
Abstract
Disruption of remyelination contributes to neurodegeneration and cognitive impairment in chronically disabled patients. Valproic acid (VPA) inhibits histone deacetylase (HDAC) function and probably promotes oligodendrocyte progenitor cell (OPC) proliferation and differentiation; however, the relevant molecular mechanisms remain unknown. Here, focal demyelinating lesions (FDLs) were generated in mice by two-point stereotactic injection of lysophosphatidylcholine (LPC) into the corpus callosum. Cognitive functions, sensorimotor abilities and histopathological changes were assessed for up to 28 days post-injury with or without VPA treatment. Primary OPCs were harvested and used to study the effect of VPA on OPC differentiation under inflammatory conditions. VPA dose-dependently attenuated learning and memory deficits and robustly protected white matter after FDL induction, as demonstrated by reductions in SMI-32 and increases in myelin basic protein staining. VPA also promoted OPC proliferation and differentiation and increased subsequent remyelination efficiency by day 28 post-FDL induction. VPA treatment did not affect HDAC1, HDAC2 or HDAC8 expression but reduced HDAC3 protein levels. In vitro, VPA improved the survival of mouse OPCs and promoted their differentiation into oligodendrocytes following lipopolysaccharide (LPS) stimulation. LPS caused OPCs to overexpress HDAC3, which translocated from the cytoplasm into the nucleus, where it directly interacted with the nuclear transcription factor PPAR-γ and negatively regulated PPAR-γ expression. VPA decreased the expression of HDAC3 and promoted remyelination and functional neurological recovery after FDL. These findings may support the use of strategies modulating HDAC3-mediated regulation of protein acetylation for the treatment of demyelination-related cognitive dysfunction.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The white matter is mainly composed of myelinated nerve fibres that play essential roles in signal transduction and communication among different brain regions [1]. At present, a large number of nervous system diseases, such as neonatal brain white matter abnormalities, Alzheimer’s disease (AD), multiple sclerosis (MS), and Huntington’s disease, are known to be related to the loss of white matter; in addition, ischemic, haemorrhagic and traumatic brain injury are related to white matter loss [2, 3]. Most patients with myelination deficiencies, such as MS, initially appear to have relapsing-remitting disease courses, but the diseases eventually evolve into secondary gradual deterioration and exhibit incomplete recovery. During central nervous system (CNS) demyelination and remyelination processes, oligodendrocytes, which are generated from endogenous oligodendrocyte precursor cells (OPCs), are not only the first direct targets of insults but also the major myelin-producing and myelin-maintaining cells [4]. It is believed that oligodendrocyte structure is important for the maintenance of neuronal integrity as well as for mediation of neuronal function. Although OPCs are crucial for myelin repair and ultimately provide neuroprotective effects [5], they usually fail to differentiate and mature in demyelinating and inflammatory environments. Previous studies have revealed that the reasons for failure may include excessive consumption of the OPC pool during remyelination or inhibition of OPC differentiation and maturation under pathological conditions [6]. Unfortunately, the recovery process for white matter in the adult brain disrupts sensorimotor function and elicits profound neurobehavioural and severe cognitive impairments. It has been shown that white matter integrity during CNS repair is important for long-term recovery after myelin damage [7]. Hence, traditional therapies targeting neurons alone are inadequate; CNS repair requires the enhancement of the remyelination process.
Some variations in the conformation of chromosomes are due to core histone acetylation, which plays a crucial role in regulating gene expression in eukaryotes [8]. Thus far, many studies on intracellular protein acetylation have shown that histone acetylation levels are decreased in various neurodegenerative diseases [9, 10]. Given this evidence, histone deacetylase (HDAC) inhibitors (HDACIs) have been developed. Numerous studies have revealed that HDACIs characteristically inhibit HDAC activity reversibly or irreversibly by binding to their catalytic centres. This mechanism has been extended to many regulators of gene transcription. In addition, HDACIs can cause excessive acetylation of target proteins under pathological conditions to correct aberrant disease states. Such excessive acetylation induces a series of biological effects, including chromosome reconstruction, cell cycle arrest, cell differentiation and transcription factor activation and inactivation [10, 11]. Furthermore, HDACI treatment reduces oligodendrocyte death in the mouse optic nerve, and it protects neurons, axons and associated glial cells from damage under oxygen and glucose deprivation while preserving white matter structural integrity [12]. A study on the pathogenesis of neurodegenerative diseases has indicated that HDACIs greatly decrease oligodendrocyte loss and myelin basic protein (MBP) reductions induced by ischaemia in the ipsilateral striatum, corpus callosum (CC) and frontal cortex [13].
Valproic acid (VPA), also called 2-propylpentanoic acid, is a short-chain fatty acid and a pan-HDACI. During recent decades, VPA has been used for the treatment of neurological diseases such as epilepsy, bipolar disorder, schizophrenia and migraine because of its neuroprotective effects [14, 15]. VPA exerts its functions via binding of its carboxylic acid group with metal ions and mainly inactivates HDAC2, HDAC3 and HDAC8 while increasing the acetylation levels of histone proteins [16]. Owing to its simple chemical structure, VPA can easily reach lesions. Furthermore, VPA can promote the proliferation, differentiation and maturation of neural stem cells after ischaemia [17]. Since oligodendrocytes are produced from OPCs or neural stem cells in adults, we hypothesized that OPCs could successfully transform into myelin-forming oligodendrocytes, strongly accelerate remyelination and delay demyelinating injury after HDAC inhibition.
The CC, which is positioned at the bottom of the cortex and is connected to the left and right hemispheres by nerve fibres, plays crucial roles in learning and memory [18, 19]. Few studies have investigated the effect of VPA-mediated HDAC inhibition on the proliferation and differentiation of OPCs during the inflammation-induced demyelination. In this study, we investigated whether VPA treatment could improve remyelination and subsequent sensorimotor and cognitive function recovery after demyelination by using a simple, reliable and inexpensive model of mouse CC demyelination that has been previously described to exhibit functional and morphological reproducibility and good validity [20]. We also addressed the potential molecular mechanisms and biological roles of individual HDAC subtypes during the process of remyelination.
Materials and Methods
Experimental Animals
Male C57BL/6 mice aged 8–10 weeks and weighing 20–25 g were provided by the Animal Center of Nantong University. The mice were given free access to standard rodent food and water and were maintained in a pathogen-free and climate-controlled environment with a temperature of 25 ± 1 °C under 12-h light/dark cycles. All experiments were conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Animal Ethics Committees of Nantong University (NU-2017-39).
Lysophosphatidylcholine-Mediated Induction of Focal Demyelinating Lesions
Focal demyelinating lesions (FDLs) were generated as described in our previous work [20, 21]. Briefly, mice were administered lysophosphatidylcholine (LPC) (9008-30-4, Sigma-Aldrich, USA; Fig. 1a) at two injection sites in the CC with a 10-μl syringe with a 33-gauge needle at a rate of 0.5 μl/min. The target sites for injection were determined according to a mouse brain atlas (coordinates, 1 mm lateral and 0.3 and 0.8 mm anterior to the bregma; 2.0 and 2.2 mm deep relative to the brain surface). Each site was injected with 10 μg of LPC in 4 μl of sterile saline. Sham-operated animals were injected with equal volumes of saline in double-point injections.

Dose-dependent protective effects of VPA on neurological performance in mice at 6 to 10 days and 24 to 28 days after FDL induction. a Establishment of the FDL model and schematic of the time points for detection. Left, identification of the two injection sites based on a mouse brain map. Right, schedule of demyelination modelling and the time points used in the different experiments. b Representative swimming traces from the Morris water maze test for the learning phase (first line) and memory phase (second line) at 6–10 days. c and d Latency to locate the submerged platform for each group of mice in the short-term and long-term Morris water maze tests. There were significant differences compared with the LPC group results in the short-term learning ability test, as shown in the left image of c. VPA at the 100 mg/kg dose improved spatial memory deficits in mice with induced FDLs when the platform was absent, as shown in the right image of c. d During postoperative days 24–28, the mice in the LPC group had weaker memory recall and spent significantly more time locating the submerged platform than the mice in the sham group. However, the mice that received VPA treatment (100 mg/kg) found the platform faster and spent more time in the same quadrant as the platform than the mice that received LPC alone. The data are displayed as the means ± SEMs, n ≥ 8 animals/group. *p < 0.05 and **p < 0.01 for the indicated comparisons or vs. the sham group; #p < 0.05 vs. the LPC group
VPA and BrdU Treatments
The HDACI VPA (as sodium valproate) was dissolved in sterile saline and administered to mice with induced FDLs at doses of 50 and 100 mg/kg body weight, according to previous reports [22, 23]. The LPC group was administered saline. Equal volumes of VPA and saline were injected intraperitoneally once a day starting 2 h after surgery and every 24 h thereafter for 5 consecutive days. According to the experimental design, all animals were randomly divided into a normal saline solution (sham) group, an LPC group, an LPC + 50 mg/kg VPA group and an LPC + 100 mg/kg VPA group. To label the newly generated cells, BrdU was dissolved in sterile saline and injected intraperitoneally (50 mg/kg) according to the schedule of the experimental design shown in Fig. 1a.
Behaviour Tests
A series of behavioural tests were used to detect spatial learning ability, memory and sensorimotor deficits; the tests were performed 5, 10 and 28 days after surgery by observers who were blinded to the treatments. These behavioural tests included the Morris water maze test, Y-maze test, open-field test, novel object recognition test and rotarod test.
Morris Water Maze Test
Cognitive function in mice was assessed with a standard hidden platform test. Before surgery, the animals were trained with 4 consecutive trials (starting from each quadrant) per day for 2 days to enable them to adapt to the environment. After training, the myelin sheath in the CC was damaged by 1% LPC treatment, and the test was performed on four consecutive days to detect learning ability by measuring the escape latency. Following the last day of the learning test, each animal was placed into the quadrant diagonal to the platform and subjected to a probe trial for evaluating memory. The probe trial was performed with the platform removed to measure quadrant preference and platform localization ability. Movement within the maze and the duration that the animal stayed in the target quadrant were recorded using a video camera linked to a tracking software.
Open-Field Test
This test was initiated on the tenth day. The open-field box was made of white opaque plexiglass with dimensions of 50 × 50 × 50 cm. Each mouse was gently placed into a corner of the box and allowed to move freely for 2 min, and then a camera was used to record the animal’s activity for 10 min. After each trial, the apparatus was thoroughly cleaned with a 75% ethanol solution to remove residual odours. For the analysis, the open-field floor was divided into two zones: the central region, which covered one-quarter of the total area, and the peripheral region. The following index parameters were measured: the total distance travelled, the number of appearances in the central region, the distance travelled, the duration of stay in each zone and the ratio of the distance travelled in the central region to the total distance.
Novel Object Recognition Test
This test was performed in the open-field arena on the tenth day. Prior to the test, each mouse was placed in the box for 5 min so it could become familiar with the environment. At the beginning of the experiment, two identical objects were placed in two corners of the box, and the animal was placed on the floor on the other side of the box and allowed to explore for 5 min. Then, the animal was placed back into its cage for 10 min. During this time, one of the objects was replaced with an object of a different colour and shape, and the other familiar object was moved to another position to guard against space bias. The mouse was then allowed to explore around the novel object for 5 min. Approaching the object or sniffing and licking within 2 cm of the object was considered exploratory behaviour. The measured parameters included the time spent exploring around the familiar object (TF), the time spent exploring around the novel object (TN) and the recognition index (RI), which was calculated as follows: RI = TN/(TN + TF). After each trial, the observer wiped the box with alcohol to eliminate odours.
Y-Maze Test
The Y-maze is also known as a three-way radiated labyrinth box. The box used in this study was made of white opaque plexiglass and shaped like the letter Y with three arms that were 35 cm long, 5 cm wide and 15 cm high. A mouse was put in the middle of the maze at the beginning of the experiment, and a camera was used to record the entries of the animal into each arm during an eight-minute period. Movement into any of the three different arms was considered correct; other activity was considered incorrect. The statistical index was calculated based on the ratio of correct movements and the number of entries.
Accelerating Rotarod Test
During the test period, motor function was measured with a rotarod apparatus that accelerated from 4 to 40 rpm at a constant rate of 1 rpm per 10 s. The amount of time that passed before each mouse fell off the rotarod device was recorded for up to 300 s in each of 3 trials. The mice received preoperative training for 3 days with 3 daily trials separated by 4-h intervals. Postoperative testing was performed in 3 trials 10 days after FDL establishment, and the mean latency to fall was analysed.
Protein Extraction and Western Blotting
After mice were anaesthetized, the damaged brain tissue was removed with bent forceps on ice and placed in 1.5 ml RNase-free centrifuge tubes. Total protein was lysed and purified using RIPA buffer (#P0013B, Beyotime, China) in the presence of the protease inhibitor phenylmethylsulfonyl fluoride (PMSF, 1 mM, ST506, Beyotime, China). The buffer contained 50 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS and appropriate concentrations of protease-inhibiting reagents, including sodium orthovanadate, sodium fluoride, EDTA and leupeptin. The samples were then fully homogenized by trituration and subjected to sonication and centrifugation at 13,000 rpm for 15 min at 4 °C; then, the supernatants were collected. The protein concentrations were determined with a BCA kit (RE232694, Thermo, USA) in a microplate reader (BioTek, VT, USA). The proteins were denatured for 5 min at 95 °C in the presence of sample buffer. A discontinuous sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) system (P0012A, Beyotime, China) was then used to separate the proteins before performing transmembrane transfer (onto PVDF membranes purchased from Millipore). Next, all membranes were blocked with blocking buffer (5% nonfat dry milk in TBS containing 0.1% Tween-20) for 2 h at room temperature and then incubated overnight at 4 °C with primary antibodies against the following proteins: HDAC1 (1:1000, #PA1-860, Thermo, USA), HDAC2 (1:1000, #ab12169, Abcam, USA), HDAC3 (1:1000, SAB4503482, Sigma-Aldrich, USA), HDAC8 (1:1000, #PA5-11243, Thermo, USA), acetyl-histone H4 (1:1000, #13944, CST, USA), acetyl-histone H3 (K12, 1:1000, #8173, CST, USA), PPAR-γ (1:2000, ab59256, Abcam, USA), β-actin (1:10000, A2228, Sigma-Aldrich, USA) and GAPDH (1:2000, G9545, Millipore, USA). The blots were then incubated with different HRP-conjugated secondary antibodies for 2 h at room temperature after washing with 0.1% TBS/Tween-20 3 times. The immunoreactive bands were visualized using a Chemiluminescent HRP Substrate Kit (1431401, Millipore, USA) and quantified with the ImageJ software to determine the differences in protein expression among the different groups.
Nuclear Protein Extraction
Nuclear protein extracts of primary OPCs were prepared using the NE-PER nuclear extraction kit (Fisher Scientific, USA), as described previously [24]. Cells were washed with ice-cold PBS and then transferred to prechilled 1.5 ml Eppendorf tubes on ice. The cells were pipetted up and down several times to disrupt cell clumps, after which the samples were centrifuged at 500×g for 5 min. The supernatant was removed from each pellet, and the pellet was resuspended in 400 μl of cytoplasmic extraction reagent I with protease inhibitors and then incubated for 10 min on ice. Subsequently, the samples were added to 22 μl of ice-cold cytoplasmic extraction reagent II before a 5-min centrifugation step at 14,000×g. The supernatant containing the cytoplasmic fraction was removed, and the nucleus-enriched pellet was resuspended in 200 μl of nuclear extraction reagent and then incubated on ice for 40 min. Following a final centrifugation step at 14,000×g for 10 min, the protein concentration in the supernatant containing the nuclear protein extract was measured with a BCA assay. The extracts were kept at − 80 °C for future use.
Tissue Section Preparation and Immunofluorescence Analysis
All procedures were performed at room temperature unless otherwise noted. Brain tissue was harvested after transcardial perfusion of a physiological saline solution containing heparin sodium followed by 0.01 M phosphate-buffered saline (PBS, pH 7.4) containing 4% paraformaldehyde. The tissues were postfixed overnight at 4 °C and then cryoprotected overnight at 4 °C in 0.01 M PBS containing 30% sucrose. Then, 25-μm-thick transverse sections were cut from frozen tissue at − 19 °C with a freezing cryostat (CM1900UV, Leica, Germany). For immunofluorescence analysis, free-floating sections were first blocked to prevent nonspecific binding for 1 h at room temperature in 0.01 M PBS containing 5% normal donkey serum (Jackson ImmunoResearch Laboratories, USA) and 0.3% Triton X-100. Then, the sections were incubated with adenomatous polyposis coil (APC) (1:200, OP80, Calbiochem, USA), oligodendrocyte transcription factor 2 (Olig2) (1:500, AB9610, Millipore, USA), NG2 (1:200, MAB5384, Millipore, USA), MBP (1:500, Abcam, Cambridge, UK), anti-nonphosphorylated neurofilament H (SMI-32; 1:500, Calbiochem, MilliporeSigma, USA) and BrdU (1:500, ab6326, Abcam, UK) antibodies at 4 °C overnight. Next, the sections were incubated with corresponding secondary antibodies conjugated with fluorophores and diluted in the same solution before being mounted with DAPI Fluoromount-G (K3215-YB35B, SouthernBiotech, USA). For BrdU immunolabelling, brain sections were incubated in 2 N HCl for 30 min at room temperature and then processed via the immunostaining protocol described above. Finally, confocal images were captured under a Leica TCS SP8 confocal microscope and analysed with the LAS X software version 2.0.
Luxol Fast Blue Staining
Luxol Fast Blue (LFB) staining was used to evaluate demyelination and remyelination levels at the lesion sites. Sections around the injection site were selected from each group and stained with an LFB kit (#KTLFB, American MasterTech, USA). The sections were then rinsed in distilled water and incubated in an LFB stain solution preheated to 60 °C for 1 h. The sections were dipped several times into 0.05% lithium carbonate followed by 70% alcohol until the grey matter and white matter could be differentiated. The sections were then washed in distilled water, dehydrated in a graded series of alcohols, cleared with three rinses in fresh xylene, mounted with permanent mounting medium, coverslipped and screened by microscopy.
Electron Microscopy
Mouse brain tissue was dissected and fixed overnight in 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4). The tissues were postfixed with 1% osmium tetroxide and embedded in Epon. Semithin (0.5 μm) and ultrathin (60 nm) sections were prepared for imaging using Reichert-Jung ultramicrotomes. Toluidine blue-stained semithin sections were examined by light microscopy (with a BX51 Olympus microscope equipped with an Optronics Microfire digital camera). The ultrathin sections were examined by transmission electron microscopy using an FEI Tecnai Spirit G2 BioTWIN microscope equipped with a bottom-mounted Eagle 4 k (16-megapixel) camera.
Quantitative Real-Time PCR
Mice under anaesthesia were perfused intracardially with precooled physiological saline solution, and their brains were quickly removed. Total RNA was isolated from the demyelinated lesions using TRIzol Reagent (15596-018, Ambion by Life Technologies) according to the manufacturer’s instructions. The RNA was then reverse transcribed to generate cDNA using a RevertAid First Strand cDNA Synthesis Kit (K1622, Thermo, USA). Quantitative real-time PCR (qRT-PCR) analysis was performed to determine the mRNA levels of HDAC1, HDAC2, HDAC3, HDAC8, TNF-α and IL1-β in brain tissue from the four groups of mice (the NS, LPC, LPC + 50 mg/kg VPA and LPC + 100 mg/kg VPA groups) using FastStart Universal SYBR Green Master Mix (04913914001, Roche Diagnostics GmbH, USA) with the primers listed in Table 1. Standard curves for the genes of interest were generated with serial dilutions of mouse cDNA. The qRT-PCR program included a step at 95 °C for 10 min and 40 cycles of 95 °C for 15 s and 60 °C for 60 s. GAPDH expression was used as a normalization control, and the ∆∆CT method was applied to compute gene expression fold changes. The data are shown as the mean ± standard error.
Primary Progenitor Cell Culture and Differentiation
Primary OPCs were harvested from postnatal day (P) 0-P2 mouse pup cortices. Briefly, the skin and skull of each pup were cut along the midline, and the brain was cut out and placed into a Petri dish with a sufficient volume of prechilled HBSS without Ca2+ or Mg2+. Afterwards, the cortices were isolated under a stereomicroscope and transferred into a 10-cm2 Petri dish containing HBSS without Ca2+ or Mg2+. All of the cortices were weighed and transferred into gentleMACS C tubes with preheated Enzyme Mix 1 (Enzyme P in Buffer X), after which Enzyme Mix 2 (Enzyme A in Buffer Y) was added. The cells were incubated in a gentleMACS Dissociator. The reagents used to prepare Enzyme Mix 1 and Enzyme mix 2 were included in a Neural Tissue Dissociation Kit with papain (130-092-628, Miltenyi Biotec, Bergisch Gladbach, Germany). OPC separation was performed with a mouse CD140a (PDGFRα) MicroBead Kit (130-101-502) according to the manufacturer’s instructions. The chemically defined OPC basal medium contained 4 mM GlutaMAX, 1 mM sodium pyruvate, 0.1% BSA, 50 μg/ml apotransferrin, 5 μg/ml insulin, 30 nM sodium selenite, 10 nM D-biotin, 10 nM hydrocortisone, 10 ng/ml PDGF-AA and 10 ng/ml bFGF. The cells were pretreated with one of three different concentrations of VPA (0.3, 1.0, or 3 mM) for 30 min. Lipopolysaccharide (LPS, L2630, Sigma-Aldrich, USA; 5 μg/ml) was then added, and the cells were cultured for an additional 24 h. After removing LPS, the OPCs were grown in differentiation medium containing one of the three concentrations of VPA for four additional consecutive days before being subjected to immunocytochemical staining for NG2 and MBP. The OPC differentiation medium contained 10 ng/ml CNTF and 5 μg/ml N-acetyl-L-cysteine (NAC). In addition, triiodothyronine (T3) can trigger a massive differentiation of cultured OPCs by binding the nuclear T3 receptor α1 [25] and 15 nM T3 was additionally added as a positive control.
Microarray
After being treated with vehicle, VPA (1 mM), LPS (5 μg/ml) or LPS + VPA for 24 h, OPCs were collected. Total RNA was isolated with an RNeasy Mini Kit (Qiagen, USA) according to the manufacturer’s instructions. Two to three replicate RNA samples were used for each treatment group. The gene expression of OPCs was assessed with an Agilent SurePrint G3 Mouse GE 8 × 60 K Microarray Kit (G4852A; CapitalBio Corp., Beijing, China).
Co-Immunoprecipitation Method
The cells were subsequently washed with PBS and lysed using lysis buffer (1 mmol/L PMSF, 5 mmol/L 2-mercaptoethanol, 2 mmol/L MgCl2, 20 mmol/L HEPES, 150 mmol/L NaCl, 10 μg/ml leupeptin and 10 μg/ml aprotinin). The lysates were then added to protein A beads (Thermo Fisher Scientific, USA), and the mixtures were incubated overnight with primary antibodies against IgG or PPAR-γ (Abcam, USA) at 4 °C. The beads were washed twice using lysis buffer to remove unbound proteins. To elute the bound proteins, the beads were then resuspended in sample buffer and boiled at 95 °C for 3 min, and subjected to immunoblotting. The corrected density for different treatment groups was calculated using the initialize value to subtract the background of IgG, and finally normalized to Input lane.
Lentiviral shRNA Infection
Lentiviral shRNA directed at mouse HDAC3 (sc-35539-V), and a lentivirus containing a control, nontargeting sequence of shRNA (sc-108080), were purchased from Santa Cruz Biotechnology (USA). OPCs were plated in a 6-well plate 24 h prior to viral infection. The medium was removed from each well and replaced with 1 ml of a mixture of complete medium with polybrene (sc-134,220, Santa Cruz Biotechnology) at a final concentration of 5 μg/ml per well. ShRNA-expressing lentiviral particles were thawed at room temperature and mixed gently, and then 2 μl of lentiviral particles (1 × 106 infectious units) was used to infect the cells in each well. The plate was swirled gently for mixing and then incubated for 24 h. Cell lysates were generated for Western blot analysis.
Statistical Analysis
All experimental values are expressed as the means ± SEMs. Two-group comparisons were performed using Student’s t tests, and multiple-group comparisons were performed using one-way ANOVA and the Student Newman-Keuls test for post hoc testing. All statistical tests were conducted in the GraphPad Prism software version 5.0. A value of p < 0.05 was determined to indicate statistical significance.
Results
VPA Improved Short- and Long-Term Cognitive Function after FDL Induction
We first performed two-point stereotactic injection of LPC into the CC, which contributed to chronic demyelination. Short-term cognitive function was monitored by the Morris water maze test after 6–10 days and by the Y-maze and novel object recognition test at 10 days after surgery. LPC induced demyelination in the CC area and further impaired short-term spatial learning and spatial memory, as revealed by significantly increased escape latency when the platform was present and reduced duration of swimming in the target quadrant when the platform was absent (Fig. 1b, c), respectively. Notably, the escape latency was greatly reduced after 100 mg/kg VPA treatment during the learning phase (Fig. 1c, p < 0.05). However, the escape latency of the mice in the LPC + 50 mg/kg VPA group was not significantly different from that of the mice in the LPC group (Fig. 1c). Therefore, VPA treatment elicited dose-dependent neuroprotective effects in the mice after FDL induction. Similar to the learning data, the memory data also suggested that the dose of 100 mg/kg VPA was the best choice for subsequent experiments (Fig. 1d, p < 0.01).
Further verifying the notable effects of VPA on mice with induced FDLs, we also found that the RI in the novel object recognition test (Fig. S1A) and the correct movement ratio in the Y-maze test were markedly greater for VPA-treated mice than for control mice (Fig. S1B and C). The percentage of time spent in the central region and the percentage of distance travelled in the central region in the open-field test were not different among groups (Fig. S1D, E and F). Similarly, LPC injection had little influence on sensorimotor function in the CC (as assessed by rotarod test), as shown in Fig. S1G.
The effects of VPA treatment on long-term cognitive deficits were examined by the Morris maze test (Fig. 1d) on postoperative days 24–28. Compared with that of the sham group, the latency of the LPC group was significantly higher in the learning test (Fig. 1d, p < 0.01), but this difference was attenuated by VPA treatment (p < 0.05 or p < 0.01). The memory of mice in the LPC + 100 mg/kg VPA group was significantly better than that of mice in the LPC group (Fig. 1d, p < 0.05). Unexpectedly, the correct movement ratio in the Y-maze test was not significantly different between the groups at day 28 after FDL induction (Fig. S1H and I).
VPA Facilitated OPC Proliferation and Differentiation In Vivo and In Vitro
To assess the effects of VPA on OPC proliferation and differentiation after demyelination, double staining was performed to identify newly generated BrdU+/NG2+ cells in primarily damaged CC tissue at day 7 post-FDL induction. Compared with those after sham surgery, the numbers of NG2+ and BrdU+/NG2+ cells in the ipsilateral CC were slightly increased at day 7 after FDL induction and were significantly increased after VPA treatment (Fig. 2a, b, c, p < 0.01). Interestingly, the number of NG2+ OPCs was increased by VPA treatment; however, there were no significant differences in the numbers of BrdU+/NG2+ cells among the three groups in the ipsilateral striatum (data not shown).

VPA promoted the proliferation and differentiation of OPCs following FDL induction. a VPA increased the number of OPCs during remyelination at 10 days after FDL induction. The images were taken at the time of proliferation and migration of oligodendrocyte lineage cells into the CC lesions, and BrdU+ and NG2+ OPCs are shown. Scale bars = 75 μm. b and c Counts of NG2+ OPCs and BrdU+/NG2+ newly generated OPCs in the CC lesion sites. d VPA induced changes in differentiating oligodendrocytes at 28 days after FDL induction. The numbers of BrdU+/APC+/Olig2+ cells were increased in demyelinating lesions following VPA treatment. Scale bars = 100 μm. e and f The total numbers of APC+/Olig2+ oligodendrocytes and the numbers of newly generated BrdU+/APC+/Olig2+ oligodendrocytes in CC regions. The data represent the means ± SEMs. n = 6 per group; *p < 0.05 and **p < 0.01 for the indicated comparisons or vs. the sham group
To observe the effect of VPA on OPC differentiation after FDL induction, the numbers of APC+/Olig2+ and BrdU+/APC+/Olig2+ cells, which represented differentiated oligodendrocytes and newly proliferating oligodendrocytes, respectively, were counted in the CC area 28 days after LPC-induced demyelination. Overall, immunofluorescence for the oligodendrocyte markers APC and Olig2 was significantly greater in the VPA group than in the LPC treatment group (Fig. 2d, e, p < 0.05). Furthermore, there were more newly proliferating oligodendrocytes in the ipsilateral CC in mice with induced FDLs than in sham-operated mice (Fig. 2d, f, p < 0.01). Compared with the LPC group, however, the VPA group exhibited greater oligodendrocyte formation from OPCs (Fig. 2d, f, p < 0.05).
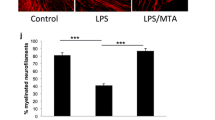
Similar results were observed with regard to the effect of VPA on the differentiation of primary cultured OPCs into oligodendrocytes after induction of inflammation by LPS in vitro. OPCs were harvested from P0-P2 mouse cortices using complete dissociation and separation kits (Fig. 3a). To enhance OPC proliferation, the cells were maintained for 7 days in chemically defined basal OPC medium. At 7 days post-plating, OPCs appeared bright under phase contrast microscopy and had typical bipolar or tripolar morphology (Fig. 3b). Furthermore, we identified cells using the early OPC marker NG2 (Fig. 3c). Different concentrations (0.3, 1 and 3 mM) of VPA were added to study the effect of VPA on OPC differentiation. MBP+ oligodendrocytes derived from a fraction of OPCs formed dramatically more complex arborization patterns after treatment with 1 mM VPA for 5 days than after subjection to the control protocol (Fig. 3d). MBP fluorescence was fairly stable in these mature oligodendrocytes under control conditions; however, LPS evoked severe loss of oligodendrocyte processes (Fig. 3e, p < 0.01). This loss of processes was alleviated by 1 mM VPA treatment (Fig. 3e, p < 0.01). However, the higher dose of 3 mM did not cause any obvious changes to these parameters (Fig. 3e).

Effect of VPA on the differentiation of primary cultured OPCs into oligodendrocytes after treatment with LPS in vitro. a Schematic view of the protocols used to obtain OPCs from P0-P2 mouse cortices and to differentiate OPCs into oligodendrocytes after treatment with VPA at different concentrations with or without LPS (5 μg/ml). b Phase microscopy images at day 7. Scale bar = 100 μm. c OPCs stained positive for NG2 with DAPI counterstaining. Scale bar = 50 μm. d Representative fields showing MBP (green)/NG2 (red) staining during differentiation in the absence or presence of VPA (0.3, 1 or 3 mM) after 24 h of LPS treatment. T3 was regarded as the positive control group. Scale bar = 30 μm. e The areas were calculated with ImageJ and are expressed in the bar graph as the means (μm2) ± SEMs for five independent experiments. **p < 0.01 for the indicated comparisons or vs. the control group. Scale bar = 50 μm
VPA Promoted White Matter Repair after FDL Induction
We further performed double immunofluorescence staining for MBP and SMI-32 (Fig. 4a) and LFB staining (Fig. 4b) to observe the integrity of myelin. Figure 4d shows that MBP levels were significantly lower in the LPC group than in the sham group, while the fluorescence intensity of SMI-32 increased after FDL induction (p < 0.01). The ratio of SMI-32 to MBP is considered to be introduced as an established measurement for demyelination [20, 24]. VPA treatment significantly reduced the ratio of SMI-32 to MBP at 28 days post-injury (Fig. 4a, d, p < 0.01), and it partially restored LFB staining (Fig. 4b), suggesting long-term remyelination of axons. Similar results were observed in Western blot analysis. The expression level of MBP was significantly lower on the experimental side than on the contralateral side in the LPC group (Fig. 4c, e, p < 0.05), revealing obvious demyelination in the ipsilateral hemisphere. However, VPA treatment inhibited MBP loss; the MBP levels were significantly higher after LPC and VPA treatment than after LPC treatment alone (Fig. 4c, e, p < 0.05). We further investigated the effects of VPA on the ultrastructure of the myelin sheath in the CC at 28 days post-FDL induction (Fig. 4f). The axons in the sham group samples were closely packed and myelinated with more abundant myelin sheaths than the axons in the treatment groups. The FDL mice showed profound injury to myelinated axons, characterized by swollen, hydropic degeneration of axons. However, VPA treatment (100 mg/kg) stimulated the regeneration of myelin in mice, in which thin, myelin sheaths appeared to support the integrity of axons and no swollen axons or spheroids were present. Quantification of the g-ratio (the ratio of axonal diameter to overall diameter of the axon plus myelin), LPC-treated mice (0.688 ± 0.024) had a significant increase compared with the sham controls (0.927 ± 0.027), and the g-ratio was significantly attenuated by L-serine treatment (0.802 ± 0.019, p < 0.01).

Repair of white matter after demyelination was promoted by VPA. a Double immunofluorescence staining for a dephosphorylated neurofilament protein (SMI-32, red) and MBP (green) in the ipsilateral CC at 28 days after sham surgery or LPC injection and VPA treatment. Under normal circumstances, SMI-32 localizes to neuronal cell bodies and neurites and is phosphorylated; once the myelin sheath is damaged and degenerated, SMI-32 is easily stained. A larger ratio of SMI-32 to MBP intensity indicates more severe demyelination. b LFB staining in the CC at 28 days after sham or LPC injection and VPA treatment. The 100 mg/kg dose of VPA enhanced the intensity of myelin staining at lesion sites; the VPA-treated group showed blue staining that was weaker than that of the control group but stronger than that of the LPC group. Scale bar = 200 μm. c Western blot analysis of MBP expression in the contralateral and ipsilateral side in each group of animals. d The degree of white matter demyelination is shown as the SMI-32/MBP intensity ratio. e MBP intensity was measured and calculated as the fold change relative to the intensity on the contralateral sham side. f High-magnification images of normal-appearing and demyelinated white matter in the CC were obtained by electron microscopy. In the normal saline solution-injected (sham) group, closely packed myelinated axons (black arrows) were abundant. After FDL, nearly complete demyelination was observed in many axons (red arrows). VPA treatment successfully induced remyelination, but a thinner layer coated the neurons in the VPA-treated group than in the sham group (green arrows). Bar = 1 μm. The values shown are the means ± SEMs from four animals per group. *p < 0.05 and **p < 0.01
HDAC3 Expression Was Increased after FDL and Decreased by VPA Treatment
The protein levels of acetylated-H3, acetylated-H4 and class 1 HDACs (HDAC1, HDAC2, HDAC3 and HDAC8) were evaluated via Western blot analysis to test the inhibitory effect of VPA on HDAC activity in the damaged CC at 5 days after FDL induction. There were no significant differences in HDAC1, HDAC2, HDAC3 and HDAC8 mRNA levels among the groups (Fig. 5a, p > 0.05). LPC treatment decreased the protein expression of acetyl-H3 and acetyl-H4 from control levels; however, VPA conspicuously increased the expression of these proteins (Fig. 5b, c, p < 0.05). As shown in Fig. 5d, e, neither HDAC1 nor HDAC2 protein expression was influenced by LPC or VPA. Unlike the qRT-PCR results, HDAC3 expression was significantly enhanced following demyelination, but its expression was prevented by VPA (Fig. 5e, p < 0.05). Interestingly and unexpectedly, HDAC8 levels were markedly lower after FDL induction than under control conditions (Fig. 5e, p < 0.01).

VPA treatment suppressed the protein expression of HDAC3 during remyelination but failed to inhibit the expression of other class I HDACs at the gene and protein levels 5 days after FDL induction in mice. a There were no significant differences in the mRNA levels of HDAC1, HDAC2, HDAC3 and HDAC8 among the groups. b Examples of acetyl-H3 and acetyl-H4 protein levels in the CCs of mice after LPC injection, as measured with Western blot analysis. c Mean values of acetyl-H3 and acetyl-H4 expression in mice. Reductions were observed in the LPC + 100 mg/kg VPA group. VPA treatment increased the protein expression of acetyl-H3 and acetyl-H4 during remyelination. d Examples of HDAC1, HDAC2, HDAC3 and HDAC8 protein expression in the CCs of mice. e Mean protein levels of HDAC1, HDAC2, HDAC3 and HDAC8 in the CC. No differences in HDAC1 and HDAC2 were observed in mice following VPA treatment. However, FDL induction significantly enhanced HDAC3 expression, and this effect was prevented by VPA treatment. Unexpectedly, HDAC8 levels were markedly lower in the LPC group than in the control group. The data are expressed as fold changes versus the sham group values and are presented as the means ± SEMs. n = 4 per group; *p < 0.05 and **p < 0.01 for the indicated comparisons or vs. the sham group
HDAC3 Regulated PPAR-γ Activity during OPC Differentiation
To elucidate the molecular mechanisms by which HDAC inhibition reinforces OPC differentiation, we performed microarray analysis of VPA that examined the differentiation of primary cultured OPCs into oligodendrocytes after treatment with LPS in vitro. The results showed that 59 myelination-related genes were upregulated by more than twofold in VPA-treated OPCs compared with OPCs treated with LPS alone. Among the 59 myelination-related genes with detectable changes, 30 were upregulated by more than ninefold. PPAR-γ was the most dramatically upregulated gene, with a greater than 17-fold increase (Fig. S2).
To validate the microarray results, we assessed the expression of PPAR-γ in vivo and in vitro. In the above well-defined FDL model of demyelination/remyelination, OPCs began to migrate to the lesions approximately 6 days after FDL induction, where they further differentiated prior to the 14th day and induced complete remyelination after 4 weeks. The expression levels of PPAR-γ gradually decreased from day 5 onward, reaching their lowest points at day 10, before gradually returning to baseline levels at day 21 after FDL induction (Fig. 6a). However, PPAR-γ levels were significantly higher in the VPA group than in the LPC group (Fig. 6b, p < 0.01).

VPA abolished HDAC3-mediated inhibition of PPAR-γ function and reduced interaction between HDAC3 and PPAR-γ. a Temporal changes in PPAR-γ expression in the CC after FDL induction in vivo. b VPA promoted in vivo PPAR-γ expression in the CC 5 days after FDL induction. c VPA modulated the spatiotemporal distribution of HDAC3 in LPS-treated OPCs in vitro. Western blot analysis of HDAC3 expression in the cytosolic and nuclear fractions of OPCs at 24 h after LPS stimulation. HDAC3 levels were decreased in the cytoplasm and increased in the nucleus after LPS stimulation. Cytosolic GSK3β expression was significantly higher in the VPA group than in the control or LPS-only group, and VPA treatment completely reversed the LPS-induced increases in nuclear HDAC3 levels. However, HDAC3 levels returned to baseline levels in all groups within 48 h. d HDAC3 directly interacted with PPAR-γ in cultured OPCs. LPS promoted the binding of HDAC-3 and PPAR-γ, but this effect was reversed by VPA treatment. e VPA suppressed the protein expression of HDAC3 but increased the protein expression of PPAR-γ in cultured OPCs at 24 h after LPS stimulation. f HDAC3 negatively regulated PPAR-γ expression in cultured OPCs. Lentivirus-mediated HDAC3 knockdown increased the protein expression of PPAR-γ. The efficiency of HDAC3 protein knockdown was confirmed by Western blot analysis at 24 h after lentiviral infection. The data are expressed as fold changes relative to the sham or control levels and are presented as the means ±SEMs. n = 4 per group; *p < 0.05 and **p < 0.01 for the indicated comparisons or vs. the LPS or control group
We further examined the spatiotemporal kinetics of HDAC3 in LPS- and VPA-treated primary cultured OPCs. HDAC3 predominantly localized to the nucleus in untreated OPCs, but it became even more concentrated in the nucleus and sparse in the cytoplasm at 24 h after LPS treatment (Fig. 6c, p < 0.01 or p < 0.05). Cytosolic HDAC3 levels were significantly higher in the VPA group than in the control and LPS-only groups, and VPA treatment almost completely reversed the LPS-induced increases in nuclear HDAC3 levels (Fig. 6c, p < 0.01 or p < 0.05). These findings suggest that VPA increases cytosolic HDAC3 levels in differentiated OPCs by promoting HDAC3 translocation to the cytoplasm. However, the expression of HDAC3 in both LPS- and VPA-treated primary OPC cultures returned to baseline by 48 h (Fig. 6c).
The above data suggest that VPA may function by regulating the HDAC3-PPAR-γ pathway. To test whether the inhibition of PPAR-γ activity that occurred upon HDAC inhibition was the consequence of an interaction between PPAR-γ and HDAC3, nuclear extracts from OPCs were immunoprecipitated with an anti-PPAR-γ antibody. A 48-kDa protein in the immunoprecipitates was recognized by an anti-HDAC3 antibody, indicating that HDAC3 directly interacted with PPAR-γ in vitro after LPS stimulation (Fig. 6d, lane 3). However, VPA treatment decreased the interaction of PPAR-γ with HDAC3 (Fig. 6d, lane 4).
Similar to the results found in vivo, the in vitro results revealed that cultured OPCs exposed to LPS for 24 h exhibited higher HDAC3 protein levels than control cells, and the LPS-induced increases were reversed by VPA treatment (Fig. 6e, p < 0.01 or p < 0.05). In contrast, PPAR-γ protein levels were decreased by LPS stimulation and were noticeably increased by VPA treatment (Fig. 6e, p < 0.01 or p < 0.05).
Given the robust preservation of PPAR-γ levels following inhibition of HDAC3 with VPA in LPS-challenged OPCs, we further tested the direct effects of HDAC3 on PPAR-γ. To this end, we infected primary cultured OPCs with lentiviral vectors (Ln) bearing shRNA targeting HDAC3 or bearing a scrambled control sequence. HDAC3 knockdown increased PPAR-γ expression (Fig. 6f, p < 0.01), suggesting that HDAC3 lies upstream of PPAR-γ signalling in the proliferation and differentiation of OPCs.
Discussion
To better simulate white matter diseases, we established a model of LPC-induced focal lesions that causes inflammation and enables study of the progression of demyelination-related processes, including the proliferation, differentiation and maturation of OPCs [26, 27]. OPCs migrate to damaged areas 6–10 days after injury; then, the OPCs differentiate and mature in 10–14 days, and remyelination occurs 1 month after injury [28, 29]. Our findings show that compared with the classic single-point LPC injection model, the two-point LPC injection model can induce not only short-term demyelination but also long-term demyelination with little remyelination in the mouse CC [20]. In this study, the FDL model group showed stable and obvious differences from the sham group and provided considerable research value for the study of proteins and molecular mechanisms.
Previous studies using BrdU/MBP to mark proliferating oligodendrocytes have revealed that HDACIs regulate the expression of MBP and have indicated that this process may be related to promotion of oligodendrocyte proliferation (12). VPA, an HDACI that directly inhibits HDACs, has been implicated in many biological activities [30]. The present study also suggests that VPA is a suitable therapeutic candidate for the prevention of demyelination-related cognitive dysfunction. We observed that VPA promoted OPC proliferation and differentiation after demyelinated lesions were induced. The electron microscopy and water maze results obtained 4 weeks after FDL induction suggested that VPA could promote oligodendrocyte proliferation and oligodendrocyte-mediated myelin repair, which could aid in the recovery of advanced cognitive functions. In vitro, VPA also improved the survival of mouse OPCs and preferentially increased OPC differentiation into oligodendrocytes after LPS stimulation. Such findings suggest that the molecular mechanism by which VPA alleviates white matter injury may involve inhibition of the activity of HDACs and increases in histone H3 and H4 gene expression and lysine acetylation (which affects gene expression) [31]. We also noticed that VPA at the highest concentration (3.0 mM) appeared to be less effective. Although the initial step of VPA action appears to be the inhibition of HDACs, various adverse effects may occur with the increase of the dose. This adverse influence was consistent with other report [32].
Importantly, we found that there were no significant changes in HDAC1, HDAC2 and HDAC8 following FDL induction; however, HDAC3 expression was upregulated in lesioned CC tissues, suggesting that HDAC3 plays an important role in LPC-induced demyelination. These findings indicate that VPA exerts neuroprotective effects probably inhibiting HDAC3 expression and activity. A similar finding was observed for HDAC3 in mouse optic nerves after being exposed to OGD [33]. The period of OGD upregulated HDAC1, HDAC2 and HDAC3 considerably, with HDAC3 being the most prominent. Consequently, HDAC 3 was prominently expressed in proximal axons and dendrites of neurons, as well as in axons of the CC [12]. Because this episode of demyelination lesion modifies HDACs expression in CC, it is reasonable to propose that the region-specific HDACs expression patterns in different white matter tracts may contribute to regional vulnerability and determine the mechanisms of injury.
We further hypothesized that VPA induces genomic reprogramming in OPCs, thereby triggering OPC differentiation. To test this hypothesis, gene-chip microarrays were performed using OPCs treated for 5 days with VPA or a vehicle. Interestingly, PPAR-γ, but not PPAR-α or PPAR-β, was upregulated by VPA. PPAR-γ is a transcription factor that regulates over 100 genes involved in inflammation, cell proliferation and differentiation [34]. Consistent with the microarray results, PPAR-γ expression was increased in OPCs upon VPA treatment or HDAC3 knockdown. Early studies have shown that PPAR-γ-agonist therapy can improve the clinical symptoms of MS, AD, spinal cord injury and stroke [35, 36]. In addition, PPAR-γ can regulate neural stem cell and OPC differentiation and gene expression to induce the differentiation of mature oligodendrocytes and decrease the expression of differentiation-related genes in glial cell lines [36, 37].
However, PPAR-γ activity is affected by posttranslational modifications, such as phosphorylation, generic ubiquitination and specific ubiquitination [38]. When ligands are absent, PPAR-γ is activated by acetylation [39]. In the current study, LPS induced overexpression of HDAC3, which translocated from the cytoplasm into the nucleus in OPCs, and directly interacted with the nuclear transcription factor PPAR-γ. Previous studies have already shown that nuclear translocation of HDAC3 is required for inhibition of transcriptional activity of PPAR-γ, whereas inhibition of HDAC3 activity results in the activation of PPAR-γ by protein acetylation [40, 41]. Multiple sites of PPAR-γ, including K289, K386, K462 and K466, are reported to be modified by acetylation [39]. In addition, PPAR-γ has been promoted to regulate neural stem cell and OPC differentiation and gene expression to induce the differentiation of mature oligodendrocytes [36]. Therefore, further studies should screen PPAR-γ functional target genes by gene-chip technology and use viruses to induce or reduce PPAR-γ expression.
Because the acetylation of histones modulates chromatin organization, the inhibition of HDACs can significantly impact the transcriptional levels of various genes [42]. HDACs activity is thought to inhibit differentiation to oligodendrocytes [43]. Consistently, VPA treatment changes global gene expression patterns in various cell types, which reduces oligodendrocyte death, and protects neurons and associated glial cells [12]. Moreover, HDAC3 deletion enhances Schwann cells remyelination and myelin thickness after peripheral nerve injury in mice [44]. Besides, this is a model of focal demyelination lesions in the CC area caused by inflammatory response, which is based on the rationale that many inflammatory diseases are paralleled by the loss of myelin. We previously reported that Scriptaid, a novel inhibitor of class I/II HDACs, inhibition of HDACs in microglia is a potential future therapy in TBI and other neurological conditions with white matter destruction [24]. Thus, we intend to detect whether VPA targets other cell types in the brain after FDL, notably microglia in future research.
We hypothesized that OPCs could successfully transform into myelin-forming oligodendrocytes, strongly accelerate remyelination and delay demyelinating injury after VPA treatment (Fig. 7). In terms of this mechanism, inflammation-induced demyelination causes OPCs to overexpress HDAC3, which translocated from the cytoplasm into the nucleus, where it then binds to the nuclear transcription factor PPAR-γ. Once deacetylated by HDAC3 due to the direct interaction, the transcription of PPAR-γ is inhibited, and PPAR-γ is potentially deactivated during remyelination. Dysregulation of HDAC3-PPAR-γ signalling in the nuclei of OPCs arrests OPCs at a premyelination stage and delays repair. The HDAC inhibitor VPA may decrease HDAC3 expression, prevent HDAC3 translocation and thus decrease HDAC3 binding to PPAR-γ. Upon recovering its activity, PPAR-γ can then promote OPC recruitment into white matter lesions via proliferation and migration, which is followed by initiation of remyelination (via differentiation of these OPCs into myelin sheath-forming oligodendrocytes) and completion of remyelination. This process aids in the regeneration of the myelin sheath on axons and ameliorates the cognitive dysfunction caused by demyelination.

Proposed mechanism underlying the control of OPC differentiation after focal white matter demyelination. Focal demyelination is induced via two-point stereotaxic injection of LPC into the mouse CC. Demyelination-triggered inflammation causes OPCs to overexpress HDAC3, which is driven from the cytoplasm into the nucleus, where it binds to the nuclear transcription factor PPAR-γ. Extensive studies have shown that PPAR-γ is essential for OPC proliferation and differentiation and contributes to remyelination of white matter. However, once PPAR-γ is bound to HDAC3, its transcription is inhibited, and PPAR-γ is potentially dysregulated during remyelination. Dysregulation of HDAC3-PPAR-γ signalling in the nuclei of OPCs results in arrest of OPCs at a premyelination stage and delays repair. The HDAC inhibitor VPA can decrease HDAC3 expression, prevent HDAC3 translocation and thus prevent HDAC3 binding to PPAR-γ. Upon recovering its activity, PPAR-γ can then induce OPC recruitment into the lesions via proliferation and migration, initiation of remyelination via the differentiation of these OPCs into myelin sheath-forming oligodendrocytes, and completion of remyelination
Data Availability
All original data and materials are available upon request.
References
Shi H, Hu X, Leak RK, Shi Y, An C, Suenaga J, Chen J, Gao Y (2015) Demyelination as a rational therapeutic target for ischemic or traumatic brain injury. Exp Neurol 272:17–25. https://doi.org/10.1016/j.expneurol.2015.03.017
Yazdi A, Baharvand H, Javan M (2015) Enhanced remyelination following lysolecithin-induced demyelination in mice under treatment with fingolimod (FTY720). Neuroscience 311:34–44. https://doi.org/10.1016/j.neuroscience.2015.10.013
Serra-de-Oliveira N, Boilesen SN, Prado de Franca Carvalho C, LeSueur-Maluf L, Zollner Rde L, Spadari RC, Medalha CC, Monteiro de Castro G (2015) Behavioural changes observed in demyelination model shares similarities with white matter abnormalities in humans. Behav Brain Res 287:265–275. https://doi.org/10.1016/j.bbr.2015.03.038
Kim S, Chung AY, Na JE, Lee SJ, Jeong SH, Kim E, Sun W, Rhyu IJ et al (2019) Myelin degeneration induced by mutant superoxide dismutase 1 accumulation promotes amyotrophic lateral sclerosis. Glia 67(10):1910–1921. https://doi.org/10.1002/glia.23669
Nave KA (2010) Myelination and support of axonal integrity by glia. Nature 468(7321):244–252. https://doi.org/10.1038/nature09614
Li X, Zhang Y, Yan Y, Ciric B, Ma CG, Gran B, Curtis M, Rostami A et al (2016) Neural stem cells engineered to express three therapeutic factors mediate recovery from chronic stage CNS autoimmunity. Molecular therapy : the journal of the American Society of Gene Therapy 24(8):1456–1469. https://doi.org/10.1038/mt.2016.104
Bennett IJ, Madden DJ (2014) Disconnected aging: cerebral white matter integrity and age-related differences in cognition. Neuroscience 276:187–205. https://doi.org/10.1016/j.neuroscience.2013.11.026
Langley B, Gensert JM, Beal MF, Ratan RR (2005) Remodeling chromatin and stress resistance in the central nervous system: histone deacetylase inhibitors as novel and broadly effective neuroprotective agents. Current drug targets CNS and neurological disorders 4(1):41–50
Yang XJ, Seto E (2007) HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 26(37):5310–5318. https://doi.org/10.1038/sj.onc.1210599
Baltan S (2012) Histone deacetylase inhibitors preserve function in aging axons. J Neurochem 123(Suppl 2):108–115. https://doi.org/10.1111/j.1471-4159.2012.07949.x
Shein NA, Grigoriadis N, Alexandrovich AG, Simeonidou C, Lourbopoulos A, Polyzoidou E, Trembovler V, Mascagni P et al (2009) Histone deacetylase inhibitor ITF2357 is neuroprotective, improves functional recovery, and induces glial apoptosis following experimental traumatic brain injury. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 23(12):4266–4275. https://doi.org/10.1096/fj.09-134700
Baltan S, Morrison RS, Murphy SP (2013) Novel protective effects of histone deacetylase inhibition on stroke and white matter ischemic injury. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics 10(4):798–807. https://doi.org/10.1007/s13311-013-0201-x
Kim HJ, Chuang DM (2014) HDAC inhibitors mitigate ischemia-induced oligodendrocyte damage: potential roles of oligodendrogenesis, VEGF, and anti-inflammation. Am J Transl Res 6(3):206–223
Jeong SG, Ohn T, Kim SH, Cho GW (2013) Valproic acid promotes neuronal differentiation by induction of neuroprogenitors in human bone-marrow mesenchymal stromal cells. Neurosci Lett 554:22–27. https://doi.org/10.1016/j.neulet.2013.08.059
Tai YT, Lee WY, Lee FP, Lin TJ, Shih CL, Wang JY, Chiu WT, Hung KS (2014) Low dose of valproate improves motor function after traumatic brain injury. Biomed Res Int 2014:980657–980658. https://doi.org/10.1155/2014/980657
Dash PK, Orsi SA, Zhang M, Grill RJ, Pati S, Zhao J, Moore AN (2010) Valproate administered after traumatic brain injury provides neuroprotection and improves cognitive function in rats. PLoS One 5(6):e11383. https://doi.org/10.1371/journal.pone.0011383
Sandvig I, Gadjanski I, Vlaski-Lafarge M, Buzanska L, Loncaric D, Sarnowska A, Rodriguez L, Sandvig A et al (2017) Strategies to enhance implantation and survival of stem cells after their injection in ischemic neural tissue. Stem Cells Dev 26(8):554–565. https://doi.org/10.1089/scd.2016.0268
Moxon-Emre I, Schlichter LC (2010) Evolution of inflammation and white matter injury in a model of transient focal ischemia. J Neuropathol Exp Neurol 69(1):1–15. https://doi.org/10.1097/NEN.0b013e3181c3ce6c
Liu XS, Chopp M, Kassis H, Jia LF, Hozeska-Solgot A, Zhang RL, Chen C, Cui YS et al (2012) Valproic acid increases white matter repair and neurogenesis after stroke. Neuroscience 220:313–321. https://doi.org/10.1016/j.neuroscience.2012.06.012
Luo Q, Ding L, Zhang N, Jiang Z, Gao C, Xue L, Peng B, Wang G (2018) A stable and easily reproducible model of focal white matter demyelination. J Neurosci Methods 307:230–239. https://doi.org/10.1016/j.jneumeth.2018.05.024
Wang G, Ding L, Gao C, Zhang N, Gan D, Sun Y, Xu L, Luo Q et al (2019) Neuroprotective effect of l-serine against white matter demyelination by harnessing and modulating inflammation in mice. Neuropharmacology 146:39–49. https://doi.org/10.1016/j.neuropharm.2018.11.025
Dehghan S, Hesaraki M, Soleimani M, Mirnajafi-Zadeh J, Fathollahi Y, Javan M (2016) Oct4 transcription factor in conjunction with valproic acid accelerates myelin repair in demyelinated optic chiasm in mice. Neuroscience 318:178–189. https://doi.org/10.1016/j.neuroscience.2016.01.028
Xuan A, Long D, Li J, Ji W, Hong L, Zhang M, Zhang W (2012) Neuroprotective effects of valproic acid following transient global ischemia in rats. Life Sci 90(11–12):463–468. https://doi.org/10.1016/j.lfs.2012.01.001
Wang G, Shi Y, Jiang X, Leak RK, Hu X, Wu Y, Pu H, Li WW et al (2015) HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3beta/PTEN/Akt axis. Proc Natl Acad Sci U S A 112(9):2853–2858. https://doi.org/10.1073/pnas.1501441112
Picou F, Fauquier T, Chatonnet F, Flamant F (2012) A bimodal influence of thyroid hormone on cerebellum oligodendrocyte differentiation. Mol Endocrinol 26(4):608–618. https://doi.org/10.1210/me.2011-1316
Kalakh S, Mouihate A (2015) The promyelinating properties of androstenediol in gliotoxin-induced demyelination in rat corpus callosum. Neuropathol Appl Neurobiol 41(7):964–982. https://doi.org/10.1111/nan.12237
Gautier HO, Evans KA, Volbracht K, James R, Sitnikov S, Lundgaard I, James F, Lao-Peregrin C et al (2015) Neuronal activity regulates remyelination via glutamate signalling to oligodendrocyte progenitors. Nat Commun 6:8518. https://doi.org/10.1038/ncomms9518
Boyd A, Zhang H, Williams A (2013) Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and mouse models. Acta Neuropathol 125(6):841–859. https://doi.org/10.1007/s00401-013-1112-y
Wegener A, Deboux C, Bachelin C, Frah M, Kerninon C, Seilhean D, Weider M, Wegner M et al (2015) Gain of Olig2 function in oligodendrocyte progenitors promotes remyelination. Brain : a journal of neurology 138(Pt 1):120–135. https://doi.org/10.1093/brain/awu375
Tartaglione AM, Cipriani C, Chiarotti F, Perrone B, Balestrieri E, Matteucci C, Sinibaldi-Vallebona P, Calamandrei G et al (2019) Early behavioral alterations and increased expression of endogenous retroviruses are inherited across generations in mice prenatally exposed to valproic acid. Mol Neurobiol 56(5):3736–3750. https://doi.org/10.1007/s12035-018-1328-x
Zhang Q, Yang F, Li X, Wang LW, Chu XG, Zhang H, Gong ZJ (2015) Trichostatin a protects against experimental acute-on-chronic liver failure in rats through regulating the acetylation of nuclear factor-kappaB. Inflammation 38(3):1364–1373. https://doi.org/10.1007/s10753-014-0108-7
Li AS, Marikawa Y (2016) Adverse effect of valproic acid on an in vitro gastrulation model entails activation of retinoic acid signaling. Reprod Toxicol 66:68–83. https://doi.org/10.1016/j.reprotox.2016.09.015
Baltan S, Bachleda A, Morrison RS, Murphy SP (2011) Expression of histone deacetylases in cellular compartments of the mouse brain and the effects of ischemia. Transl Stroke Res 2(3):411–423. https://doi.org/10.1007/s12975-011-0087-z
Al-Ghadban S, Diaz ZT, Singer HJ, Mert KB, Bunnell BA (2020) Increase in leptin and PPAR-gamma gene expression in lipedema adipocytes differentiated in vitro from adipose-derived stem cells. Cells 9(2). https://doi.org/10.3390/cells9020430
Cheng HS, Tan WR, Low ZS, Marvalim C, Lee JYH, Tan NS (2019) Exploration and development of PPAR modulators in health and disease: an update of clinical evidence. Int J Mol Sci 20(20). https://doi.org/10.3390/ijms20205055
Villapol S (2018) Roles of peroxisome proliferator-activated receptor gamma on brain and peripheral inflammation. Cell Mol Neurobiol 38(1):121–132. https://doi.org/10.1007/s10571-017-0554-5
Kanakasabai S, Pestereva E, Chearwae W, Gupta SK, Ansari S, Bright JJ (2012) PPARgamma agonists promote oligodendrocyte differentiation of neural stem cells by modulating stemness and differentiation genes. PLoS One 7(11):e50500. https://doi.org/10.1371/journal.pone.0050500
Brunmeir R, Xu F (2018) Functional regulation of PPARs through post-translational modifications. Int J Mol Sci 19(6). https://doi.org/10.3390/ijms19061738
Jiang X, Ye X, Guo W, Lu H, Gao Z (2014) Inhibition of HDAC3 promotes ligand-independent PPARgamma activation by protein acetylation. J Mol Endocrinol 53(2):191–200. https://doi.org/10.1530/JME-14-0066
Gao Z, He Q, Peng B, Chiao PJ, Ye J (2006) Regulation of nuclear translocation of HDAC3 by IkappaBalpha is required for tumor necrosis factor inhibition of peroxisome proliferator-activated receptor gamma function. J Biol Chem 281(7):4540–4547. https://doi.org/10.1074/jbc.M507784200
Fajas L, Egler V, Reiter R, Hansen J, Kristiansen K, Debril MB, Miard S, Auwerx J (2002) The retinoblastoma-histone deacetylase 3 complex inhibits PPARgamma and adipocyte differentiation. Dev Cell 3(6):903–910. https://doi.org/10.1016/s1534-5807(02)00360-x
Yoon S, Eom GH (2016) HDAC and HDAC inhibitor: from cancer to cardiovascular diseases. Chonnam medical journal 52(1):1–11. https://doi.org/10.4068/cmj.2016.52.1.1
Humphrey GW, Wang YH, Hirai T, Padmanabhan R, Panchision DM, Newell LF, McKay RD, Howard BH (2008) Complementary roles for histone deacetylases 1, 2, and 3 in differentiation of pluripotent stem cells. Differentiation; research in biological diversity 76(4):348–356. https://doi.org/10.1111/j.1432-0436.2007.00232.x
He X, Zhang L, Queme LF, Liu X, Lu A, Waclaw RR, Dong X, Zhou W et al (2018) A histone deacetylase 3-dependent pathway delimits peripheral myelin growth and functional regeneration. Nat Med 24(3):338–351. https://doi.org/10.1038/nm.4483
Acknowledgements
This work was supported by the Chinese Natural Science Foundation (Grants 81873924, 81471257) and sponsored by Nantong Science and Technology Project (MS12018030, MS12018048). Dr. Wang’s work has also been funded by Qing Lan Project of Jiangsu Province of China.
Availability of Data and Materials
All original data are available upon request.
Author information
Authors and Affiliations
Contributions
G.H.W. and Z.L.J. conceived, organized and supervised the work; L.Z.D., J.M.Z. and L.S.Y. performed the experiments; Y.C.S., D.Q.G., L.H.X. and Q.Q.L. contributed to the analysis of data; G.H.W. prepared, wrote and revised the manuscript.
Corresponding authors
Ethics declarations
All experiments were conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Animal Ethics Committees of Nantong University (NU-2017-39).
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Fig. S1
Effect of VPA on spatial exploration and emotion in mice after FDL induction. (A) Proportions of time spent by mice exploring the novel object vs. both objects (RI values) in the novel object recognition test. (B–C) Results of the Y-maze test, (D–F) open-field test and (G) rotarod test performed 10 days after FDL induction. (H–I) Results of the Y-maze test performed 28 days after FDL induction. The data are presented as the means ± SEMs, n ≥ 8 mice/group. *p < 0.05 and **p < 0.01 for the indicated comparisons. (PNG 926 kb)
Fig. S2
Microarray analysis of the effect of VPA on the differentiation of primary cultured OPCs into oligodendrocytes after treatment with LPS in vitro. Gene-chip microarray analyses were performed using OPCs treated with vehicle, VPA (1 mM), LPS (5 μg/ml) or LPS + VPA for 24 h. The table lists several functional genes that showed at least 9-fold changes after VPA treatment based on two independent microarray assays. Interestingly, PPAR-γ, but not PPAR-α or PPAR-β, was upregulated by VPA. PPAR-γ is a transcription factor that regulates over 100 genes involved in inflammation, cell proliferation and differentiation. (PNG 1150 kb)
Rights and permissions
About this article

Cite this article
Ding, L., Zhou, J., Ye, L. et al. PPAR-γ Is Critical for HDAC3-Mediated Control of Oligodendrocyte Progenitor Cell Proliferation and Differentiation after Focal Demyelination. Mol Neurobiol 57, 4810–4824 (2020). https://doi.org/10.1007/s12035-020-02060-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-02060-8