNS, respectively) of PD patients [8, 9]. Extensive comparisons between normal and PD brains revealed that neuronal loci affected by α-Syn aggregation proceed in a relatively stereotypic manner, progressing from the brainstem to the cortex during the evolution of the disease [6], which has allowed for the implementation of grading to diagnose the severity of the disease when considering the neuroanatomical alterations resulting from α-Syn aggregation [6, 10]. The original locations of α-Syn aggregation in the CNS are the dorsal motor nucleus in the brainstem and the olfactory bulb. As the disease progresses, α-Syn aggregates appear in the pontine tegmentum, followed by the amygdala, and finally reach the temporal cortex and the neocortex at later disease stages [6].
As mentioned above, at the neuropathological level, PD is characterized by degeneration of dopaminergic neurons located in the SNpc, and the accumulation of α-Syn in LB and LN. While the progressive loss of dopaminergic neurons in the SNpc and the subsequent dopaminergic denervation in the forebrain are the main features of motor alterations in PD [11], the non-dopaminergic neuronal loss is associated with non-motor PD symptoms [12, 13].
Abnormal aggregates of α-Syn are prevalent in the syndromes known as synucleinopathies [14]. α-Syn aggregation at the cellular level can be observed in several forms of inclusions: (1) deposits of α-Syn in neuronal LB and LN in PD (as already mentioned), and dementia with LB, as well as in a considerable number of other pathological conditions; (2) protein deposits in oligodendroglia in multiple system atrophy (MSA); and (3) α-Syn inclusions in axonal spheroids in neuroaxonal dystrophies [14]. It is noteworthy that synucleinopathies are largely heterogeneous in regard to their clinical features; for instance, while MSA has been described to display several clinical phenotypes linked to different anatomical localization of lesions, PD has been linked to four different clinical phenotypes. Moreover, despite the challenges in diagnosing DLB compared with other synucleinopathies, evidence suggests common clinical features across various pathological forms (typical DLB, DLB with Alzheimer’s disease (AD), and AD with amygdala predominant Lewy pathology) [14]. Therefore, heterogeneity among synucleinopathies is closely associated with their pathological phenotypes.
α-Syn pathology in PD patients leads to a wide spectrum of toxic effects in the PNS [15]. The first LB identified outside the CNS was found in the enteric nervous system (ENS) [16, 17]. α-Syn aggregates have also been identified in the gastrointestinal system, the spinal cord, the sympathetic ganglia, and the vagus nerve [18]. The presence of α-Syn aggregates outside the basal ganglia provides a potential mechanism for non-motor symptoms associated with PD [2], and a promising biomarker. For instance, gastrointestinal dysfunction is one of the most common non-motor symptoms associated with PD, starting several years before the beginning of motor or behavior alterations [19]. In agreement, the most consistent localization of α-Syn in the ENS appears to be from the esophagus to the rectum [15]. It is noteworthy that α-Syn aggregates appear in the ENS years before the pathological signs develop in the CNS, parallel to gastrointestinal symptoms [18, 20], suggesting that α-Syn aggregations in neurons from the ENS are the main cause of gastric alterations.
Pharmacological Treatments for PD
The pharmacological treatments available for PD are designed to reduce the symptoms, but not the progression of the disease. There are several drugs in use at the clinical level for treatment at different stages of the pathological condition. Among them, the most relevant include carbidopa/levodopa, inhibitors of monoamine oxidase B (MAO-B), DA agonists, and anticholinergic agents. Table 1 summarizes the main features of some of these agents.
Biological Functions of α-Syn
α-Syn assumes several physiological functions given its presynaptic localization [27] and its ability to bind biological membranes; these functions include optimal neurotransmitter release, in which the protein regulates coupling and fusion of vesicles [28,29,30,31]. α-Syn has also been associated with exo- and endocytosis of synaptic vesicles [32]. In addition, neuroprotective and antiapoptotic properties [33], as well as synaptic plasticity, have been attributed to this protein [34]. Neuronal protection against oxidative stress [35] and preservation of mitochondrial function in neurons [36] have also been shown to be part of α-Syn functions. An active role of α-Syn in axonal transport by its direct interaction with microtubules has also been described [37]. Although all these important functions highlight the relevance of α-Syn to neuronal physiology, its modification, misfolding, and further aggregation are responsible for several pathological events, as will be described below.
α-Syn and Neuronal Damage in PD and Other Neuropathies
Neurons are morphologically different from other cells in several aspects, including compartamentalization: they possess dendrites, a unique cell body (neuronal soma), an axon, and several synaptic terminals (Fig. 1). In the cytoplasmic domain, several organelles are interconnected, forming a complex system of structures that display a wide variety of enzymatic characteristics. Lysosomes and mitochondria are accompanied and supported by vesicular structures and protein filaments forming the cytoskeleton [38], with all these structures playing specific roles in the transmission of electric and chemical signals [39].

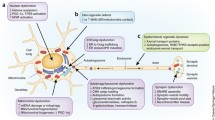
Alpha-synuclein: pathological progression in synucleinopathies. In the left side, a representation of a typical neuron (1). A detail of the neuronal nucleus (2) is also shown. The gene codifying for α-Syn (SNCA) is located in chromosome 4. SNCA is composed by six exons, from which the last five codify for α-Syn. The domains and mutations of α-Syn are also depicted. Misfolded α-Syn induces endoplasmic reticulum (ER) stress (3). In addition, misfolded α-Syn is transported by vesicles producing fragmentation of organels such as the Golgi complex (GC) (4). At the mitochondrial level (5), α-Syn produces dysfunction by fragmentation after binding to membranes. The interaction of α-Syn with complex I reduces mitochondrial activity. In lysosomes (6), α-Syn is degraded by autophagy (CMA), or in the cytosolic domain by the ubiquitin–proteasome system (UPS); however, these degradative systems may present autophagic abnormalities linked to the pathology of Parkinson’s disease (PD), leading to the cytosolic accumulation of α-Syn toxic species. In turn, this process leads to the formation of Lewy bodies (LB) as dimers, oligomers, fibrils, and β-sheets (protofibrils). At the cytoskeleton (7), the presence of α-Syn aggregates produces structural impairment and compromised dynamics in axonal transport. At the synaptic membrane (SM) (8) oligomeric α-Syn plays a multifunctional role, forming transmembrane rings (pores) capable of compromising the membrane integrity, disrupting Ca2+ homeostasis, and signaling. Various structures of the protein are shown (9): UPS is unable to degrade misfolded α-Syn (9a); dimers (9b); oligomers (9c); amyloid fibers (9d); β-sheets (9e); dense nucleus LB (9f1); halo lacking LB (9f2); pale LB (9f3)
At the cellular level, native α-Syn is present in synaptic terminals, organelles, and neuronal nuclei [27, 40]. While its physiological functions in each subcellular compartment remain poorly understood, the expression of pathological forms of α-Syn has been associated with aberrant cellular responses. In turn, the cytotoxic effects of α-Syn fibrils may cause oxidative stress, altered axonal transport, mitochondrial and synaptic dysfunction [41, 42], Golgi complex (GC) fragmentation, endoplasmic reticulum (ER) stress, plasmatic membrane, and cytoskeleton alterations, as well as disruption of lysosomal-autophagy and proteasome-ubiquitin degradation systems [43, 44].
The accumulation of toxic α-Syn species in presynaptic terminals is responsible for synaptic dysfunction and synaptotoxicity, leading to cell death by degeneration (Fig. 4). Since the cellular and molecular mechanisms inherent to PD and other synucleinopathies underlie α-Syn aggregation, and the precise role of the protein in neurodegeneration has yet to be fully characterized, the aim of this review is to update the reader known α-Syn gene mutations codifying for the protein and its different structural forms, providing crystalographic structural representations of some of these forms, and describing the consequences of α-Syn misfolding and aggregation. We also describe post-translational modifications and correlate them with morphological and functional alterations produced by oligomers in cellular organelles, with emphasis on altered synthesis in the rough endoplasmic reticulum (RER), GC fragmentation, inhibition of degradative systems, damage of neural organelles, and defects in cytoskeleton dynamics (Fig. 1). By discusing these approaches, we aim to provide an integrative view of the molecular processes involved in the pathogenesis of PD and other synucleinopathies.
α-Syn Gene Mutations
The gene encoding α-Syn is located in choromosome 4; this single gene is known as SNCA (4q23) and consists of 7 axons (5 of which are encoder) [45] (Fig. 1). The gene may present genomic duplication and triplication, as well as nonsense mutations such as A30P, A53T, and E46K [46, 47] (Fig. 1). These three mutations mainly affect the N-terminal domain of α-Syn. A30P and A53T stimulate the formation of protofibrils, leading α-Syn small aggregates to larger inclusions [48]. Rospigliosi et al. [49] reported that the E46K mutation enhances the positive charge of the N-terminal domain, thus modifying the net charge of the protein and promoting the interaction contacts of the C-terminal with the N-terminal domain. In contrast, A30P and A53T mutations do not alter the charge of the N-terminal domain, nor promote N-terminal contacts with C-terminal domain. Several reports have demonstrated that the three mutations affect in different manners the properties and functions of α-Syn [50, 51]. Moreover, since the discovery of the A53T mutation, three additional mutations on the SNCA gene identified as H50Q [52], G51D [53], and A53E [54] have been related to idiopathic PD [55]. Genome-wide association studies have consistently revealed highly significant regions of genetic variation around the SNCA gene that contribute to PD risk factors. Recently, Chang et al. [56] carried out the genome-wide association analysis identifying 17 novel risk loci.
α-Syn Interaction with Nuclear DNA
Although the name “synuclein” indicates the primary synaptic and nuclear distribution of this protein [27], its function within the nucleus has yet to be appreciated. α-Syn seems to modulate the physical properties of DNA. Experiments using wild-type α-Syn in nanofluids revealed that its binding to DNA is mainly stimulated by electrostatic interactions, resulting in a gradual increase in DNA length [57]. One of the key aspects in the interaction between α-Syn and the nucleus lies in its import from the cytoplasmic domain, evidenced by the crucial role of nuclear pore complex in α-Syn translocation across the nuclear membrane [58] (Fig. 1). When α-Syn and karyopherin alpha 6 (a nuclear adapter protein) were labeled in SH-SY5Y cells and fetal primary cortical neurons from mice to evaluate their physical interaction using FRET, it was found that these two proteins interact once sumoylated in a process that is required for its nuclear transport. In addition, although nuclear co-localization of α-Syn and DNA has been described (Fig. 1), the precise role of α-Syn in the nucleus remains controversial since a nuclear synchronism responsible for negatively regulating the repair of DNA genes responsible for cell cycle has been reported. In this regard, Pinho et al. [59] described specific α-Syn phosphorylation sites and the nuclear localization of different α-Syn forms affecting gene expression responsible for neurotoxicity.
α-Syn Structure, Aggregations, and LB
α-Syn Chemical and Molecular Structure
Several groups have gathered structural information on α-Syn at the molecular level. α-Syn is a soluble protein highly expressed and conserved [60]. The protein is expressed in different isoforms composed by 98, 112, 126, or 140 amino acids [61, 62], with the latter being the most studied; this isoform possesses a molecular weight of 14 kDa and its 140 amino acids are distributed into three domains: (i) the frequently acetylated N-terminal domain, comprising the first 60 amino acids, is positively charged by lysine residues and displays an established series of KTKEGV repeats, forming an α-helix for binding with membrane lipids, thus reducing the formation of β structures [63,64,65]; (ii) the highly hydrophobic non-amyloid β component (or NAC), comprising amino acids 61 to 95 from the central region, which is fundamental for protein aggregation [65, 66]; and (iii) the highly acidic C-terminal domain, which is negatively charged by glutamic and aspartic acids, comprising amino acids 96 to 140, and is prone to lack of stable structure (Fig. 1). In this last domain, several post-translational modifications can be found [65].
Several α-Syn structures have been determined by electron diffraction, nuclear magnetic resonance (NMR), and cryomicroscopy (Cryo-EM); these structures have already been deposited in the Protein Data Bank (PBD). Among them, the monomeric α-Syn structure bound to micelles can be obtained by NMR (PDB ID 1XQ8) [67]. As mentioned before, this monomeric structure contains three domains: N-terminal, NAC, and C-terminal domains (highlighted in different colors in Fig. 2). The three main mutations seem to be located in the N-terminal domain, although this structure is not mutated. Figure 2A shows where the mutations might be located in A30, E46, and A53 (orange color) [68], providing two β-hairpin folding mechanisms through molecular dynamics’ simulations; it was observed that A30P and A53T mutations accelerate the formation of β-hairpins, favoring the part of the core that constitutes the α-Syn fibrils, and undergoing stabilization through hydrophobic contacts and hydrogen bonds. However, E46K is responsible for enhanced aggregation with respect to A30P and A53T [69]. These authors also studied the effects of several mutations, mainly A30P, E46K, and A53T, on the rates of lipid-induced aggregation; they emphasized the influence of a singular mutation on different steps of α-Syn aggregation. Furthermore, a short segment of residues (68 to 78) of the NAC domain (also highlighted in cyan color in Fig. 2A) plays a major role in the aggregation and cytotoxicity of α-Syn. This short segment, known as NACore (Fig. 2B), was obtained by micro-electron diffraction (PDB ID 4RIL) [70]. Soon thereafter [71], the first tridimensional model of full-length α-Syn fibril obtained by solid state NMR (PDB ID 2N0A) was reported (Fig. 2C). The fibril core is formed by a β-strand set, and is stabilized by non-covalent interactions, mainly hydrophobic contacts and hydrogen bonds. More recently [72], a structure containing α-Syn cytotoxic fibrils from residues 1 to 121 (PDB ID 6H6B), which is composed by two connected protofilaments (Fig. 2D), was determined by Cryo-EM. Finally, the same group determined new α-Syn fibril structures referred to as polymorphous 2A (PDB ID 6RT0) and 2B (PDB ID 6RTB) (Figs. 2E and F, respectively) [73].

α-Syn structures. A Monomeric α-Syn structure obtained by NMR (PDB ID 1XQ8) [67]. B Segment of the non-amyloid β component obtained by electron diffraction (PDB ID 4RIL) [70]. C First tridimensional model of α-Syn fibers determined by solid state NMR (PDB ID 2N0A) [71]. D Structure containing cytotoxic α-Syn fibrils (PDB ID 6H6B) [72]. E α-Syn fibril polymorphic 2A structure (PDB ID 6RT0) [73]. F α-Syn fibril polymorphic 2B structure (PDB ID 6RTB) [73]
The last three structures are formed by two protofilaments exhibiting a staggered β-strand fold which could cause formation, growth, and stability of α-Syn cytotoxic fibrils (Figs. 2D, E, and F).
α-Syn Aggregation and LB
In the cytosolic domain, α-Syn monomers are expressed as polypeptidic chains [74] organized as β-sheets or protofibrils. These monomers undergo conformational changes that interact to form two types of dimers: antiparallel (not propagating) and parallel (propagating) dimers. These processes occur in the cytoplasmic domain or in association with membranes. Propagating α-Syn dimers can be enlarged through the addition of displayed monomers to generate oligomers and ring-like oligomers. Cytoplasmic α-Syn oligomers are enlarged by the addition of soluble monomers, first forming small amyloid fibrils, and later larger fibrils [66] (Fig. 1: 9b, 9c, and 9d). During fibrillogenesis and α-Syn aggregation, the intermediate species (oligomers and amyloid fibrils) are highly toxic [75]. The accumulation of amyloid fibrils leads to the formation of intracellular inclusions known as LB [76]; however, recent studies have shown that the acetylated N-terminal of α-Syn is conserved in conditions of a disordered monomer, compared to oligomerization in neuronal cells under physiological conditions [77, 78]. The accumulated membrane-bound monomers undergo conformational changes to form intermediates rich in β-sheets (protofibrils), and then associate with ring-like oligomers or amyloid fibrils [79]. Oligomeric α-Syn simultaneously localized to the cytosolic domain and the membrane, plays multifunctional roles, forming transmembrane pore-like rings capable of breaching membrane integrity, thus altering intracellular Ca2+ homeostasis and signaling [66] [80].
Although LB are intracellular inclusions composed mainly of α-Syn fibrils [76, 81], they also comprise other proteins, such as ubiquitin, parkin, neurofilaments (NF) [76], leucine-rich kinase 2 (LRRK2) [82], histone deacetylase 6 (HDAC6) [83], and the endosomal sorting related protein charged multivesicular body protein 2B (CHMP2B) [84]. Histologically, these structures appear as spherical bodies displacing all other cell components; three varieties have been described: the first one is an eosinophilic inclusion with a dense core radiating fibrils [85], the second form lacks the halo [86], and the third one is a pale body composed by eosinophilic round granules, which are believed to be precursors of LB (Fig. 1: 9f1, 9f2, and 9f3) [87].
About 90% of α-Syn in LB is phosphorylated at Ser129 (pS129-α-Syn) in the C-terminal region. It has been demonstrated that modifications at these sites inhibit α-Syn aggregation [88, 89]. Similarly, tyrosine phosphorylation at Y125, Y133, and Y135 is associated with suppression of aggregation and α-Syn toxicity [90, 91].
Neurotoxic Association of α-Syn with Neuronal Membranes
ER Stress and PD
α-Syn synthesis takes place in the ER. Folding, in cooperation with chaperones, constitutes the first step of the transport mechanism of the RER-GC-lysosome complex [92]. ER is a membranous organelle that projects into the cytoplasm as an extension of the external nuclear membrane; it forms flattened closed coats known as cisterns, which conform an interconnected tubular net [93]. The ER is classified as smooth endoplasmic reticulum (SER) and RER, according to its morphology. The ER establishes several sites of contact with various organelles such as mitochondria, endosomes, the endo-lysosomic system, and the cytoplasmic membrane. Its association with mitochondrial membranes allows for the exchange of Ca2+ and lipids [38]. The ER also constitutes the main site of intracellular storage and regulation of Ca2+, and mediates several key signals in various cellular processes, including appropriate protein folding [94].
Altered RER function, such as altered Ca2+ levels, increased oxidative stress, and dysfunctional N-glycosylation, may trigger stress and promote the activation of signaling pathways grouped under the term UPR (unfolded protein response) in an attempt to squash the stress in its course, thus rescuing cells and restoring RER homeostasis. However, neurodegenerative disorders include the accumulation of endogenous mutant misfolded proteins [95] and a decrease in Ca2+ reserves in the lumen. Chaperons, such as HSPA5 (heat-shock protein 5) and CALR (calreticulin), are located in the lumen of the RER. Since they require high levels of Ca2+ [96], the exhaustion of this cation in deposits leads chaperons to be inactive, thus altering protein folding processes, and resulting in the accumulation of α-Syn oligomers in the lumen. In addition, the stress induced by α-Syn overexpression (gene duplications and triplications) may facilitate the increase in oligomer concentrations. Combined, these studies indicate that ER stress induced by oligomers is involved in PD pathology [97] by inhibiting vesicular trafficking from RER to GC [98], thus triggering chronic stress-induced cell death [99] (Figs. 1 and 3).

The unfolded protein response (UPR) pathway. UPR is controlled by three transducers: IRE1, ATF6α, and PERK (signaling pathways appear detailed by color lines in the scheme). Under physiological conditions, IRE1, ATF6α, and PERK associate with each other and remain inactive by the complex BIP/GRP78, which resides in the ER. In contrast, under ER stress conditions, BIP/GRP78 dissociates, then allowing the activation of the UPR. ATF6 is transported from ER to GC, where it undergoes proteolitic cleavage by proteases S1P/S2P. The cytosolic ATF6 fragment (active ATF6α) is finally translocated into the nucleus where it acts as a transcription factor for the genes required for endoplasmic reticulum-associated degradation (ERAD) and modulates the transcription of XBP1. ER stress also activates PERK, which phosphorylates prostaglandin F2α to attenuate protein translation. Simultaneously, ATF4 translation stimulates the expression of ER chaperones and other genes controlling autophagy, redox activity, and the metabolism of nutrients. Under conditions of severe ER stress, the PERK-ATF4 complex regulates proapoptotic genes, including CHOP, thus leading to programmed cell death. In turn, once activated, IRE1 coordinates the alternative splicing of XBP1, which upregulates genes codifying for ER chaperones, ERAD components, and proteins involved in the pathway of lipid biosynthesis. ER stress also induces mitochondrial fragmentation and mitophagy in a PERK/ATF4 pathway-dependent manner. In turn, ATF4 acts as a transcription factor for the gene codifying for parkin, which is recruited in mitochondrial membranes by PINK1. The PINK1-parkin complex regulates mitophagy, though under pathological conditions, the system is deregulated, leading to cell dysfunction. The α-Syn-chaperone complex binds the lysosomal membrane and interacts with the LAMP-2A receptor for chaperone-mediated autophagy (CMA); then, under physiological conditions, α-Syn translocates into the lysosomal domain to be degraded. Soluble α-Syn is also directed to the UPS, crossing through the proteasomal pore to be degraded into small peptides. However, alterations in the function of cell organelles and or disruption of the described signaling pathways may induce changes in the protein structure and function, leading to the production of toxic forms of α-Syn. In addition, misfolded, aggregated, and/or mutated forms of α-Syn (appearing in the scheme in several places as an aggregated structure and denoted at the left bottom as “α-Syn aggregation”) interact with several organelles, affecting their function and signaling
GC Fragmentation of PD
α-Syn synthesized in normal conditions in the RER is transported to the GC by conventional mechanisms (RER-GC-lysosome); α-Syn travels within vesicles to the GC. This organelle is formed by cisterns stacked in dichthyosomes which are joined together through tubular interconnections, each one showing two distinct surfaces: cis (entrance) and trans (exit). These special compartments are strongly associated with each other and are formed by a network of tubular and cistern structures: cis Golgi network (CGN) and trans Golgi network (TGN), forming a single complex. The GC is responsible for transporting, modifying, and packing proteins. The location of the GC depends on microtubules’ organization [38, 100]. One common feature of neurodegenerative disorders is the considerable number of neurons showing fragmented GC [101,102,103], resulting in a loss of vesicles, probably due to the formation of α-Syn oligomers [103, 104], with ensuing alterations in axonal transport (Figs. 1 and 3). Since this organelle is the central hub for vesicular trafficking and an important center for the integration of several signaling pathways, it makes sense that some regulatory proteins are altered; among them, Rab1, a small GTPase regulating the RER transport to GC, is overexpressed in surviving neurons [105].
Intracellular Protein Degradation Systems: Chaperone-Mediated Autophagy (CMA) and Ubiquitin–Proteasome System (UPS)
As mentioned above, protein exportation occurs in the RER-GC, while degradation is carried out by lysosomes, which are spherical organelles delimited by a single membrane. Lysosomes contain more than 60 different acidic hydrolases for the degradation of misfolded proteins and old organelles, and more than 50 proteins for membrane recognition [106]. In neurons, due to their morphology, metabolic characteristics, and post-mitotic status, the autophagy-lysosomal system is particularly vulnerable to the deregulation of degradative protein systems. This is a crucial process for the preservation of intracellular homeostasis, which is maintained by two independent but complementary systems: the CMA system and the UPS, both receiving the names of their final destinations: lysosome and proteasome, respectively (Figs. 1 and 3).
α-Syn contains a recognition site for CMA known as the KFERQ sequence, which is sensed by the cytosolic HSC70 chaperone; the α-Syn-chaperone complex binds the lysosomal membrane, interacting with LAMP-2A receptor at CMA, followed by α-Syn translocation into the lysosomal lumen for its degradation [107] (Fig. 3). However, alterations in α-Syn due to mutations or post-translational modifications may compromise the α-Syn exchange in CMA. Some mutant forms of α-Syn in PD (A30P and A53T) do not undergo efficient degradation through CMA; instead, these mutants can bind to LAMP-2A on the lysosomal surface with high affinity, but they do not internalize into lysosomes, thus avoiding degradation. In addition, the CMA system is inhibited by the decreased capacity of these α-Syn forms to be degraded by this pathway, thus favoring the increase in these soluble forms in the cytosolic domain and stimulating the formation of oligomeric protofibril intermediates, which progress to cytotoxic insoluble α-Syn fibrils [108] (Fig. 3).
On the other hand, the UPS, independently of lysosomes, acts in the cytosol by polyubiquitinating proteins to be degraded by proteasome. Unfolded or misfolded soluble proteins are directed to the UPS, cross the proteasomal pore, and undergo degradation into short peptides [109] (Fig. 3). It has been observed that mutant α-Syn found in dopaminergic cells may induce functional alterations in proteasomal 20/26S protein [110]. Other studies have demonstrated that α-Syn overexpression leads to early catalytic disruption of proteasome 26S, with a consequent dysfunction of UPS. This alteration has been associated with the selective accumulation of phosphorylated α-Syn (p-α-Syn) at S129 in neurons, which in turn is related with an enhanced toxicity [111] responsible for dopaminergic degeneration. The dysfunction of any of the cellular degradative systems, or both, has been implicated in the onset and progression of PD.
Cross-linking α-Syn Toxicity, Autophagy, ER Stress, and PD
Mutations in the gene responsible for α-Syn encoding, the SNCA gene, are linked to autosomal-dominant forms of PD. These mutations include A53T, duplication, and/or triplication [112]. Autophagy is one of the most effective α-Syn degradation systems [113]. All forms (wild-type, mutant, phosphorylated, and oligomeric) of α-Syn can activate ER stress, thereby promoting autophagy; however, they can also block autophagy induction by impairing autophagosome maturation, fusion with lysosomes, and lysosomal biogenesis or function [112]. Autophagy can be promoted by both wild-type and A53T mutant α-Syn by upregulating the Beclin-1 and microtubule-associated protein 1A/1B-light chain 3 (LC3) expression [114, 115]. In contrast, enhanced expression of wild-type α-Syn decreases autophagy by inhibiting Ras-related protein Rab-1A (RAB1A), thus leading to decreased formation of autophagosomes [116]. It is known that dopaminergic neurodegeneration is protected by RAB1 overexpression in several PD animal models [98, 117, 118]. In addition, both wild-type and A53T α-Syn increase mTOR activity and decrease autophagy [119, 120]. Animal models overexpressing α-Syn display impaired autophagy by sequestering the transcription factor EB (TFEB) in the cytoplasm and accumulating p62 and microtubule-associated protein 1A/1B-light chain 3-phosphatidylethanolamine conjugate (LC3-II) proteins [115]. In addition, PC12 cells loaded with A53T α-Syn exhibit autophagic-vesicular structures and reduced lysosomal hydrolysis [121]. Highly neurotoxic phosphorylated α-Syn species, also known as “pα-Syn*,” are present in primary neurons cultured in the presence of α-Syn fibrils. The accumulation of these species results from incomplete autophagic degradation of α-Syn [122], hence contributing to the cross-talk between ER stress, mitochondrial fission, and mitophagy. It is noteworthy that while wild-type α-Syn contains a KFERQ sequence allowing its degradation by the CMA pathway (already mentioned above), pathogenic α-Syn mutants act as CMA uptake inhibitors [123]. Moreover, it has been shown that adeno-associated virus (AAV)-induced overexpression of A53T α-Syn in neurons, or A53T α-Syn in transgenic mice, can reduce CMA-mediated proteolysis of other substrates [124, 125]. Wild-type α-Syn can be also modified by DA, resulting in impaired CMA proteolysis, which resembles the one produced by mutant α-Syn [126]. Altogether, the evidence described above contributes to better understanding of the role of α-Syn modifications and mutations in nigrostriatal pathogenesis in PD.
Cross-linking α-Syn Toxicity, UPS, Mitochondrial Dysfunction, and UPR
Degradation of aggregated, misfolded, damaged, and mutant proteins is carried out by the UPS through a highly regulated process involving the tagging of the target protein by polyubiquitination and further degradation by the 26S proteasome [127]. UPS dysfunction is inherent to all forms of PD, which has been inferred from sporadic, toxin-induced studies as well as genetic forms of PD [128]. Experimental evidence demonstrates that proteasomal inhibition induces dopaminergic degeneration and α-Syn both under in vitro and in vivo conditions in several PD models [129, 130]. An active mitochondrial role in this process has been suggested since mtDNA depletion is tightly linked to UPS downregulation and α-Syn oligomerization [131], while reduced ATP production impairs proteasomal function [132]. It is also known that mutations in the gene encoding the neuromodulatory protein parkin are responsible for impaired E3 ubiquitinprotein ligase activity, thus reducing the mitochondrial quality control and accumulating parkin-specific substrates such as α-Syn [133], thus leading to impairment of dopaminergic neurons [134]. Remodeling the protein profile of the outer mitochondrial membrane by UPS-associated parkin is essential for the preservation of mitophagy, thus supporting a link between the UPS and autophagy [135]. PINK1 also participates in the UPS-parkin interaction, increasing the degradation of misfolded proteins [136]. Therefore, it is generally accepted that the stimulation of the UPS activity and the preservation of mitochondrial activity reduce the risk of α-Syn aggregation and inclusion by the protein clearance [134] (Fig. 3).
The use of cell models of PD with biochemical reconstruction assays has demonstrated that α-Syn inhibits the processing of the transcription factor ATF6 either directly through physical interactions or indirectly by a restricted incorporation of COPII (vesicles associated to the transport of RER and GC). Decrease ATF6 signaling was accompanied by impaired degradative ER function (ERAD) and an increased proapoptotic signaling [137]. In addition, ER stress and UPR activation have been observed in cell and murine models of PD accompanied by α-Syn aggregation. Both the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and the 6-hydroxydopamine (6-OHDA) PD models cause upregulation of the C/EBP homologous protein (CHOP) and ATF4, pointing to ER stress, while CHOP deletion can prevent the neurodegeneration induced by 6-OHDA. Congruently, post mortem brain samples of PD patients show the presence of several markers of ER stress and UPR activation due to increased levels of misfolded α-Syn, such as phosphorylated ERK [138] (Fig. 3).
Post-translational Modifications
Several α-Syn post-translational modifications include phosphorylation, oxidation, acetylation, ubiquitination, glycation, glycosylation, proteolysis, and nitration, mainly occurring at the carboxy-terminal domain and resulting in changes in the net charge and protein structure. These modifications lead to alterations in the binding affinity for other proteins and lipids, and the subsequent changes in hydrophobicity. Which of these post-translational modifications are physiologically relevant, and which of them emerge from the pathology associated to a synucleinopathy, has yet to be elucidated. In the interim, it seems clear that α-Syn structure and function may be significantly altered by the following post-translational modifications:
-
a
Phosphorylation is one of the most common post-translational modifications and plays a major role in the function of several target proteins. A common phosphorylation occurs at α-Syn Ser129 [139], being relevant to neurodegeneration in PD.
-
b
Acetylation of α-Syn amino-terminal augments its propensity for helical folding, its affinity for membranes, and its resistance to aggregation mediated by binding of the acetyl group to the amino group at the first amino acid [140, 141].
-
c
Sumoylation of α-Syn is modified by small ubiquitin-related modifier 1 (SUMO1); monosumation occurs at a single site, and this process seems to inhibit α-Syn aggregation by increasing its solubility [142, 143].
-
d
Glycation: advanced glycation end products (AGEs) and α-Syn are both found in the brain of PD patients. Indeed, AGEs co-localize with α-Syn in LB in the CNS [144]. It has been reported that glycation of α-Syn reduces monoubiquitination, as well as degradation via proteasome and autophagy in cellular models of PD [145]. The intracellular accumulation of α-Syn precedes the accumulation of LB, while the formation of extracellular AGEs accelerates the intracellular process of LB formation [146]. Glycation with D-ribose generates the formation of molten globule-like aggregates, leading to oxidative stress and toxicity [147].
-
e
Glycosylation: α-Syn is post-translationally modified by O-glycosyl-N-acetylation (O-GlcNAc); however, the consequences of this process remain unknown. The O-GlcNAc modification plays a major role in the prevention of aggregation and toxicity of extracellular α-Syn fibers [148].
-
f
Truncation: another modification of a-Syn is proteolysis, which is caused by a truncation on this protein, removing the acidic terminal. In vitro studies have used enzymes involved in α-Syn proteolysis such as neurosin, cathepsin D, and metaloproteases, among several others. Here, we highlight the role of neurosin, a serine proteinase whose viral administration has been shown to promote α-Syn degradation and co-localization in LB. Recently [149], it has been reported that neurosin truncates α-Syn beyond the residue 80, thus inhibiting polymerization; however, if the protein is truncated beyond the residue 97, it tends to polymerize. On the basis of these findings, it has been assumed that neurosin constitutes a therapeutic target in PD.
-
g
Nitration of α-Syn may occur in most of filaments and insoluble fractions from the affected brain regions in synucleinopathies [150]. Four tyrosine residues (Y39, Y125, Y133, and Y136) are susceptible to nitration [151,152,153], with Y39 nitration accelerating α-Syn oligomerization. In addition, it has been shown that both monomeric and dimeric forms of nitrated α-Syn may accelerate the formation of fibrils, as well as the additional fibrillation of unmodified α-Syn. The specific incorporation of 3-nitrotyrosine in several regions of α-Syn [152] suggests that different species of nitrated α-Syn display distinct aggregation properties. In addition, it has been demonstrated that the intermolecular interactions between N- and C-terminal regions of α-Syn play a crucial role in mediating nitration-induced α-Syn oligomerization [152]. Moreover, there is greater nitration of tyrosine residues in positions 125 and 136 (Y125/136) than in the nitrated residues in position 39 of tyrosine (Y39) in early-onset PD [154]. α-Syn residues may be nitrated in a differential manner, causing diverse effects; for instance, nitration located in Y39 is responsible for a reduced binding of this protein to vesicles and a decrease in the rate of protein degradation [155].
-
h
Oxidative and nitrative stress: PD and other synucleinopathies are neurodegenerative disorders characterized by oxidative stress and neuroinflammation [156]. As any other biological substrate, α-Syn is susceptible to attack and modification by reactive oxygen species (ROS). These modifications include the formation of a complex with 4-hydroxy-2-nonenal (4-HNE-α-Syn), its nitration (n-α-Syn), and its oxidation (o-α-Syn), all of which have been implicated in the stimulation of various forms of protein oligomerization, with the 4-HNE-α-Syn modification being selectively toxic to neurons [157]. Consequently, oxidative stress has been shown to affect patterns of α-Syn aggregation and membrane binding, thus affecting mitochondrial function, neurotransmitter recycling, protein degradation, and trafficking [156]. These observations have led to the hypothesis that under physiological conditions, α-Syn might act as a free radical scavenger prior to the onset of synucleinopathies. Indeed, some of these modifications have been shown to be responsible for augmented ROS formation, thus contributing to a toxic feedback loop, contributing to neuronal damage [157]. Finally, environmental factors favoring oxidative and nitrative α-Syn modifications include exposure to heavy metals, pesticides, and polycations, to name a few [158].
α-Syn Interactions with Mitochondrial Membranes
Mitochondria are double-membrane organelles involved in the oxidation of metabolites, ATP generation, oxidative phosphorylation, and electron transport chain activation. When α-Syn binds to both mitochondrial membranes (inner mitochondrial membrane (IMM) and outer mitochondrial membrane (OMM)), it leads to mitochondrial dysfunction. This effect is induced by α-Syn binding at sites enriched with cardiolipin (CL), similar in content to that of synaptic vesicles. The biophysical characteristics of mitochondrial membranes facilitate the binding of α-Syn oligomers due to the CL content [159], which may lead to IMM and EMM permeation. The insertion of β-sheet oligomers is responsible for the formation of a toroidal protein-lipid pore [160] (Fig. 3), causing membrane rupture and cell death. It has been shown that α-Syn, in its α-helix conformation, binds to IMM at high concentrations [161] (Fig. 3). In addition, oligomers bound to CL form a triple complex with cytochrome C (Fig. 3), acting as substrates for the peroxidase activity of cytochrome; in turn, this interaction favors the permeation of mitochondrial membranes, also contributing to oxidative stress in dopaminergic neurons [162].
Increased levels of α-Syn bound to mitochondria have also been associated to a reduced activity of complex I and a subsequent increase in ROS, reducing the respiratory activity and inducing oxidative damage to mitochondrial DNA in PD. Another consequence of the α-Syn-CL complex formation is the loss of function of the ADP/ATP carrier, leading to a precipitous fall in the mitochondrial membrane potential (Δψm), thus compromising the respiratory function [163, 164].
Cytoskeletal Alterations in PD
The neuronal cytoskeleton comprises a delicate intracellular net of filaments located in the cytoplasmic matrix, where three forms of filaments are distinguished: intermediate filaments (IF) or NF (10 nm diameter), actin-based microfilaments (FA) (6 nm diameter), and tubulin-based microtubules (24 nm diameter) [165].
Neurons possess a NF network [166] which carries out several functions, such as the definition of the axonal caliber, signaling and nerve conduction [167], regulation of synaptic vesicles transport, synaptic plasticity modulation, cell shaping, and intracellular signaling and transcription [168]. In addition, NF organize the cellular environment, and determine nuclear and organelle positioning. The activity of NF is modulated by post-translational modifications [169]. An abnormal display of assembly and accumulation of NF is associated with the onset of neurodegenerative diseases. Although the precise mechanism of NF aggregation remains unknown, hyperphosphorylation is considered one of the main triggering factors [170]. When axonal damage occurs in neurodegenerative disorders, NF are released and secreted into the cerebrospinal fluid (CSF), and circulate in the blood. Therefore, detection of NF in blood is considered a useful marker of axonal damage with diagnostic value for a variety of acute and chronic neurological disorders [171].
Another protein involved in neuronal degeneration is tubulin, a structural component of microtubules, which provide structural support and organelle positioning to neurons [172], as well as vesicular and mRNA transport [173, 174]. Alterations in microtubule stability, such as variations in the levels of tubulin and depolymerization, may lead to a deficient maturation of α-Syn protofibrils, causing neurotoxicity [175] due to deregulation of proteins associated with microtubules and hyperphosphorylation of tau protein. This modification facilitates the release of tau from microtubules, affecting axonal transport after the hyperphosphorylated protein is aggregated. In PD, several alterations induced by α-Syn have been observed in the cytoskeleton dynamics of microtubules [176, 177], causing axon degeneration [178].
Analogous to NF, actin is abundant in the presynaptic terminals, where it plays a role in the organization and mobilization of synaptic vesicles, exocytosis, and endocytosis [179]. Actin fibers form a dense filamentous net located near the cytoplasmic membrane, which undergoes reorganization upon cellular stimulation to allow access of vesicles to their fusion sites [180]. In neurons, actin deregulation may have implications for cell morphology and function [181]. Both actin and cofilin-actin rods may excessively accumulate affecting synapses and ensuing neurodegeneration [182, 183]. α-Syn interacts directly with actin to regulate the dynamics and function of the latter, which are altered by pathogenic mutation of A30P [184], thus compromising vesicular release. Given its cellular localization, actin might function as a physical barrier for exocytosis upon binding to synaptic vesicles and block their mobilization, since this protein plays a crucial role in amassing the molecular complexes needed to facilitate the fusion of vesicles to neurotransmitter release sites [185] (Fig. 4).

Schematic representation of a typical synapse in physiological and pathological conditions. In 4a, the synapse is highlighted by the green circle. In 4b, the synaptic function is mediated by the soluble N-ethylmaleimide sensitive factor-attachment protein receptor (SNARE) complex (the synaptic vesicle has been magnified for better visualization). In the presynaptic membrane, the vesicle associated membrane protein 2 (VAMP2) is shown inserted in the vesicle in red. Also in the presynaptic membrane, the proteins syntaxin and synaptosomal-associated protein 25 (SNAP-25) are shown in green, whereas complexin, which stabilizes the complex [204], is shown in light blue. In 4c, the approach of synaptic and vesicular membranes is regulated by VAMP2, syntaxin, and SNAP-25. In 4d, membrane fusion and neurotransmitter release are mediated by Munc13 and Munc18 proteins [225, 226]. In 4e, the accumulation of toxic α-Syn species bound to VAMP2 in presynaptic terminals causes synaptic dysfunction or synaptotoxicity, preventing the release of neurotransmitters. In 4f, cytoskeletal impairment is mediated by the accumulation of toxics α-Syn species
α-Syn Interaction with the Plasma Membrane
The plasma membrane is a separate structure that protects cells from exogenous chemical components in the environment, regulating the cells’ perimeter. This structure is composed of a phospholipidic bilayer (5–6 nm thick) with proteins (ionic channels and diverse receptors) [38]. Specifically, the neuronal membrane plays a key role in synaptic functions since it is able to transmit electric and chemical signals [186].
Toxic α-Syn species, specifically oligomers, may induce alterations in plasma membrane [28], causing synaptic dysfunction and other insults. Several studies have demostrated that the combination of lipids negatively charged with phosphatidic acid (PA) is sufficient to trigger the α-Syn oligomer binding to plasma membrane, leading to membrane permeation [187, 188]. α-Syn monomers may also interact with vesicles containing phosphatidylglycerol (FG), including deep insertion into membranes, resulting in their rupture [189]. The plasma membrane may also thicken upon interaction with β-helix oligomers [190], forming ring-like structures similar to pores [80] that compromise the cellular structural stability and its function (Fig. 1).
α-Syn has been shown to bind membranes at N-terminal and NAC sites [191]. The binding of the α-helix protein to membranes occurs in two distinct steps: first, the protein anchors to amino acid residues 3 to 25 at the N-terminal region. Next, a fraction of the α-helix binds to residues 26 to 97, which in turn determines the affinity for the membrane, whereas the C-terminal domain presents a weak binding [192, 193]. A computational modeling study revealed that α-Syn rapidly assembles with the lipid bilayer due to its high propensity to aggregate; in this model, the interaction α-Syn–membranes is posited to play a critical role during the aggregation process [194].
Vesicular Transport, Membrane Fusion, Neurotransmission, Synapse (SNARE Proteins), Synaptotoxicity, and Neurotoxicity
The vesicular trafficking in secretory and endocytic pathways includes the formation, translocation, anchoring, and fusion of the plasmatic membrane [195]. The formation of vesicles is a budding process originated in the GC and mediated by COPI, COPII, and clathrin, as well as the small GTPases Sar1 and Arf [196,197,198]. The vesicular transport is carried out by the protein rails inherent to the cytoskeleton, reaching the plasmatic membrane [199]. The anchoring proteins and Rab regulate coupling of vesicles to the plasmatic membrane [199, 200].
The fusion of vesicles with the membrane constitutes the final step of their transport system; this step is mediated by a family of proteins known as SNARE (soluble N-ethylmaleimide sensitive factor-attachment protein receptor) [201, 202]. The specific binding of vesicular SNARE (v-SNARE) with target SNARE (t-SNARE) at the membrane forms the SNARE complex, which mediates the fusion of the vesicle membrane with the plasmatic membrane [203, 204]. The neurotransmitter release requires not only of SNARE proteins but also of Munc18 and Munc13 proteins (Fig. 4).
It is known that α-Syn facilitates the vesicular coupling to plasmatic membrane via the formation of the SNARE complex [205,206,207]. It is noteworthy that α-Syn can facilitate the binding of secretory vesicles with the plasmatic membrane. Through modeling, the N-terminal of α-Syn binds to the plasma membrane, while simultaneously, its C-terminal interacts with the vesicle associated membrane protein 2 (VAMP2), forming a membrane-vesicle bridge to facilitate coupling [207]. Another proposal suggests that α-Syn-helix complex, spreading between synaptic vesicles and the membrane, may facilitate membrane-vesicles coupling [208] (Fig. 4).
The largest α-Syn oligomers preferentially bind to the N-terminal of VAMP2, thus inhibiting the formation of the SNARE complex and blocking the vesicles coupling [28], contradicting the concept that α-Syn multimers bind the membrane to block the SNARE complex formation [206]. In addition, mutants linked to PD, including A30P and A53T, are toxic for exocytosis [209]. Collectively, α-Syn seems to play a crucial role in coupling, priming steps, and exocytotic fusion, probably serving as an atypical chaperone that facilitates SNARE assembly [205, 206, 210].
The continuous cycle of assembly and disassembly of the SNARE complex is essential for neurotransmitter release, implying constant conformational changes of SNARE proteins. Some studies have demonstrated that α-Syn and cysteine string protein α (CSPα) share the chaperon function endowed with the assessment of the stability of the SNARE complex [211]. It is noteworthy that in the absence of CSPα activity, neurodegenerative processes emerge.
Neurotransmitter release requires the fusion of synaptic vesicles to the plasmatic membrane and the subsequent release of their content to the synaptic cleft [186]. The neuronal communication occurs at the moment in which neurotransmitters are released, cross the synaptic cleft, and bind to membrane receptors of the neighboring postsynaptic terminal, transforming the chemical signal into a new electric signal. Several proteins mediate and control the process of fusion with high precision [212] (Fig. 4).
The accumulation of α-Syn toxic species in presynaptic terminals causes synaptopathy (synaptic dysfunction or synaptotoxicity), further leading to cell death by neurodegeneration. Under physiological conditions, α-Syn functions as monomer in presynaptic terminals, whereas the oligomeric forms of α-Syn are considered the most toxic species [75]. This suggests that the magnitude of the pathology can be related not only with the formation and accumulation of oligomers but also with their structural and functional properties [213]. As the result of excessive accumulation, synaptic transmission is affected, leading to the loss of neural connectivity. Yet to be demonstrated is whether distinct α-Syn oligomeric species exert differential toxic mechanisms with distinct signatures of compromised neurotransmission at the synaptic terminal (Fig. 4e).
Toxic Extracellular α-Syn Aggregates
α-Syn is present in the extracellular domain of cells overexpressing α-Syn, suggesting that its release is independent of cell death. α-Syn aggregates intra- or extracellularly in various aggregation forms [40]. Exosomes represent a vehicle for the release of excess protein; next, α-Syn is endocytosed by microglia and astrocytes [214]. Several brain cell types, such as glia, are capable of phagocyting protein aggregates [215, 216]. Astrocytes have been identified as key mediators in the elimination of extracellular α-Syn. These α-Syn toxic species can be internalized by astrocytes and readily degraded by endo-lysosomal processing [217,218,219]; however, after extensive endocytosis of α-Syn aggregates, astrocytes may develop high amounts of intracellular deposits while the protein degradation capacity is reduced [217]. Since astrocytes do not express the native form of α-Syn [220], this suggests that the aggregated species are released from affected neurons and then endocytosed by astrocytes to limit the propagation of neuropathological events. It has been shown that aggregated species (oligomers and fibrils) exhibit a more pronounced accumulation in cell receptors with a compromised lysosomal activity, accompanied by accumulation of expanded lysosomes. Extracellular α-Syn plays a role in pathological conditions as it may cause cellular toxicity and dysfunctional synaptic transmission. It has also been suggested that α-Syn propagation from cell to cell plays a key role in the progression of synucleinopathies; however, the main mechanism regulating the extracellular levels of α-Syn has yet to be fully elucidated [221].
Conclusion
The structural integrity and functional stability of organelles constitute a sine qua non condition for adequate neuronal viability and ability to respond to adverse conditions. Mutated α-Syn associates with several cellular organelles while being trafficked in cells, causing toxicity by compromising cellular integrity and altering synaptic transmission, protein trafficking, and energy production. At the clinical level, and of relevance for therapeutic design, differences between synucleinopathies are also accompanied by distinct neuroanatomical localization of α-Syn deposits: while PD and LBD are characterized by α-Syn aggregates located in the neuronal soma and neurites [8], MSA displays α-Syn deposits in myelin produced by oligodendrocytes [222]. Given the major role α-Syn plays in the etiology of PD and other neurodegenerative disorders, future research should be directed at fully characterizing α-Syn function/dysfunction, which will be instrumental for the understanding of the molecular mechanisms underlying its neurotoxic profile and facilitate the design for novel therapies for synucleinopathies.
A final consideration for the evidence discussed in this review is Braak’s hypothesis, which suggests that a given environmental factor or pathogen is responsible for sporadic PD when it penetrates the body via the nasal cavity, reaching the gut and initiating LP from the digestive tract [223]. In turn, LP might be responsible for α-Syn aggregation in the CNS, reaching the SNpc via olfactory bulbs and the vagus nerve. While there is experimental and clinical evidence supporting Braak’s hypothesis, it is important to mention that not all PD patients adhere to Braak’s hypothesis since some do not develop LP in the vagal nerve or the ENS, though they develop LP in brain regions [224]. Whether or not the onset and development of PD and other synucleinopathies are subordinated to the pattern of LP expression, the pathogenic role of α-Syn mutation, misfolding, and aggregation remains a key factor for the understanding of these and other neurodegenerative disorders.
Availability of Data and Material
Not applicable.
Code Availability
Not applicable.
Abbreviations
- 6-OHDA:
-
6-Hydroxydopamine
- α-Syn:
-
Alpha-Synuclein
- AGEs:
-
Advanced glycation end products
- ATF:
-
Cyclic AMP-dependent transcription factor
- AVV:
-
Adeno-associated virus
- CMA:
-
Chaperone-mediated autophagy
- CNS:
-
Central nervous system
- CHMP2B:
-
Charged multivesicular body protein 2B
- CHOP:
-
C/EBP homologous protein
- CL:
-
Cardiolipin
- Cryo-EM:
-
Cryomicroscopy
- CSPα:
-
Cysteine string protein α
- DLB:
-
Dementia with Lewy bodies
- EMM:
-
External mitochondrial membrane
- ENS:
-
Enteric nervous system
- ERK:
-
Extracellular signal-regulated kinase
- GC:
-
Golgi complex
- HDAC6:
-
Histone deacetylase 6
- HNE:
-
4-Hydroxy-2-nonenal
- IF:
-
Intermediate filaments
- IMM:
-
Internal mitochondrial membrane
- LB:
-
Lewy bodies
- LC3:
-
Microtubule-associated protein 1A/1B-light chain 3
- LC3-II:
-
LC3-phosphatidylethanolamine conjugate
- LN:
-
Lewy neurites
- LP:
-
Lewy pathology
- LRRK2:
-
Leucine-rich kinase 2
- MPTP:
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- NF:
-
Neurofilaments
- NMR:
-
Nuclear magnetic resonance
- O-GlcNAc:
-
O-glycosyl-N-acetylation
- PBD:
-
Protein Data Bank
- PD:
-
Parkinson’s disease
- RAB1A:
-
Ras-related protein Rab-1A
- RER:
-
Rough endoplasmic reticulum
- ROS:
-
Reactive oxygen species
- SER:
-
Smooth endoplasmic reticulum
- SNpc:
-
Substantia nigra pars compacta
- SNAP-25:
-
Synaptosomal-associated protein 25
- SNARE:
-
Soluble N-ethylmaleimide sensitive factor-attachment protein receptor
- SUMO1:
-
Small ubiquitin-related modifier 1
- TFEB:
-
Transcription factor EB
- UPR:
-
Unfolded protein response
- UPS:
-
Ubiquitine-proteasome system
- VAMP2:
-
Vesicle associated membrane protein 2
References
Hatano T, Kubo S, Sato S, Hattori N (2009) Pathogenesis of familial Parkinson’s disease: new insights based on monogenic forms of Parkinson’s disease. J Neurochem 111(5):1075–1093. https://doi.org/10.1111/j.1471-4159.2009.06403.x
Chaudhuri KR, Healy DG, Schapira AHV (2006) Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol 5(3):235–245. https://doi.org/10.1016/S1474-4422(06)70373-8
Parkkinen L, Pirttilä T, Alafuzoff I (2008) Applicability of current staging/categorization of α-synuclein pathology and their clinical relevance. Acta Neuropathol 115(4):399–407. https://doi.org/10.1007/s00401-008-0346-6
Parkkinen L, Kauppinen T, Pirttilä T, Autere JM, Alafuzoff I (2005) α-Synuclein pathology does not predict extrapyramidal symptoms or dementia. Ann Neurol 57(1):82–91. https://doi.org/10.1002/ana.20321
Milber JM, Noorigian JV, Morley JF (2012) Lewy pathology is not the first sign of degeneration in vulnerable neurons in Parkinson disease. Neurology 79(24):2307–2314. https://doi.org/10.1212/WNL.0b013e318278fe32
Braak H, Del Tredici K, Rüb U, De Vos RAI, Jansen ENH, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24(2):197–211. https://doi.org/10.1016/s0197-4580(02)00065-9
Goedert M, Jakes R, Spillantini MG (2017) The synucleinopathies: twenty years on. J Parkinsons Dis 7(s1):S51–S69. https://doi.org/10.3233/JPD-179005
Spillantini MG, Schmidt ML, Lee VM-Y, Trojanowki JQ, Jakes R, Goedert M (1997) α-Synuclein in Lewy bodies. Nature 388(6645):839–840. https://doi.org/10.1038/42166
Bendor JT, Logan TP, Edwards RH (2013) The function of α-synuclein. Neuron 79(6):1044–1066. https://doi.org/10.1016/j.neuron.2013.09.004
Marui W, Iseki E, Nakai T, Miura S, Kato M, Uéda K, Kosaka K (2002) Progression and staging of Lewy pathology in brains from patients with dementia with Lewy bodies. J Neurol Sci 195(2):153–159. https://doi.org/10.1016/s0022-510x(02)00006-0
Jovanovic VM, Salti A, Tilleman H, Zega K, Jukic MM, Zou H, Friedel RH, Prakash N, Blaess S, Edenhofer F, Brodski C (2018) BMP/SMAD pathway promotes neurogenesis of midbrain dopaminergic neurons in vivo and in human induced pluripotent and neural stem cells. J Neurosci 38(7):1662–1676. https://doi.org/10.1523/JNEUROSCI.1540-17.2018
Obeso JA, Rodriguez-Oroz MC, Goetz CG, Marin C, Kordower JH, Rodriguez M, Hirsch EC, Farrer M, Schapira AHV, Halliday G (2010) Missing pieces in the Parkinson’s disease puzzle. Nat Med 16(6):653–661. https://doi.org/10.1038/nm.2165
Surmeier DJ, Obeso JA, Halliday GM (2017) Parkinson’s disease is not simply a prion disorder. J Neurosci 37(41):9799–9807. https://doi.org/10.1523/JNEUROSCI.1787-16.2017
Halliday GM, Holton JL, Revesz T, Dickson DW (2011) Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol 122(2):187–204. https://doi.org/10.1007/s00401-011-0852-9
Braak H, de Vos RAI, Bohl J, Del Tredici K (2006) Gastric α-synuclein immunoreactive inclusions in meissner’s and auerbach’s plexuses in cases staged for parkinson’s disease-related brain pathology. Neurosci Lett 396(1):67–72. https://doi.org/10.1016/j.neulet.2005.11.012
Wakabayashi K, Takahashi H, Ohama E, Ikuta F (1990) Parkinson’s disease: an immunohistochemical study of Lewy body-containing neurons in the enteric nervous system. Acta Neuropathol 79(6):581–583. https://doi.org/10.1007/BF00294234
Wakabayashi K, Takahashi H, Ohama E, Takeda S, Ikuta F (1993) Lewy bodies in the visceral autonomic nervous system in Parkinson’s disease. Adv Neurol 60: 609–612. https://pubmed.ncbi.nlm.nih.gov/8420198
Beach TG, Adler CH, Sue LI, Vedders L, Lue L, White CL III, Akiyama H, Caviness JN, Shill HA (2010) Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol 119:689–702. https://doi.org/10.1007/s00401-010-0664-3
Pfeiffer RF (2003) Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol 2(2):107–116. https://doi.org/10.1016/S1474-4422(03)00307-7
Shannon KM, Keshavarzian A, Mutlu EM, Dodiya HB, Daian D, Jaglin JA, Kordower JH (2012) Alpha-synuclein in colonic submucosa in early untreated Parkinson’s disease. Mov Disord 27(6):709–715. https://doi.org/10.1002/mds.23838
Fox SH, Katzenschlager R, Lim SY, Barton B, De Brie RMA, Seppi K, Coelho M, Sampaio C, MDSEBMC, (2018) International Parkinson and movement disorder society evidence-based medicine review: update on treatments for the motor symptoms of Parkinson’s disease. Mov Disord 33(8):1248–1266. https://doi.org/10.1002/mds.27372
Wolters Kluwer Clinical Drug Information (2019) Inc Lexicomp [On line]. Available: https://online.lexi.com [Last acces: 5–12–2019]
Halli-Tierney AD, Lufer J, Carroll DD (2020) Parkinson disease. Am Fam Physician 102(11):679–691
Homayoun H (2018) Parkinson disease. Ann Intern Med 169(5):33–48. https://doi.org/10.7326/AITC201809040
Okun MS (2017) Management of Parkinson disease in 2017: personalized approaches for patient-specific needs. JAMA 318(9):791–792. https://doi.org/10.1001/jama.2017.7914
Stocchi F, Hersh BP, Scott BL, Nausieda PA, Giorgi L, Ease PDMSI (2008) Ropinirole 24-hour prolonged release and ropinirole immediate release in early Parkinson’s disease: a randomized, double-blind, non-inferiority crossover study. Curr Med Res Opin 24:2883–2895. https://doi.org/10.1185/03007990802387130
Maroteaux L, Campanelli JT, Scheller RH (1988) Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci 8(8):2804–2815. https://doi.org/10.1523/JNEUROSCI.08-08-02804.1988
Choi BK, Choi MG, Kim JY, Yang Y, Lai Y, Kweon DH, Lee NK, Shin YK (2013) Large α-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc Natl Acad Sci USA 110(10):4087–4092. https://doi.org/10.1073/pnas.1218424110
Ghiglieri V, Calabrese V, Calabrese P (2018) Alpha-synuclein: from early synaptic dysfunction to neurodegeneration. Front Neurol 9:295. https://doi.org/10.3389/fneur.2018.00295
Thayanidhi N, Helm JR, Nycz DC, Bentley M, Liang Y, Hay JC (2010) α-synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol Biol Cell 21(11):1783–1908. https://doi.org/10.1091/mbc.e09-09-0801
Oaks AW, Marsh-Armstrong N, Jones JM, Credle JJ, Sidhu A (2013) Synucleins antagonize endoplasmic reticulum function to modulate dopamine transporter trafficking. PLoS ONE 8(8):e70872. https://doi.org/10.1371/journal.pone.0070872
Sulzer D, Edwards RH (2019) The physiological role of α-synuclein and its relationship to Parkinson’s disease. J Neurochem 150:475–486. https://doi.org/10.1111/jnc.14810
Alves da Costa C, Paitel E, Vincent B, Checler F (2002) α-Synuclein lowers p53-dependent apoptotic response of neuronal cells. J Biol Chem 277(52):50980–50984. https://doi.org/10.1074/jbc.M207825200
Ullman O, Fisher CK, Stultz CM (2011) Explaining the structural plasticity of α-synuclein. J Am Chem Soc 133(48):19536–19546. https://doi.org/10.1021/ja208657z
Menges S, Minakaki G, Schaefer PM, Meixner P, Prots I, Schlötzer U, Friedland K, Winner B, Outeiro TF, Winklhofer KF, von Arnim CAF, Xiang W, Winkler J, Klucken J (2017) Alpha-synuclein prevents the formation of spherical mitochondria and apoptosis under oxidative stress. Sci Rep 7(1):42942. https://doi.org/10.1038/srep42942
Faustini G, Marchesan E, Zonta L, Bono F, Bottani E, Longhena F, Ziviani E, Valerio A, Bellucci A (2019) Alpha-synuclein preserves mitochondrial fusion and function in neuronal cells oxidative medicine and cellular longevity. Oxid Med Cell Longev 2019:4246350. https://doi.org/10.1155/2019/4246350
Cartelli D, Aliverti A, Barbiroli A, Santambrogio C, Ragg EM, Casagrande FVM, Cantele F, Beltramone S, Marangon J, De Gregorio C, Pandini V, Emanuele M, Chieregatti E, Pieraccini S, Holmqvist S, Bubacco L, Roybon L, Pezzoli G, Grandori R et al (2016) α-Synuclein is a novel microtubule dynamase. Sci Rep 6(1):33289. https://doi.org/10.1038/srep33289
Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2002) Molecular biology of the cell. Garland Science, New York (ISBN-10: 0-8153-3218-1)
de Waegh SM, Lee VMY, Brady ST (1992) Local modulation of neurofilament phosphorylation, axonal caliber, and slow axonal transport by myelinating Schwann. Cell 68(3):451–463. https://doi.org/10.1016/0092-8674(92)90183-D
Lee HJ, Bae EJ, Lee SJ (2014) Extracellular α-synuclein-a novel and crucial factor in Lewy body diseases. Nat Rev Neurol 10(2):92–98. https://doi.org/10.1038/nrneurol.2013.275
Irwin DJ, Lee VM-Y, Trojanowski JQ (2013) Parkinson’s disease dementia: convergence of α-synuclein, tau and amyloid-β pathologies. Nat Rev Neurosci 14(9):626–636. https://doi.org/10.1038/nrn3549
Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, Schrag AE, Lang AE (2017) Parkinson disease. Nat Rev Dis Primers 3:17013. https://doi.org/10.1038/nrdp.2017.13
Kaushik S (2015) Cuervo AM (2015) Proteostasis and aging. Nat Med 21(12):1406–1415. https://doi.org/10.1038/nm.4001
Brundin P, Melki R (2017) Prying into the prion hypothesis for Parkinson’s disease. J Neurosci 37(41):9808–9818. https://doi.org/10.1523/JNEUROSCI.1788-16.2017
Chen X, Rohan de Silva HA, Pettenati MJ, Nagesh P, George PS, Roses AD, Xia Y, Horsburgh K, Uéda K, Saitoh T (1995) The human NACP/α-synuclein gene: chromosome assignment to 4q21.3–q22 and TaqI RFLP analysis. Genomics 26(2):425–427. https://doi.org/10.1016/0888-7543(95)80237-G
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 276(5321):2045–2047. https://doi.org/10.1126/science.276.5321.2045
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn K (2003) α-Synuclein locus triplication causes Parkinson’s disease. Science 302(5646):841. https://doi.org/10.1126/science.1090278
Rochet JC, Hay BA, Guo M (2012) Molecular insights into Parkinson’s disease. Prog Mol Biol Transl Sci 107:125–188. https://doi.org/10.1016/B978-0-12-385883-2.00011-4
Rospigliosi CC, McClendon S, Schmid AW, Ramlall TF, Barré P, Lashuel HA, Eliezer D (2009) E46K Parkinson’s-linked mutation enhances C-terminal-to-N-terminal contacts in α-synuclein. J Mol Biol 388(5):1022–1032. https://doi.org/10.1016/j.jmb.2009.03.065
Harada R, Kobayashi N, Kim J, Nakamura C, Han SW, Ikebukuro K, Sode K (2009) The effect of amino acid substitution in the imperfect repeat sequences of α-synuclein on fibrillation. Biochim Biophys Acta Mol Basis Dis 1792(10):998–1003. https://doi.org/10.1016/j.bbadis.2009.06.010
Anderson VL, Ramlall TF, Rospigliosi CC, Webb WW (2010) Eleizer D (2010) Identification of a helical intermediate in trifluoroethanol-induced alpha-synuclein aggregation. Proc Natl Acad Sci USA 107(44):18850–18855. https://doi.org/10.1073/pnas.1012336107
Appel S, Vilarino C, Encarnacion M, Sherman H, Yu I, Shah B, Weir D, Thompson C, Szu C, Trinh J, Aasly JO, Rajput A, Rajput AH, Stoessl AJ, Farrer MJ (2013) Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord 28(6):811–813. https://doi.org/10.1002/mds.25421
Lesage S, Anheim M, Letournel F, Bousset L, Honoré A, Rozas N, Pieri L, Madiona K, Dürr MR, Verny C, Brice A (2013) G51D α-synuclein mutation causes a novel parkinsonian–pyramidal syndrome. Ann Neurol 73:459–471. https://doi.org/10.1002/ana.23894
Proukakis C, Dudzik CG, Brier T, MacKay DS, Millhauser GL, Houlden F, Schapira AH (2013) A novel α-synuclein missense mutation in Parkinson disease. Neurology 80(11):1062–1064. https://doi.org/10.1212/WNL.0b013e31828727ba
Simón J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan C, Lichtner P, Scholz SW, Hernandez DG, Krüger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M et al (2019) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 41(12):1308–1312. https://doi.org/10.1038/ng.487
Chang D, Nalls M, Hallgrímsdóttir I, Hunkapiller J, van der Brug M, Cai F, Kerchner GA, Ayalon G, Bingol B, Sheng M, Hinds D, Behrens TW, Singleton AB, Bhangale TR, Graham RR (2017) A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 49(10):1511–1516. https://doi.org/10.1038/ng.3955
Jiang K, Rocha S, Westling A, Sriram KK, Dorfman KD, Wittung P, Westerlund F (2018) Alpha-synuclein modulates the physical properties of DNA. Chem Eur 24:15685–15690. https://doi.org/10.1002/chem.201803933
Ryu S, Baek I, Liew H (2019) Sumoylated α-synuclein translocates into the nucleus by karyopherin α6. Mol Cell Toxicol 15(1):103–109. https://doi.org/10.1007/s13273-019-0012-1
Pinho R, Paiva I, Jerčić KG, Fonseca L, Gerhardt E, Fahlbusch C, Garcia P, Kerimoglu C, Pavlou MA, Villar A, Szegő É, Lopes da Fonseca T, Fischle W, Schwamborn JC, Meyer T, Kügler S, Ferrer I, Attems J, Fischer A, Becker S, Zweckstetter M et al (2019) Nuclear localization and phosphorylation modulate pathological effects of alpha-synuclein. Hum Mol Genet 28:31–50. https://doi.org/10.1093/hmg/ddy326
Jakes R, Spillantini MG, Goedert M (1994) Identification of two distinct synucleins from human brain. FEBS Lett 345(1):27–32. https://doi.org/10.1016/0014-5793(94)00395-5
Bungeroth M, Appenzeller S, Regulin A, Völker W, Lorenzen I, Grötzinger J, Pendziwiat M, Kuhlenbäumer G (2014) Differential aggregation properties of alpha-synuclein isoforms. Neurobiol Aging 35(8):1913–1919. https://doi.org/10.1016/j.neurobiolaging.2014.02.009
Gantiva M (2019) Obtención y caracterización parcial de un mutante de la proteína recombinante α-sinucleína, y producción de su anticuerpo policlonal. Tesis, Universidad Nacional de Colombia Facultad Medicina Instituto de Genética. https://repositorio.unal.edu.co/handle/unal/75758
Trexler AJ, Rhoades E (2012) N-terminal acetylation is critical for forming α-helical oligomer of α-synuclein. Protein Sci 21(5):601–605. https://doi.org/10.1002/pro.2056
Dikiy I, Eliezer D (2014) N-terminal acetylation stabilizes N-terminal helicity in lipid and micelle-bound. α-Synuclein and increases its affinity for physiological membranes. J Biol Chem 289(6):3652–3665. https://doi.org/10.1074/jbc.M113.512459
Mor DE, Daniels MJ, Ischiropoulos H (2019) The usual suspects, dopamine and alpha-synuclein, conspire to cause neurodegeneration. Mov Disord 34:167–179. https://doi.org/10.1002/mds.27607
Lashuel HA, Overk CR, Oueslati A, Masliah E (2013) The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci 14(1):38–48. https://doi.org/10.1038/nrn3406
Ulmer TS, Bax A, Cole NB, Nussbaum RL (2005) Structure and dynamics of micelle-bound human α-synuclein. J Biol Chem 280(10):9595–9603. https://doi.org/10.1074/jbc.M411805200
Yu H, Han W, Ma W, Schulten K (2015) Transient β-hairpin formation in α-synuclein monomer revealed by coarse-grained molecular dynamics simulation. J Chem Phys 143(24):243142. https://doi.org/10.1063/1.4936910
Flagmeier P, Meisl G, Vendruscolo M, Knowles TPJ, Dobson CH, Buell AK, Galvagnion C (2016) Mutations associated with familial Parkinson’s disease alter the initiation and amplification steps of α-synuclein aggregation. Proc Natl Acad Sci USA 113(37):10328–10333. https://doi.org/10.1073/pnas.1604645113
Rodriguez JA, Ivanova MI, Sawaya MR, Cascio D, Reyes FE, Shi D, Sangwan S, Guenther EL, Johnson LM, Zhang M, Jiang L, Arbing MA, Nanneng BL, Hattne J, Whitelegge J, Brewster AS, Messerschmidt M, Boutet S, Sauter NK, Gonen T, Eisenberg DS (2015) Structure of the toxic core of α-synuclein from invisible crystals. Nature 525(7570):486–490. https://doi.org/10.1038/nature15368
Tuttle MD, Comellas G, Nieuwkoop AJ, Covell DJ, Berthold DA, Kloepper KD, Courtney JM, Kim JK, Barclay AM, Kendall A, Wan W, Stubbs G, Schwieters CD, Lee VMY, George JM, Rienstra CM (2016) Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat Struct Mol Biol 23(5):409–415. https://doi.org/10.1038/nsmb.3194
Guerrero-Ferreira R, Taylor NMI, Mona D, Ringler P, Lauer ME, Riek R, Britschgi M, Stahlberg H (2018) Cryo-EM structure of alpha-synuclein fibrils. Elife 7:e36402. https://doi.org/10.7554/eLife.36402
Guerrero-Ferreira R, Taylor NMI, Arteni AA, Kumari P, Mona D, Ringler P, Britschgi M, Lauer ME, Makky A, Verasdonck J, Riek R, Melkyi R, Meier BH, Böckmann A, Bousset L, Stahlberg H (2019) Two new polymorphic structures of human full-length alpha-synuclein fibrils solved by cryo-electron microscopy. Elife 8:e48907. https://doi.org/10.7554/eLife.48907
Fauvet B, Fares MB, Samuel F, Dikiy I, Tandon A, Eliezer D, Lashuel HA (2012) Characterization of semisynthetic and naturally Nα-acetylated α-synuclein in vitro and in intact cells: implications for aggregation and cellular properties of α-synuclein. J Biol Chem 287(34):28243–28262. https://doi.org/10.1074/jbc.M112.383711
Ingelsson M (2016) Alpha-synuclein oligomers—neurotoxic molecules in Parkinson’s disease and other lewy body disorders. Front Neurosci 10:408. https://doi.org/10.3389/fnins.2016.00408
Shults CW (2006) Lewy bodies. Proc Natl Acad Sci USA 103(6):1661–1668. https://doi.org/10.1073/pnas.0509567103
Theillet FX, Binolfi A, Bekei B, Martorana A, May H, Stuiver M, Verzini S, Lorenz D, van Rossum M, Goldfarb D (2016) Selenko P (2016) Structural disorder of monomeric alpha-synuclein persists in mammalian cells. Nature 530(7588):45–50. https://doi.org/10.1038/nature16531
Medeiros AT, Soll LG, Tessari I, Bubacco L, Morgan JR (2017) α-Synuclein fission during clathrin-mediated synaptic vesicle recycling. Front Cell Neurosci 11:388. https://doi.org/10.3389/fncel.2017.00388
Wang W, Nguyen LTT, Burlak C, Chegini F, Guo F, Chataway T, Ju S, Fisher OS, Miller DW, Datta D, Wu F, Wu CX, Landeru A, Wells JA, Cookson MR, Boxer MB, Thomas CJ, Ping W, Ringe D, Petsko GA, Hoang QQ (2016) Caspase-1 causes truncation and aggregation of the Parkinson’s disease-associated protein α-synuclein. Proc Natl Acad Sci USA 113(34):9587–9592. https://doi.org/10.1073/pnas.1610099113
Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T, Lansbury PT Jr (2002) α-Synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol 322(5):1089–1102. https://doi.org/10.1016/S0022-2836(02)00735-0
Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M (1998) Filamentous α-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett 251(3):205–208. https://doi.org/10.1016/S0304-3940(98)00504-7
Vitte J, Traver S, De Paula AM, Lesage S, Rovelli G, Corti O, Duyckaerts DA (2010) Leucine-rich repeat kinase 2 is associated with the endoplasmic reticulum in dopaminergic neurons and accumulates in the core of Lewy bodies in Parkinson disease. J Neuropathol Exp Neurol 69(9):959–972. https://doi.org/10.1097/NEN.0b013e3181efc01c
Miki Y, Mori F, Tanji K, Kakita A, Takahashi H, Wakabayashi K (2011) Accumulation of histone deacetylase 6, an aggresome-related protein, is specific to Lewy bodies and glial cytoplasmic inclusions. Neuropathology 31:561–568. https://doi.org/10.1111/j.1440-1789.2011.01200.x
Tanikawa S, Mori F, Tanji K, Kakita A, Takahashi H (2012) Wakabayashi K (2012) Endosomal sorting related protein CHMP2B is localized in Lewy bodies and glial cytoplasmic inclusions in α-synucleinopathy. Neurosci Lett 527(1):16–21. https://doi.org/10.1016/j.neulet.2012.08.035
Forno LS (1996) Neuropathology of Parkinson’s disease. J Neuropathol Exp Neurol 55(3):259–272. https://doi.org/10.1097/00005072-199603000-00001
Gómez-Tortosa E, Newell K, Irizarry MC, Sanders JL, Hyman BT (2000) α-synuclein immunoreactivity in dementia with Lewy bodies: morphological staging and comparison with ubiquitin immunostaining. Acta Neuropathol 99(4):352–357. https://doi.org/10.1007/s004010051135
Kanazawa T, Adachi E, Orimo S, Nakamura A, Mizusawa H, Uchihara T (2012) Pale neurites, premature α-synuclein aggregates with centripetal extension from axon collaterals. Brain Pathol 22(1):67–78. https://doi.org/10.1111/j.1750-3639.2011.00509.x
Anderson JP, Walker DE, Goldstein JM, De Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M, Diep L, Keim P, Shen X, Chataway T, Schlossmacher MG, Seubert P, Schenk D, Sinha S, Ping W, Chilcote TJ (2006) Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic Lewy body disease. J Biol Chem 281(40):29739–29752. https://doi.org/10.1074/jbc.M600933200
Paleologou KE, Oueslati A, Shakked G, Rospigliosi CC, Kim HY, Lamberto GR, Fernandez CO, Schmid A, Chegini F, Ping W, Chiappe D, Moniatte M, Schneider BL, Aebischer P, Eliezer D, Zweckstetter M, Masliah E, Lashuel HA (2010) Phosphorylation at S87 is enhanced in synucleinopathies, inhibits α-synuclein oligomerization, and influences synuclein-membrane interactions. Neurosci 30(9):3184–3198. https://doi.org/10.1523/JNEUROSCI.5922-09.2010
Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T (2002) α-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 4(2):160–164. https://doi.org/10.1038/ncb748
Chen L, Feany MB (2005) α-Synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci 8(5):657–663. https://doi.org/10.1038/nn1443
Buchberger A, Bukau B, Sommer T (2010) Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell 40(2):238–252. https://doi.org/10.1016/j.molcel.2010.10.001
Shibata Y, Voeltz GK, Rapoport TA (2006) Rough sheets and smooth tubules. Cell 126(3):435–439. https://doi.org/10.1016/j.cell.2006.07.019
Berridge MJ (2002) The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32(5–6):235–249. https://doi.org/10.1016/S0143416002001823
Sweeney P, Park H, Baumann M, Dunlop J, Frydman F, Kopito R, McCampbell A, Leblanc G, Venkateswaran A, Nurmi A, Hodgson R (2017) Protein misfolding in neurodegenerative diseases: implications and strategies. Transl Neurodegener 6:6. https://doi.org/10.1186/s40035-017-0077-5
Lucero HA, Kaminer B (1999) The role of calcium on the activity of ERcalcistorin/protein-disulfide isomerase and the significance of the C-terminal and its calcium binding. A comparison with mammalian protein-disulfide isomerase. J Biol Chem 274(4):3243–3251. https://doi.org/10.1074/jbc.274.5.3243
Ma Y, Hendershot LM (2004) ER chaperone functions during normal and stress conditions. J Chem Neuroanatomy 28(1–2):51–65. https://doi.org/10.1016/j.jchemneu.2003.08.007
Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill K, Bhull B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, Lindquist S (2006) Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 313(5785):324–328. https://doi.org/10.1126/science.1129462
Colla E, Coune P, Liu Y, Pletnikova O, Troncoso JC, Iwatsubo T, Schneider BL, Lee MK (2012) Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J Neurosci 32:3306–3320. https://doi.org/10.1523/JNEUROSCI.5367-11.2012
Mironov AA, Beznoussenko GV (2011) Molecular mechanisms responsible for formation of Golgi ribbon. Histol Histopathol 26:117–133. https://doi.org/10.14670/HH-26.117
Gonatas NK, Stieber A, Gonatas JO (2006) Fragmentation of the Golgi apparatus in neurodegenerative diseases and cell death. J Neurol Sci 246:21–30. https://doi.org/10.1016/j.jns.2006.01.019
Fan J, Hu Z, Zeng L, Lu W, Tang X, Zhang J, Li T (2008) Golgi apparatus and neurodegenerative diseases. Int J Dev Neurosci 26:523–534. https://doi.org/10.1016/j.ijdevneu.2008.05.006
Martínez JA, Tomás M, Martínez N, Martínez E (2019) Golgi Fragmentation in neurodegenerative diseases: is there a common cause? Cells 8:748. https://doi.org/10.3390/cells8070748
Haase G, Rabouille C (2015) Golgi fragmentation in ALS motor neurons. New mechanisms targeting microtubules, tethers, and transport vesicles. Front Neurosci 9:448. https://doi.org/10.3389/fnins.2015.00448
Tomás M, Martínez-Alonso E, Martínez-Martínez N, Cara-Esteban M, Martínez-Menarguéz JA (2021) Fragmentation of the Golgi complex of dopaminergic neurons in human substantia nigra: New cytopathological findings in Parkinson’s disease. Histol Histopathol 36(1):47–60. https://doi.org/10.14670/HH-18-270
Xu H, Ren D (2015) Lysosomal physiology. Annu Rev Physiol 77:57–80. https://doi.org/10.1146/annurev-physiol-021014-071649
Dice JF, Terlecky SR, Chiang HL, Olson TS, Isenman LD, Short SR, Freundlieb S, Terlecky LJ (1990) A selective pathway for degradation of cytosolic proteins by lysosomes. Semin Cell Biol 1:449–455
Kaushik S, Cuervo AM (2018) The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol 19:365–381. https://doi.org/10.1038/s41580-018-0001-6
Ciechanover A, Kwon YT (2015) Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med 47:e147. https://doi.org/10.1038/emm.2014.117
Zondler L, Kostka M, Garidel P, Heinzelmann U, Hengerer B, Mayer B, Hengere JH, Gillardon F, Danzer KM (2017) Proteasome impairment by alpha-synuclein. PLoS ONE 12:e0184040. https://doi.org/10.1371/journal.pone.0184040
McKinnon C, De Snoo ML, Gondard E, Neudorfer C, Chau H, Ngana SG, O’Hara DM, Brotchie JM, Koprich JB, Lozano AM, Kalia LV, Kalia SK (2020) Early-onset impairment of the ubiquitin-proteasome system in dopaminergic neurons caused by α-synuclein. Acta Neuropathol Commun 8:17. https://doi.org/10.1186/s40478-020-0894-0
Ren H, Zhai W, Lu X, Wang G (2021) The cross-links of endoplasmic reticulum stress, autophagy, and neurodegeneration in Parkinson’s disease. Front Aging Neurosci 13(691881):1–18. https://doi.org/10.3389/fnagi.2021.691881
Hou X, Watzlawik JO, Fiesel FC, Springer W (2020) Autophagy in Parkinson’s disease. J Mol Biol 432:2651–2672. https://doi.org/10.1016/j.jmb.2020.01.037
Yu WH (2009) DoradoB, Figueroa HY, Wang L, Planel E, Cookson MR, Clark LN, Duff KE (2009) Metabolic activity determines efficacy of macroautophagic clearance of pathological oligomeric alpha-synuclein. Am J Pathol 175(2):736–747. https://doi.org/10.2353/ajpath.2009.080928
Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Bjorklund A (2013) TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc Natl Acad Sci USA 110(19):E1817–E1826. https://doi.org/10.1073/pnas.1305623110
Winslow AR, Chen CW, Corrochano S, Acevedo A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, Brown S, O’Kane CJ, Rubinsztein DC (2010) Alpha-synuclein impairs macroautophagy: implications for Parkinson’s disease. J Cell Biol 190(6):1023–1037. https://doi.org/10.1083/jcb.201003122
Gitler AD, Bevis BJ, Shorter J, Strathearn KE, Hamamichi S, Su LJ, Caldwell KA, Caldwell GA, Rochet JC, McCaffery M, Barlowe C, Lindquist S (2008) The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc Natl Acad Sci USA 105(1):145–150. https://doi.org/10.1073/pnas.0710685105
Coune PG, Schneider BL, Aebischer P (2012) Parkinson’s disease: gene therapies. Cold Spring Harb Perspect Med 2(4):a009431. https://doi.org/10.1101/cshperspect.a009431
Jiang TF, Zhang YJ, Zhou HY, Wang HM, Tian LP, Liu J, Ding JQ, Chen SD (2013) Curcumin ameliorates the neurodegenerative pathology in A53T alpha-synuclein cell model of Parkinson’s disease through the downregulation of mTOR/p70S6K signaling and the recovery of macroautophagy. J Neuroimmune Pharmacol 8(1):356–369. https://doi.org/10.1007/s11481-012-9431-7
Gao S, Duan C, Gao G, Wang X, Yang H (2015) Alpha-synuclein overexpression negatively regulates insulin receptor substrate 1 by activating mTORC1/S6K1 signaling. Int J Biochem Cell Biol 64:25–33. https://doi.org/10.1016/j.biocel.2015.03.006
Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA (2001) Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci 21(24):9549–9560. https://doi.org/10.1523/JNEUROSCI.21-24-09549.2001
Grassi D, Howard S, Zhou M, Diaz N, Urban NT, Guerrero D, Kamasawa N, Volpicelli LA, LoGrasso P, Lasmézas CI (2018) Identification of a highly neurotoxic alpha-synuclein species inducing mitochondrial damage and mitophagy in Parkinson’s disease. Proc Natl Acad Sci USA 115(11):E2634–E2643. https://doi.org/10.1073/pnas.1713849115
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D (2004) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305(5688):1292–1295. https://doi.org/10.1126/science.1101738
Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L (2009) Aberrant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS ONE 4(5):e5515. https://doi.org/10.1371/journal.pone.0005515
Malkus KA, Ischiropoulos H (2012) Regional deficiencies in chaperone-mediated autophagy underlie alpha-synuclein aggregation and neurodegeneration. Neurobiol Dis 46(3):732–744. https://doi.org/10.1016/j.nbd.2012.03.017
Martinez M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV, Hodara R, Fredenburg R, Wu DC, Follenzi A, Dauer W, Przedborski S, Ischiropoulos H, Lansbury PT, Sulzer D, Cuervo AM (2008) Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest 118(2):777–788. https://doi.org/10.1172/JCI32806
Goldberg AL (2003) Protein degradation and protection against misfolded or damaged proteins. Nature 426:895–899. https://doi.org/10.1038/nature02263
McNaught KS, Belizaire R, Isacson O, Jenner P, Olanow CW (2003) Altered proteasomal function in sporadic Parkinson’s disease. Exp Neurol 179:38–46. https://doi.org/10.1006/exnr.2002.8050
McNaught KS, Perl DP, Brownell AL, Olanow CW (2004) Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson’s disease. Ann Neurol 56:149–162. https://doi.org/10.1002/ana.20186
Wang XF, Li S, Chou AP, Bronstein JM (2006) Inhibitory effects of pesticides on proteasome activity: implication in Parkinson’s disease. Neurobiol Dis 23:198–205. https://doi.org/10.1016/j.nbd.2006.02.012
Esteves AR, Arduino DM, Swerdlow RH, Oliveira CR, Cardoso SM (2010) Microtubule depolymerization potentiates alpha-synuclein oligomerization. Front Aging Neurosci 1:5. https://doi.org/10.3389/neuro.24.005.2009
Liu W, Vives-Bauza C, Acin-Perez R, Yamamoto A, Tan Y, Li Y, Magrane J, Stavarache MA, Shaffer S, Chang S, Kaplitt MG, Huang XY, Beal MF, Manfredi G, Li C (2009) PINK1 defect causes mitochondrial dysfunction, proteasomal deficit and alpha-synuclein aggregation in cell culture models of Parkinson’s disease. PLoS ONE 4:e4597. https://doi.org/10.1371/journal.pone.0004597
Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ (2001) Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson’s disease. Science 293:263–269. https://doi.org/10.1126/science.1060627
Calì T, Ottolini D, Brini M (2011) Mitochondria, calcium, and endoplasmic reticulum stress in Parkinson’s disease. BioFactors 37(3):228–240. https://doi.org/10.1002/biof.159
Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC (2011) Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet 20:1726–1737. https://doi.org/10.1093/hmg/ddr048
Deas E, Plun-Favreau H, Wood NW (2009) PINK1 function in health and disease. EMBO Mol Med 1:152–165. https://doi.org/10.1002/emmm.200900024
Credle J, Forcelli PA, Delannoy M, Oaks AW, Permaul E, Berry DL, Duka V, Wills J, Sidhu A (2015) α-Synuclein-mediated inhibition of ATF6 processing into COPII vesicles disrupts UPR signaling in Parkinson’s disease. Neurobiol Dis 76:112–125. https://doi.org/10.1016/j.nbd.2015.02.005
Luna E, Luk KC (2015) Bent out of shape: α-Synuclein misfolding and the convergence of pathogenic pathways in Parkinson’s disease. FEBS Lett 589(24):3749–3759. https://doi.org/10.1016/j.febslet.2015.10.023
Sato H, Kato T, Arawaka S (2013) The role of Ser129 phosphorylation of α-synuclein in neurodegeneration of Parkinson’s disease: a review of in vivo models. Rev Neurosci 24:115–123. https://doi.org/10.1515/revneuro-2012-0071
Kang L, Moriarty GM, Woods LA, Ashcroft AE, Radford SE, Baum J (2012) N-terminal acetylation of alpha-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Sci 21:911–917. https://doi.org/10.1002/pro.2088
Bartels T, Kim NC, Luth ES, Selkoe D (2014) N-alpha-acetylation of α-synuclein increases its helical folding propensity, GM1 binding specificity and resistance to aggregation. PLoS ONE 9:e103727. https://doi.org/10.1371/journal.pone.0103727
Krumova P, Meulmeester E, Garrido M, Tirard M, Hsiao HH, Bossis G, Urlaub H, Zweckstetter M, Kügler S, Melchior F, Bähr M, Weishaupt JH (2011) Sumoylation inhibits α-synuclein aggregation and toxicity. J Cell Biol 194:49–60. https://doi.org/10.1083/jcb.201010117
Shahpasandzadeh H, Popova B, Kleinknecht A, Fraser PE, Outeiro TF, Braus GH (2014) Interplay between sumoylation and phosphorylation for protection against α-synuclein inclusions. J Biol Chem 289:31224–31240. https://doi.org/10.1074/jbc.M114.559237
Münch G, Lüth HJ, Wong A, Arendt T, Hirsch E, Ravid R, Riederer P (2000) Crosslinking of α-synuclein by advanced glycation endproducts—an early pathophysiological step in Lewy body formation? J Chem Neuroanat 20:253–257. https://doi.org/10.1016/s0891-0618(00)00096-x
Miranda HV, Szegő EM, Oliveira LMA, Breda C, Darendelioglu E, Oliveira RM, Ferreira DG, Gomes MA, Rott R, Oliveira M, Munari F, Enguita FJ, Simoes T, Rodrigues EF, Heinrich M, Martins IC, Zamolo I, Riess O, Cordeiro C, Ponces A et al (2017) Glycation potentiates α-synuclein-associated neurodegeneration in synucleinopathies. Brain 140:1399–1419. https://doi.org/10.1093/brain/awx056
Shaikh S, Nicholson LF (2008) Advanced glycation end products induce in vitro cross-linking of α-synuclein and accelerate the process of intracellular inclusion body formation. J Neurosci Res 86:2071–2082. https://doi.org/10.1002/jnr.21644
Chen L, Wei Y, Wang X, He R (2010) Ribosylation rapidly induces α-synuclein to form highly cytotoxic molten globules of advanced glycation end products. PLoS ONE 5:e9052. https://doi.org/10.1371/journal.pone.0009052
Levine PM, Galesic A, Balana AT, Mahul AL, Navarro MX, De Leon CA, Lashue HA, Pratt MR (2019) α-Synuclein O-GlcNAcylation alters aggregation and toxicity, revealing certain residues as potential inhibitors of Parkinson’s disease. Proc Natl Acad Sci USA 116:1511–1519. https://doi.org/10.1073/pnas.1808845116
Zhang J, Li X, Li J-D (2019) The roles of post-translational modifications on α-synuclein in the pathogenesis of Parkinson’s diseases. Front Neurosci 13:381. https://doi.org/10.3389/fnins.2019.00381
Giasson BI, Duda JE, Murray IVJ, Chen Q, Souza JM, Hurtig HI (2000) Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science 290:985–989. https://doi.org/10.1126/science.290.5493.985
Sevcsik E, Trexler AJ, Dunn JM, Rhoades E (2011) Allostery in a disordered protein: oxidative modifications to α-synuclein act distally to regulate membrane binding. J Am Chem Soc 133:7152–7158. https://doi.org/10.1021/ja2009554
Burai R, Ait-Bouziad N, Chiki A, Lashuel H (2015) Elucidating the role of site-specific nitration of α-synuclein in the pathogenesis of Parkinson’s disease via protein semisynthesis and mutagenesis. J Am Chem Soc 137:5041–5052. https://doi.org/10.1021/ja5131726
Danielson SR, Held JM, Schilling B, Oo M, Gibson BW, Andersen JK (2009) Preferentially increased nitration of α-synuclein at tyrosine-39 in a cellular oxidative model of Parkinson’s disease. Anal Chem 81:7823–7828. https://doi.org/10.1021/ac901176t
Fernández E, García-Moreno J-M, de Pablos AM, Chacón J (2013) May the evaluation of nitrosative stress through selective increase of 3-nitrotyrosine proteins other than nitroalbumin and dominant tyrosine-125/136 nitrosylation of serum α-synuclein serve for diagnosis of sporadic Parkinson’s disease? Antioxid Redox Signal 19:912–918. https://doi.org/10.1089/ars.2013.5250
Hodara R, Norris EH, Giasson BI, Mishizen AJ, Lynch DR, Lee VMY, Ischiropoulos H (2004) Functional consequences of α-synuclein tyrosine nitration: diminished binding to lipid vesicles and increased fibril formation. J Biol Chem 279:47746–47753. https://doi.org/10.1074/jbc.M408906200
Schildknecht S, Gerding HR, Karreman C et al (2013) Oxidative and nitrative alpha-synuclein modifications and proteostatic stress: implications for disease mechanisms and interventions in synucleinopathies. J Neurochem 125:491–511. https://doi.org/10.1111/jnc.12226
Xiang W, Schlachetzki JCM, Helling S et al (2013) Oxidative stress-induced posttranslational modifications of alpha-synuclein: specific modification of alpha-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol Cell Neurosci 54:71–83. https://doi.org/10.1016/j.mcn.2013.01.004
Fink AL (2006) The aggregation and fibrillation of α-synuclein. Acc Chem Res 39:628–634. https://doi.org/10.1021/ar050073t
Dudek J (2017) Role of cardiolipin in mitochondrial signaling pathways. Front Cell Dev Biol 5:90. https://doi.org/10.3389/fcell.2017.00090
Ghio S, Camilleri A, Caruana M, Ruf VC, Schmidt F, Leonov A, Ryazanov S, Griesinger C, Cauchi RJ, Kamp F, Giese A, Vassallo N (2019) Cardiolipin promotes pore-forming activity of alpha-synuclein oligomers in mitochondrial membranes. ACS Chem Neurosci 10:3815–3829. https://doi.org/10.1021/acschemneuro.9b00320
Robotta M, Gerding HR, Vogel A, Hauser K, Schildknecht S, Karreman C, Leist M, Subramaniam V, Drescher M (2014) Alpha-synuclein binds to the inner membrane of mitochondria in an α-helical conformation. Chem Bio Chem 15:2499–2502. https://doi.org/10.1002/cbic.201402281
Bayır H, Kapralov AA, Jiang J, Huang Z, Tyurina YY, Tyurin VA, Zhao Q, Belikova NA, Vlasova II, Maeda A, Zhu J, Na HM, Mastroberardino PG, Sparvero LJ, Amoscato AA, Chu CT, Greenamyre JT, Kagan VE (2009) Peroxidase mechanism of lipid-dependent cross-linking of synuclein with cytochrome C. J Biol Chem 284:15951–15969. https://doi.org/10.1074/jbc.M900418200
Beyer K, Nuscher B (1996) Specific cardiolipin binding interferes with labeling of sulfhydryl residues in the adenosine diphosphate/adenosine triphosphate carrier protein from beef heart mitochondria. Biochemistry 35:15784–15790. https://doi.org/10.1021/bi9610055
Claypool SM, Oktay Y, Boontheung P, Loo JA, Koehler CM (2008) Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J Cell Biol 182:937–950. https://doi.org/10.1083/jcb.200801152
Muñoz-Lasso DC, Romá-Mateo C, Pallardó FV, Gonzalez P (2020) Much more than a scaffold: cytoskeletal proteins in neurological disorders. Cells 9:358. https://doi.org/10.3390/cells9020358
Petzold A (2005) Neurofilament phosphoforms: surrogate markers for axonal injury, degeneration and loss. J Neurol Sci 233:183–198. https://doi.org/10.1016/j.jns.2005.03.015
Sakaguchi T, Okada M, Kitamura T, Kawasaki K (1993) Reduced diameter and conduction velocity of myelinated fibers in the sciatic nerve of a neurofilament-deficient mutant quail. Neurosci Lett 153:65–68. https://doi.org/10.1016/0304-3940(93)90078-y
Dong DLY, Xu ZS, Hart GW, Cleveland DW (1996) Cytoplasmic O-GlcNAc modification of the head domain and the KSP repeat motif of the neurofilament protein neurofilament-H. J Biol Chem 271:20845–20852. https://doi.org/10.1074/jbc.271.34.20845
Villalón E, Barry DM, Byers N, Frizzi K, Jones MR, Landayan DS, Dale JM, Downer NL, Calcutt NA, García ML (2018) Internode length is reduced during myelination and remyelination by neurofilament medium phosphorylation in motor axons. Exp Neurol 306:158–168. https://doi.org/10.1016/j.expneurol.2018.05.009
Sihag RK, Inagaki M, Yamaguchi T, Shea TB, Pant HC (2007) Role of phosphorylation on the structural dynamics and function of types III and IV intermediate filaments. Exp Cell Res 313:2098–2109. https://doi.org/10.1016/j.yexcr.2007.04.010
Bridel C, van Wieringen WN, Zetterberg H, Tijms BM, Teunissen CE (2019) Diagnostic value of cerebrospinal fluid neurofilament light protein in neurology: a systematic review and meta-analysis. JAMA Neurol 76:1035–1048. https://doi.org/10.1001/jamaneurol.2019.1534
Melkov A, Abdu U (2018) Regulation of long-distance transport of mitochondria along microtubules. Cell Mol Life Sci 75:163–176. https://doi.org/10.1007/s00018-017-2590-1
Khaitlina SY (2014) Intracellular transport based on actin polymerization. Biochemistry Moscow 79:917–927. https://doi.org/10.1134/S0006297914090089
Gumy LF, Katrukha EA, Kapitein LC, Hoogenraad CC (2014) New insights into mRNA trafficking in axons. Dev Neurobiol 74:233–244. https://doi.org/10.1002/dneu.22121
Lee HJ, Lee SJ (2002) Characterization of cytoplasmic alpha-synuclein aggregates. Fibril formation is tightly linked to the inclusion-forming process in cells. J Biol Chem 277:48976–48983. https://doi.org/10.1074/jbc.M208192200
Carnwath T, Mohammed R, Tsiang D (2018) The direct and indirect effects of α-synuclein on microtubule stability in the pathogenesis of Parkinson’s disease. Neuropsychiatr Dis Treat 14:1685–1695. https://doi.org/10.2147/NDT.S166322
Prots I, Grosch J, Brazdis RM, Simmnacher K, Veber V, Havlicek S, Hannappel C, Krach F, Krumbiegel M, Schütz O, Reis A, Wrasidlo W, Galasko DR, Groemer TW, Masliah E, Schlötzer U, Xiang W, Winkler J, Winner B (2018) α-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc Natl Acad Sci USA 115:7813–7818. https://doi.org/10.1073/pnas.1713129115
Qiang L, Yu W, Andreadis A, Luo M, Baas PW (2006) Tau protects microtubules in the axon from severing by katanin. J Neurosci 26:3120–3129. https://doi.org/10.1523/JNEUROSCI.5392-05.2006
Rust MB, Maritzen T (2015) Relevance of presynaptic actin dynamics for synapse function and mouse behavior. Exp Cell Res 335:165–171. https://doi.org/10.1016/j.yexcr.2014.12.020
Esposito A, Dohm CP, Kermer P, Bähr M, Wouters FS (2007) α-Synuclein and its disease-related mutants interact differentially with the microtubule protein tau and associate with the actin cytoskeleton. Neurobiol Dis 26:521–531. https://doi.org/10.1016/j.nbd.2007.01.014
Zhang W, Benson DL (2001) Stages of synapse development defined by dependence on F-actin. J Neurosci 21:5169–5181. https://doi.org/10.1523/jneurosci.21-14-05169.2001
Minamide LS, Striegl AM, Boyle JA, Meberg PJ, Bamburg JR (2000) Neurodegenerative stimuli induce persistent ADF/cofilin-actin-rods that disrupt distal neurite function. Nat Cell Biol 2:628–636. https://doi.org/10.1038/35023579
Munsie LN, Truant R (2012) The role of the cofilin-actin rod stress response in neurodegenerative diseases uncovers potential new drug targets. BioArchitecture 2:204–208. https://doi.org/10.4161/bioa.22549
Sousa VL, Bellani S, Giannandrea M, Yousuf M, Valtorta F, Meldolesi J, Chieregatti E (2009) α-synuclein and its A30P mutant affect actin cytoskeletal structure and dynamics. Mol Biol Cell 20:3619–3771. https://doi.org/10.1091/mbc.e08-03-0302
Eira J, Santos CS, Mendes M, Almeida M (2016) The cytoskeleton as a novel therapeutic target for old neurodegenerative disorders. Prog Neurobiol 141:61–82. https://doi.org/10.1016/j.pneurobio.2016.04.007
Kaeser PS, Regehr WG (2017) The readily releasable pool of synaptic vesicles. Curr Opin Neurobiol 43:63–70. https://doi.org/10.1016/j.conb.2016.12.012
Stöckl MT, Zijlstra N, Subramaniam V (2013) α-synuclein oligomers: an amyloid pore? Insights into mechanisms of α-synuclein oligomer-lipid interactions. Mol Neurobiol 47(2):613–621. https://doi.org/10.1007/s12035-012-8331-4
van Rooijen BD, Claessens MMAE, Subramaniam V (2009) Lipid bilayer disruption by oligomeric α-synuclein depends on bilayer charge and accessibility of the hydrophobic core. Biochim Biophys Acta Biomembr 1788:1271–1278. https://doi.org/10.1016/j.bbamem.2009.03.010
Hannestad JK, Rocha S, Agnarsson B, Zhdanov VP, Wittung P, Höök F (2020) Single-vesicle imaging reveals lipid-selective and stepwise membrane disruption by monomeric α-synuclein. Proc Natl Acad Sci USA 25:14178–14186. https://doi.org/10.1073/pnas.1914670117
Fusco G, Chen SW, Williamson PTF, Cascella R, Perni M, Jarvis JA, Cecchi C, Vendruscolo M, Chiti F, Cremades N, Ying L, Dobson CM, De Simone A (2017) Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science 358:1440–1443. https://doi.org/10.1126/science.aan6160
Bussell R Jr, Eliezer D (2003) A structural and functional role for 11-mer repeats in α-synuclein and other exchangeable lipid binding proteins. J Mol Biol 329:763–778. https://doi.org/10.1016/S0022-2836(03)00520-5
Bartels T, Ahlstrom LS, Leftin A, Kamp F, Haass C, Brown MF, Beyer K (2010) The N-terminus of the intrinsically disordered protein α-synuclein triggers membrane binding and helix folding. Biophys J 99:2116–2124. https://doi.org/10.1016/j.bpj.2010.06.035
Fusco G, De Simone A, Gopinath T, Vostrikov V, Vendruscolo M, Dobson CM, Veglia G (2014) Direct observation of the three regions in α-synuclein that determine its membrane-bound behaviour. Nat Commun 5:3827. https://doi.org/10.1038/ncomms4827
Zhengjian LV, Hashemi M, Banerjee S, Zargoski K, Rochet JC, Lyubchenko YL (2019) Assembly of α-synuclein aggregates on phospholipid bilayers. Biochim Biophys Acta Proteins Proteom 1867:802–812. https://doi.org/10.1016/j.bbapap.2019.06.006
Cai H, Reinisch K, Ferro-Novick S (2007) Coats, tethers, rabs, and SNAREs work together to mediate the intracellular destination of a transport vesicle. Dev Cell 12:671–682. https://doi.org/10.1016/j.devcel.2007.04.005
Bonifacino JS, Lippincott-Schwartz J (2003) Coat proteins: shaping membrane transport. Nat Rev Mol Cell Biol 4:409–414. https://doi.org/10.1038/nrm1099
Bonifacino JS, Glick BS (2004) The mechanisms of vesicle budding and fusion. Cell 116:153–166. https://doi.org/10.1016/s0092-8674(03)01079-1
Bremser M, Nickel W, Schweikert M, Ravazzola M, Amherdt M, Hughes CA, Söllner T, Rothman J, Wieland F (1999) Coupling of coat assembly and vesicle budding to packaging of putative cargo receptors. Cell 96:495–506. https://doi.org/10.1016/S0092-8674(00)80654-6
Kamal A, Goldstein LSB (2000) Connecting vesicle transport to the cytoskeleton. Curr Opin Cell Biol 12:503–508. https://doi.org/10.1016/s0955-0674(00)00123-x
Verhage M, Sørensen JB (2008) Vesicle docking in regulated exocytosis. Traffic 9:1414–1424. https://doi.org/10.1111/j.1600-0854.2008.00759.x
Hong W (2005) SNAREs and traffic. Biochim Biophys Acta Mol Cell Res 1744:120–144. https://doi.org/10.1016/j.bbamcr.2005.03.014
Chen YA, Scheller RH (2001) SNARE-mediated membrane fusion. Nat Rev Mol Cell Biol 2:98–106. https://doi.org/10.1038/35052017
Söllner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE (1993) A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 75:409–418. https://doi.org/10.1016/0092-8674(93)90376-2
Nichols BJ, Ungermann C, Pelham HR, Wickner WT, Haas A (1997) Homotypic vacuolar fusion mediated by t- and v-SNAREs. Nature 387:199–202. https://doi.org/10.1038/387199a0
Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC (2010) α-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329:1663–1667. https://doi.org/10.1126/science.1195227
Burré J, Sharma M, Südhof TC (2014) α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci USA 111:E4274–E4283. https://doi.org/10.1073/pnas.1416598111
Lou X, Kim J, Hawk BJ, Shi YK (2017) α-Synuclein may cross-bridge v-SNARE and acidic phospholipids to facilitate SNARE-dependent vesicle docking. Biochem J 474:2039–2049. https://doi.org/10.1042/BCJ20170200
Georgieva ER, Ramlall TF, Borbat PP, Freed JH, Eliezer D (2010) The lipid-binding domain of wild type and mutant alpha-synuclein: compactness and interconversion between the broken and extended helix forms. J Biol Chem 285:28261–28274. https://doi.org/10.1074/jbc.M110.157214
Logan T, Bendor J, Toupin C, Thorn K, Edwards RH (2017) α-Synuclein promotes dilation of the exocytotic fusion pore. Nat Neurosci 20:681–689. https://doi.org/10.1038/nn.4529
Chandra S, Gallardo G, Fernández R, Schlüter OM, Südhof TC (2005) α-Synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123:383–396. https://doi.org/10.1016/j.cell.2005.09.028
Hohl TM, Parlati F, Wimmer C, Rothman JE, Söllner TH, Engelhardt H (1998) Arrangement of subunits in 20S particles consisting ofNSF, SNAPs, and SNARE complexes. Mol Cell 2:539–548. https://doi.org/10.1016/s1097-2765(00)80153-7
Baker RW, Hughson FM (2016) Chaperoning SNARE assembly and disassembly. Nat Rev Mol Cell 17:465–479. https://doi.org/10.1038/nrm.2016.65
Tosatto L, Horrocks MH, Dear AJ, Knowles TPJ, Dalla M, Cremades N, Dobson CM, Klenerman D (2015) Single-molecule FRET studies on alpha-synuclein oligomerization of Parkinson’s disease genetically related mutants. Sci Rep 5:16696. https://doi.org/10.1038/srep16696
Danzer KM, Kranich LR, Ruf WP, Cagsal O, Winslow AR, Zhu L, Vanderburg CR, McLean PJ (2012) Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegeneration 7:42. https://doi.org/10.1186/1750-1326-7-42
Filippini A, Gennarelli M, Russo I (2019) α-Synuclein and glia in Parkinson’s disease: a beneficial or a detrimental duet for the endo-lysosomal system? Cell Mol Neurobiol 39:161–168. https://doi.org/10.1007/s10571-019-00649-9
Jung YJ, Chung WS (2018) Phagocytic roles of glial cells in healthy and diseased brains. Biomol Ther 26:350–357. https://doi.org/10.4062/biomolther.2017.133
Lindström V, Gustafsson G, Sanders LH, Howlett EH, Sigvardson J, Kasrayan A, Ingelsson M, Bergström J, Erlandsson A (2017) Extensive uptake of α-synuclein oligomers in astrocytes results in sustained intracellular deposits and mitochondrial damage. Mol Cell Neurosci 82:143–156. https://doi.org/10.1016/j.mcn.2017.04.009
Loria F, Vargas JY, Bousset L, Syan S, Salles A, Melki R, Zurzolo C (2017) α-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol 134:789–808. https://doi.org/10.1007/s00401-017-1746-2
Sacino AN, Brooks MM, Chakrabarty P, Saha K, Khoshbouei H, Golde TE, Giasson BI (2017) Proteolysis of α-synuclein fibrils in the lysosomal pathway limits induction of inclusion pathology. J Neurochem 140:662–678. https://doi.org/10.1111/jnc.13743
Aflaki E, Stubblefield BK, McGlinchey RP, McMahon RP, Ory DS, Sidransky E (2020) A characterization of Gaucher iPS-derived astrocytes: potential implications for Parkinson’s disease. Neurobiol Dis 134:104647. https://doi.org/10.1016/j.nbd.2019.104647
Volpicelli LA, Gamble KL, Schultheiss CE, Riddle DM, West AB, Lee VMY (2014) Formation of α-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol Biol Cell 25:3987–4204. https://doi.org/10.1091/mbc.E14-02-0741
Tu PH, Galvin JE, Baba M, Giasson B, Tomita T, Leight S, Nakajo S, Iwatsubo T, Trojanowski JQ, Lee VMY (1998) Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble α-synuclein. Ann Neurol 44:415–422. https://doi.org/10.1002/ana.410440324
Braak H, Del Tredici K, Bratzke H, Hamm-Clement J, Sandmann-Keil D, Rüb U (2002) Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson’s disease (preclinical and clinical stages). J Neurol 249:1–5. https://doi.org/10.1007/s00415-002-1301-4
Rietdijk CD, Perez-Pardo P, Garssen J, van Wezel RJA, Kraneveld AD (2017) Exploring Braak’s hypothesis of Parkinson’s disease. Front Neurol 8:37. https://doi.org/10.3389/fneur.2017.00037
Meng J (1856) Wang J (2015) Role of SNARE proteins in tumourigenesis and their potential as targets for novel ant-cancer therapeutics. Biochim Biophys Acta Rev Cancer 1:1–12. https://doi.org/10.1016/j.bbcan.2015.04.002
McNew JA, Sogaard M, Lampen NM, Machida S, Ye RR, Lacomis PT, Rothman JE, Söllner TH (1997) Ykt6p, a prenylated SNARE essential for endoplasmic reticulum-Golgi transport. J Biol Chem 272(28):17776–17783. https://doi.org/10.1074/jbc.272.28.17776
Acknowledgements
The authors appreciate the valuable support of Jorge Armando Florez-Ospina for artwork and M.Sci. Teresa Gómez-Quintero for references’ ordering.
Funding
This work was supported by grants R01ES03771 and R01ES10563 from the National Institute of Environmental Health Sciences (NIEHS) to MA.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, information collection, and analysis were performed by Iris N. Serratos, Elizabeth Hernández-Pérez, and Carolina Campos. All authors integrated information. The first draft of the manuscript was written by Iris N. Serratos, Elizabeth Hernández-Pérez, and Carolina Campos. Michael Aschner and Abel Santamaría translated, complemented, corrected, and edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Highlights
1. α-Synuclein (α-Syn) plays important physiological roles in the brain
2. α-Syn is also involved in the pathogenesis of synucleinopathies
3. Conformational changes of α-Syn undergo post-translational modifications
4. Mutated α-Syn aberrantly interacts with membranes and organelles
5. Protein misfolding and aggregation trigger cellular dysfunction and pathology
Rights and permissions
About this article

Cite this article
Serratos, I.N., Hernández-Pérez, E., Campos, C. et al. An Update on the Critical Role of α-Synuclein in Parkinson’s Disease and Other Synucleinopathies: from Tissue to Cellular and Molecular Levels. Mol Neurobiol 59, 620–642 (2022). https://doi.org/10.1007/s12035-021-02596-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-021-02596-3