
Abstract
Activation of microglia and mitochondrial dysfunction are two major contributors to the pathogenesis of sepsis-associated brain dysfunction. Mitochondrial dysfunction can alter the immunological profile of microglia favoring to a pro-inflammatory phenotype. Mitochondrial transplantation, as an emerging mitochondria-targeted therapy, possesses considerable therapeutic potential in various central nervous system injuries or diseases. However, the effects of mitochondrial transplantation on microglial polarization and neuroprotection after sepsis remain unclear. In this study, lipopolysaccharide (LPS)/interferon-γ (IFN-γ) and interleukin-4 (IL-4)/interleukin-13 (IL-13) were used to induce different phenotypes of BV2 microglial cells. We observed that mitochondrial content and function were enhanced in IL-4-/IL-13-stimulated microglia. In vitro, mitochondria treatment conferred neuroprotection by enhancing microglial polarization from the M1 phenotype to the M2 phenotype and suppressing microglial-derived inflammatory cytokine release. Furthermore, microglial phenotypes and behavior tests were assessed after mice were subjected to sepsis by cecal ligation and puncture (CLP) followed by intracerebroventricular injection of exogenous functional mitochondria. We found that mitochondrial transplantation induced microglial M2 rather than M1 response 24 h after sepsis. Mitochondrial transplantation improved behavioral deficits by increasing the latency time in inhibitory avoidance test and decreasing the number of crossing and rearing in the test session of open field test 10 days after CLP onset. These findings indicate that mitochondrial transplantation promotes the phenotypic conversion of microglia and improves cognitive impairment in sepsis survivors, supporting the potential use of exogenous mitochondrial transplantation therapy that may be a potential therapeutic opportunity for sepsis-associated brain dysfunction.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Sepsis is characterized by various organ dysfunctions and overwhelming systemic inflammatory responses [1, 2]. It has been well known that the central nervous system (CNS) is one of the first organs affected during sepsis [3], and neuroinflammation occurs early in the systemic inflammation [4]. Many clinical evidences indicate that sepsis survivors often develop prolonged form of cognitive dysfunction [5,6,7] in which neuroinflammation is a crucial step [8]. Although considerable advances have been made on the treatment of sepsis, cognitive impairment after sepsis remains a major cause of mortality and can be further aggravated by constant detrimental neurological responses [9]. To date, little is known on the exact mechanisms involved in brain dysfunction during sepsis; further attention and effective therapies are needed to be directed toward the neurological impairment in sepsis.
Microglial activation is one of the major changes during sepsis [10,11,12]. Activated microglia with amplified cytokine production might result in prolonged detrimental effects by negatively affecting neighboring neurons [13]. Two major microglial subpopulations, classically activated (M1) phenotype and alternatively activated (M2) phenotype, have been recognized and exerted either detrimental or beneficial effect on neurological outcomes [14, 15]. It is classically believed that the promotion of M2 microglia mediated anti-inflammatory effects while the reduction of M1 microglia–associated pro-inflammatory mediators may facilitate the resolution of uncontrolled inflammation and improve the prognosis of sepsis-associated brain dysfunction [16].
Mitochondria are critical for cellular metabolism and function [17]. Brain mitochondrial dysfunction occurs early after sepsis, which is temporally related to cognitive impairment [18, 19]. Furthermore, increasing evidences demonstrate that mitochondrial dysfunction in activated microglia has been observed in several animal models of CNS diseases [20, 21]. In a recent study, Baik et al. demonstrated that M1 microglia metabolisms are reprogrammed from oxidative phosphorylation to aerobic glycolysis during the inflammatory process, indicating the changes of mitochondrial function in M1 microglia [22]. Indeed, mitochondrial dysfunction will change the immunological phenotypes of microglia, which hamper the alternative activation of M2 microglia that associated with tissue repair and anti-inflammatory effects [23]. Fortunately, the delivery of exogenous functional mitochondria into damaged tissue has been proposed as a pivotal mechanism for the restoration of the function of damaged cells [24, 25]. In addition, the transfer of mitochondria from an allogeneic cell can restore cellular metabolic gene expression, metabolite profiles, and global energetic [26]. Mitochondrial transplantation, which aims to restore mitochondrial functions and control mitochondrial mass, has been considered as a potential master key for the treatment of various neurological diseases [27, 28]. However, whether the protective effect of exogenous mitochondria by inducing M2 microglial polarization on sepsis-associated brain dysfunction has not yet been well studied.
Therefore, we hypothesized that exogenous mitochondrial transplantation could effectively drive microglial phenotypic conversion and then ameliorate inflammatory responses and further improve cognitive impairment after sepsis. Here, we examined mitochondrial content and function in both lipopolysaccharide (LPS)-/interferon-γ (IFN-γ)- and interleukin-4 (IL-4)-/interleukin-13 (IL-13)-stimulated BV2 murine microglial cell lines and found that mitochondrial status was restored in IL-4-/IL-13-stimulated microglia. We explored the ability of exogenous transplanted mitochondria to modulate microglial phenotypic conversion both in vivo and in vitro. In addition, the effects of exogenous mitochondria on microglia-mediated inflammation and neuronal survival were also investigated. Furthermore, we assessed the impacts of transplanted functional mitochondria on long-term cognitive outcomes in the mice model of cecal ligation and puncture-induced sepsis to support the potential of transplanted mitochondria for the improvement of cognitive decline after sepsis.
Materials and Methods
Animals
All animal experiments were approved by the Institutional Animal Care and Use Committees of Xi’an Jiaotong University (Xi’an, Shaanxi, China). Male C57BL/6 mice (6 to 8 weeks old) were obtained from Experimental Animal Center of Xi’an Jiaotong University (Xi’an, Shaanxi, China) and housed in specific pathogen-free cages with a constant temperature of 21 ± 1 °C at 12-h light–dark cycle and free access to food and water.
Mitochondrial Isolation and Fluorescent Labeling
The mitochondrial isolation was the same as previous studies [29, 30]. Briefly, the pectoralis major was used to obtain allogeneic biopsies of muscle samples through a biopsy punch. The samples were washed with ice-cold homogenization buffer (300 mM sucrose, 10 mM HEPES, and 1 mM EGTA, pH 7.4) and then homogenized using Dounce tissue grinders. After homogenization, subtilisin A protease (Sigma-Aldrich, St. Louis, MO, USA) was added for 5 min and then filtered across 40-μm Falcon Cell Strainers (Thermo Fisher Scientific, Waltham, MA, USA). The filtrate was centrifuged at 600×g for 10 min at 4 °C. The supernatant was collected and filtered across a 10-μm pluriSelect mesh (PluriSelect, San Diego, CA, USA). Then, the filtrate was centrifuged at 9000×g for 10 min at 4 °C. The number of mitochondria was determined as previously described [29].
Cell Culture and Treatment
Mouse BV2 microglial cell line and HT22 hippocampal cell line were cultured in Dulbecco’s modified Eagle’s medium (HyClone, Logan, UT, USA) with 10% fetal bovine serum (Gibco, Rockville, MD, USA) and 1% penicillin/streptomycin (Gibco, Rockville, MD, USA) in a humidified atmosphere of 95% air and 5% CO2 at 37 °C. Microglial polarization into M1 and M2 states was induced according to the described protocol [31, 32].
Primary mouse microglial cells were isolated from the whole brain of postnatal day 1 C57BL/6 mouse pups. Briefly, the meninges were removed, and single-cell suspensions were generated by trypsinization and trituration. Mixed glial cultures were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. After 7–10 days, microglia were harvested from the mixed glial cultures by using a rocking device and were allowed to rest for 24 h prior to treatment.
To generate M1 microglia, cells were treated with LPS (100 ng/ml, Sigma-Aldrich, St. Louis, MO, USA) and recombinant mouse IFN-γ (10 ng/ml, PeproTech, Rocky Hill, NJ, USA) for 24 h. To generate M2 microglia, cells were treated with IL-4 (20 ng/ml, PeproTech, Rocky Hill, NJ, USA) and IL-13 (20 ng/ml, PeproTech, Rocky Hill, NJ, USA) for 24 h.
Mitochondrial Transplantation Protocol In vitro
About 3 × 106 intact mitochondria from C57BL/6 donor mice skeletal muscle were immediately added into different stimulated microglia media. LPS-/IFN-γ-stimulated microglia were treated with mitochondria for 24, 48, or 72 h. The control + mitochondria (ctrl + mito) group was given equivalent numbers of mitochondria for 24, 48, or 72 h without stimulating microglia using LPS/IFN-γ. After each treatment, cells were harvest for further testing.
Cellular MitoTracker Red Fluorescence Staining
After microglia were stimulated using LPS/IFN-γ and IL-4/IL-13, respectively, cells were incubated with MitoTracker Red CMXRos dye (M7512, Thermo Fisher Scientific, Waltham, MA, USA) for 20 min at a concentration of 200 nM. Then, cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, Sigma-Aldrich, St. Louis, MO, USA). The fluorescence microscopy of staining cells was observed by a confocal microscope system (TCS SP8 STED 3×, Leica, Germany) with excitation wavelength 579 nm and emission wavelength 599 nm.
mtDNA Quantitation
The levels of mtDNA copy number were assessed by quantitative polymerase chain reaction (qPCR) using TB Green Premix Ex Taq™ II Kit (TaKaRa, Kusatsu, Shiga, Japan) and the ABI StepOne plus Real-Time PCR System (Applied Biosystems). Cell total DNA was isolated using QIAamp DNA Mini Kit (Qiagen, Valencia, CA, USA). The mtDNA contents were determined as mtDNA-encoded nicotinamide adenine dinucleotide dehydrogenase subunit 1 (ND1) gene and normalized against the nuclear-encoded POU class 5 homeobox 1 gene as in a previous study [33]. Relative quantification of mtDNA copy number was calculated using the comparative-Ct method (ΔΔCt method). Specific primers were synthesized by the Shenggong (Wuhan, China) as follows: ND1: forward 5′-GTGACGTTGACATCCGTAAAGA-3′ and reverse 5′-GTAACAGTCCGCCTAGAAGCAC-3′, POU class 5 homeobox 1: forward 5′-AGAGTATGAGGCTACAGGGAC-3′ and reverse 5′-CAGAGCAGTGACGGGAACAGA-3′.
Mitochondrial Membrane Potential
The mitochondrial membrane potential was assessed by flow cytometry using JC-1 mitochondrial membrane potential detection assay kit (Beyotime, Nantong, China). Microglia were seeded at a density of 5 × 105 cells/well in 6-well plates. After incubating for 24 h, the cells were treated with different agent combinations for 24 h to induce corresponding phenotypes. The cells were collected and resuspended in 0.5 ml complete medium and then added 0.5 ml JC-1 working solution followed by incubating at 37 °C for 20 min in the dark according to the manufacturer’s instructions. After washing with buffer solution, cells were analyzed by flow cytometry.
ATP Determination
Intracellular ATP levels were measured using a firefly luciferase-based ATP assay kit (Beyotime, Nantong, China) according to the manufacturer’s instructions. After treatments, microglia were lysed and centrifuged at 12,000×g for 5 min. Supernatants were mixed with ATP detection working dilution in a black 96-well plate. Luminescence was measured using a Varioskan® Flash microplate reader (Thermo Fisher Scientific, Inc., Rockford, IL). Standard curves were also generated, and the protein concentration of each treatment groups was determined using the bicinchoninic acid method. Total ATP levels were expressed as micromoles per milligram of protein.
Direction of Transplanted Mitochondria In vitro
To observe isolated mitochondria transfer into M1 microglia, microglia were seeded on glass coverslips in 24-well plates and stimulated using LPS/IFN-γ. The prepared mitochondria stained with MitoTracker Red CMXRos dye (M7512, Thermo Fisher Scientific, Waltham, MA, USA) were immediately co-incubated with LPS-/IFN-γ-stimulated microglia for 24 h and proceed with the following immunocytochemistry experiments. The images were captured using a Nikon A1 confocal microscope (Nikon, Tokyo, Japan).
Immunocytochemistry
After control microglia and LPS-/IFN-γ-stimulated microglia were treated with mitochondria, cells were washed in phosphate-buffered saline (PBS), fixed for 20 min in 4% paraformaldehyde at room temperature, and were permeabilized in PBS containing 0.1% Triton X-100 for 15 min, followed by blocking in PBS with 1.5% bovine serum albumin (BSA) for 60 min. Then, they were incubated for 24 h at 4 °C with the following primary antibodies: an anti-Arg-1 rabbit antibody (1:50 dilution, Abcam, San Francisco, CA, USA) and an anti-iNOS rabbit antibody (1:50 dilution, Abcam, San Francisco, CA, USA). To visualize the transfer of mitochondria into LPS-/IFN-γ-stimulated microglia, cells were incubated with an anti-Iba-1 mouse antibody (1:100 dilution, Santa Cruz Biotechnology, USA) only. After three 10-min rinses in PBS, cells were incubated with Alexa Fluor 488–labeled goat anti-rabbit IgG (1:200 dilution, Invitrogen, Carlsbad, CA, USA) and Alexa Fluor 488–labeled goat anti-mouse IgG (1:200 dilution, Invitrogen, Carlsbad, CA, USA) for 2 h at room temperature in the dark and counterstained with DAPI (Sigma-Aldrich, St. Louis, MO, USA). Cells without the addition of primary antibody served as negative controls. Images were performed under a fluorescence microscope (Carl Zeiss, Jena, Germany) or Nikon A1 confocal microscope (Nikon, Tokyo, Japan). Fluorescence intensity of cells was measured using the ImageJ software.
Enzyme-Linked Immunosorbent Assay
The supernatant of different treated microglia was rapidly harvested. Tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), IL-4, interleukin-10 (IL-10), and transforming growth factor-β (TGF-β) levels were quantified with specific enzyme-linked immunosorbent (ELISA) assay kits for mice according to the manufacturer’s instructions (eBioscience, San Diego, CA, USA).
Co-culturing HT22 Cells and BV2 Cells Using the Transwell System
Here we use a transwell contact-independent neuron-microglia co-culture system. LPS-/IFN-γ-stimulated BV2 cells (5 × 104/well) with or without mitochondria treatment for 24 h were seeded in transwell inserts, and HT22 cells (2 × 105/well) were cultured in 24-well culture plates in the lower compartment. To evaluate the effects of the BV2 cells on the survival of the HT22 cells, we divided the cells into three groups: (1) control group—BV2 cells were seeded on transwell inserts; (2) LPS/IFN-γ group—BV2 cells were stimulated with LPS/IFN-γ for 24 h; (3) LPS/IFN-γ + mito group—LPS-/IFN-γ-stimulated BV2 cells were treated with mitochondria for 24 h. Then, microglia were co-cultured with HT22 cells in a humidified incubator at 37 °C for 24 h.
Determination of Apoptotic Rate by Flow Cytometry
After HT22 cells co-culture with microglia for 24 h, HT22 cells were collected, washed twice with cold PBS, resuspended at a density of 5 × 105 cells/ml, and then stained with 5 μl of Annexin V-FITC and 5 μl of PI for 15 min at room temperature in the dark according to the manufacturer’s instructions. Cell apoptosis was analyzed by using flow cytometry (Calibur, BD Biosciences, USA).
Cell Proliferation and Cytotoxicity Assay
After HT22 cells were co-cultured with microglia for 24 h, the HT22 cells were washed and evaluated via the CCK-8 assay. CCK-8 solution was added to each well of the plate according to manufacturer’s instructions (Beyotime, Nantong, China), which was then incubated for 4 h. Finally, the absorbance was measured at 450 nm using a microplate reader.
LDH Release Assay
LDH release was measured using an LDH Cytotoxicity Assay Kit (Beyotime, Nantong, China) according to the manufacturer’s instructions [34]. In brief, LDH activity in the supernatants of HT22 cells after co-culture with different treated microglia was collected. The blank control was the wells without cells but containing 200 μl of culture medium, and the maximum control was the wells containing 10% lactate release reagent. OD was measured at a wavelength of 490 using a Varioskan® Flash microplate reader (Thermo Fisher Scientific, Inc., Rockford, IL). LDH release was calculated by the following: Cytotoxicity (%) = [Experimental ODs − Blank ODs] / [Maximum LDH release ODs − Blank ODs] × 100.
CLP Model
Mice were subjected to cecal ligation and puncture (CLP) model as previously described with some additional modifications [35]. Briefly, mice were anesthetized using an inhalation of isoflurane and the midline laparotomy was performed. A longitudinal 1-cm incision was then made in the peritoneum. The cecum was exteriorized and ligated using a 4-0 silk suture at half of the distance between the basis of the cecum and the distal pole. Then, a 21-gauge needle was used to perforate with through-and-through puncture and the cecum was squeezed to extrude a small amount of feces. The cecum was returned to the peritoneal cavity. In the sham group, the cecum was taken out and returned without ligation or puncture. Survival rate and body weight were evaluated over a 10-day period with assessment every 12 h after treatment. In addition, mice were resuscitated with pre-warmed saline (50 ml/kg) subcutaneously (s.c.) immediately after CLP. All animals received antibiotics (ceftriaxone at 30 mg/kg and clindamycin 25 mg/kg) every 12 h s.c. for a total of 3 days to minimize incidence of infection [36].
Mitochondrial Transplantation Protocol In vivo
Mice were randomly divided into the following groups: sham + vehicle, sham + mito, CLP + vehicle, and CLP + mito. The CLP + vehicle and CLP + mito underwent CLP and received an ICV injection of isolated mitochondria or isometric control solution immediately after CLP. Freshly prepared mitochondria or equivalent amount of vehicle was injected into the left lateral ventricle (0.4 mm posterior, 1 mm lateral, and 2.5 mm ventral to the bregma) over 10 min, and the needle was left in place afterwards for another 10 min. No animals died after transplantation. Mice received a unilateral injection with 4 × 106/5 μl mitochondrial particles or only 5 μl of vehicle solvent. During the whole procedure, the temperature of rectal was monitored and maintained at 37.0–37.5 °C by surface heating or cooling until the mice recovered from anesthesia.
Direction of Transplanted Mitochondria In vivo
The isolated mitochondria were staining with 0.1 μM MitoTracker Red CMXRos for 20 min. The ICV injection of labeled mitochondria was given as previously described. Then, the brains were fixed in 4% paraformaldehyde for 24 h after operation. To verify the presence of exogenous mitochondria in hippocampal microglia, the images were captured using a Nikon A1 confocal microscope (Nikon, Tokyo, Japan).
Immunohistochemistry
At 24 h after operation onset, the mice were anesthetized and transcardially perfused with normal saline and 4% paraformaldehyde, and their brains were removed and post-fixed overnight in 4% paraformaldehyde in PBS at 4 °C. After dehydration in 20% and 30% sucrose, the brains were sectioned (13 μm) using a CryoStar NX50 cryostat (Thermo Scientific, San Jose, CA, USA). The sections were washed three times with PBS and permeabilized with 0.3% Triton X-100 in PBS for 15 min at room temperature. After the sections were washed with PBS, they were blocked for 60 min with 2% BSA in PBS; then, the sections were incubated overnight at 4 °C with primary antibody: an anti-Iba-1 mouse antibody (1:100 dilution, Santa Cruz Biotechnology, USA). After three washes times with PBS, sections were incubated with secondary antibody Alexa Fluor 488–labeled goat anti-mouse IgG (1:200 dilution, Invitrogen, Carlsbad, CA, USA) for 2 h at room temperature. Nuclei were counterstained with DAPI (Servicebio Technology Co., Ltd., Wuhan, Hubei, China). Fluorescence images were captured using a Nikon A1 confocal microscope (Nikon, Tokyo, Japan).
Western Blot Analysis
The hippocampus was rapidly isolated on ice at 24 h after operation, and western blot was performed as previous described [31]. The following primary antibodies to iNOS (1:1000, Abcam, San Francisco, CA, USA), IL-6 (1:1000, Proteintech, Wuhan, China), CD206 (1:1000, Proteintech, Wuhan, China), arginase-1 (Arg-1, 1:1000, Abcam, San Francisco, CA, USA), and β-actin (1:1000, Proteintech, Wuhan, China) were used. The immunoreactive bands were detected by enhanced chemiluminescence detection kit (Pierce, Rockford, IL). Images were captured using a chemiluminescence system (Bio-Rad, Hercules, CA, USA) and analyzed with QuantityOne software (Bio-Rad Laboratories).
Behavioral Tests
All behavioral tests were performed between 9:00 am and 4:00 pm during the light cycle in a sound-isolated room. To avoid interference of fecal boli and urination from other mice, the trial site was thoroughly cleaned after each test. All mice were evaluated in the open field test and the step-down inhibitory avoidance test 10 days after CLP.
Open Field Test
The training session and the test session were used to assess motor performance and non-associative memory respectively as previous study [8, 37]. During the test session, each trial lasted for 300 s, the number of lines crossed on the floor of the arena (number of crossing), and the number of rearing (elevation on rear paws) was recorded. After 24 h, the mice were submitted again in the test session. The behaviors were recorded by an open field system (Chengdu Taimeng Software Co. Ltd., Sichuan, China). The decreases in the number of crossing and rearing between the two sessions represented the retention of habituation memory.
Step-down Inhibitory Avoidance Test
The test was performed as previously described [8]. For training, mice were placed on a platform in the left corner of an inhibitory avoidance box. When mice stepped down on the grid with all four paws, they received a 0.3-mA, 2.0-s foot shock. After 24 h of the training session, mice were placed on the platform again and their latency to step down on the grid with all four paws was recorded. A maximum time of 180 s was used in the test session. The test trial latency of the test session was used to evaluate the aversive memory and was recorded by a step-down inhibitory avoidance system (Chengdu Taimeng Software Co. Ltd., Sichuan, China).
Statistical Analysis
Comparisons of differences in continuous variables were conducted using two-tailed unpaired Student’s t test or one-way ANOVA test followed by Tukey’s test for multiple comparisons. Data are presented as mean ± SD. Kaplan–Meier survival curves were analyzed using the log-rank test. The changes of body weight were determined by two-way ANOVA with two-factor repeated measures ANOVA. The mean latency of step-down inhibitory avoidance test was expressed as median and interquartile ranges using Mann–Whitney U test. Data were analyzed using GraphPad Prism (GraphPad Software 8.0, Inc., San Diego, CA, USA) and SPSS 20.0 (SPSS, Inc., Chicago, IL, USA). P < 0.05 was considered statistically significant.
Results
Mitochondrial Content and Function Were Restored in M2 Microglia
To verify the content and function of mitochondria in different phenotypes of microglia, LPS and IFN-γ as M1 triggers and IL-4 and IL-13 as M2 triggers were used to stimulate BV2 microglia. The protein expression of induced nitric oxide synthase (iNOS) (P = 0.0002, control vs. LPS/IFN-γ; Fig. 1a) was enhanced by LPS/IFN-γ treatment in microglia. In contrast, the expression of Arg-1 was dramatically increased in IL-4-/IL-13-stimulated microglia (P < 0.0001, control vs. IL-4/IL-13; Fig. 1b). We also found that the expression of mitochondrial biomarker COX IV was decreased after LPS/IFN-γ treatment (P = 0.0208, control vs. LPS/IFN-γ) and increased in the presence of IL-4/IL-13 (P = 0.0058, LPS/IFN-γ vs. IL-4/IL-13; Fig. 1c). Consistently, we used MitoTracker Red CMXRos, a probe that is dependent on mitochondrial membrane potential and its staining correlates with mitochondrial function. MitoTracker fluorescence intensity was decreased in LPS-/IFN-γ-stimulated microglia (P = 0.0475, control vs. LPS/IFN-γ) and markedly increased in IL-4/IL-13 treatment group (P = 0.0099; Fig. 1d, e) compared with those in LPS/IFN-γ treatment group, which indicated that M2 microglia contained more functional mitochondria. Additionally, the mtDNA copy number, normalized against the nuclear DNA, was reduced after LPS/IFN-γ treatment (P = 0.0309, control vs. LPS/IFN-γ) and increased IL-4/IL-13 treatment group (P = 0.0145, LPS/IFN-γ vs. IL-4/IL-13; Fig. 1f). These data indicated that M2 microglia presented more mitochondria content and more functional mitochondria than M1 detrimental phenotype. To determine the mitochondrial energy supply in different phenotypes of microglia, we tested the intracellular ATP levels. The results demonstrated that the intracellular ATP levels were obviously increased in IL-4-/IL-13-stimulated microglia compared with the LPS/IFN-γ group (P = 0.0016; Fig. 1g). To further explore the integrity and function of mitochondria in different phenotypes of microglia, we measured the mitochondrial membrane potential by flow cytometry. As shown in Fig. 1 h and i, mitochondria stained with JC-1 had higher red fluorescence signal in IL-4-/IL-13-stimulated microglia (P = 0.0063) than LPS-/IFN-γ-stimulated microglia, which indicated that the function and integrity of mitochondria were collapsed in M1 phenotype. These results demonstrate that mitochondrial content and function are restored in M2 beneficial microglial phenotype.

Mitochondrial content and function in different BV2 microglial phenotypes. The M1 microglia were induced by LPS plus IFN-γ, and the M2 microglia were induced by IL-4 plus IL-13. a Quantitative analysis showed that LPS/IFN-γ treatment increased iNOS expression (n = 3). b IL-4/IL-13 treatment increased Arg-1 expression (n = 3). c IL-4/IL-13 treatment increased COX IV expression (n = 4). d, e MitoTracker Red CMXRos fluorescence staining and its fluorescence intensity showed the increased mitochondrial content in IL-4/IL-13 treatment group (n = 5). f Mitochondrial DNA (mtDNA) copy number of different stimulated microglia was assessed by quantitative polymerase chain reaction (n = 3). g Intracellular ATP levels in different stimulated microglia (n = 5). h Flow cytometry analysis of the mitochondrial membrane potential of different stimulated microglia stained with JC-1. FL1 represents JC-1 green, and FL2 represents JC-1 red. In the normal cells, JC-1 aggregates and emits red fluorescence, but when the mitochondrial membrane potential collapsed, JC-1 is not able to accumulate in the mitochondria and emits green fluorescence. i Quantitation of the JC-1 red/JC-1 green ratio showed increased mitochondrial membrane potential after IL-4/IL-13 treatment (n = 6). Data are expressed as mean ± SD. *P < 0.05, ***P < 0.001 versus the control group; #P < 0.05, ##P < 0.01, ####P < 0.0001 versus the LPS/IFN-γ group, by one-way ANOVA and Tukey’s test
Mitochondrial Transplantation Tended to Prime Microglia Toward M2 Phenotype In vitro
We first confirmed whether transplanted mitochondria could be internalized into LPS-/IFN-γ-stimulated microglia. Confocal microscopy images suggested that MitoTracker Red CMXRos–labeled mitochondria were internalized into LPS-/IFN-γ-stimulated BV2 microglia (Fig. 2a). To further establish a direct link between exogenous mitochondria and the activation state of microglia, isolated mitochondria were applied for 24, 48, and 72 h to LPS-/IFN-γ-stimulated microglia, and protein expressions were analyzed after mitochondria treatment. As seen in Fig. 2b, the expression levels of iNOS (P < 0.0001) and IL-6 (P = 0.0101) were increased after LPS/IFN-γ stimulation, while iNOS (P < 0.0001, P = 0.0009, P < 0.0001, respectively) and IL-6 (P = 0.0085, P = 0.0062, P = 0.0260, respectively) protein levels were significantly decreased after the stimulated microglia exposure to mitochondria for 24, 48, and 72 h. In contrast, the expression of Arg-1 (P = 0.0014, P = 0.0016, P = 0.0210, respectively) was dramatically increased at 24, 48, and 72 h. Meanwhile, the level of CD206 was only increased at 24 and 48 h (P = 0.0030, P = 0.0354, respectively; Fig. 2c). Thus, exogenous mitochondria applied for 24 h were sufficient to induce the phenotypic switch of microglia.

Mitochondrial transplantation promoted the phenotypic conversion of microglia. a Confocal microscopy of MitoTracker Red CMXRos–labeled mitochondria was internalized into LPS-/IFN-γ-stimulated BV2 microglia. b Quantitative analysis of iNOS and IL-6 expressions in LPS-/IFN-γ-stimulated BV2 microglia treated with or without mitochondria for 24, 48, and 72 h (n = 3). c Quantitative analysis of Arg-1 and CD206 expressions in LPS-/IFN-γ-stimulated BV2 microglia treated with or without mitochondria for 24, 48, and 72 h (n = 3). Data are expressed as mean ± SD. *P < 0.05, ****P < 0.0001 versus the control group; #P < 0.05, ##P < 0.01, ###P < 0.001, and ####P < 0.0001 versus the LPS/IFN-γ group, by one-way ANOVA and Tukey’s test
Accordingly, the immunofluorescence assay about the primary microglial cells also showed that iNOS protein level (P < 0.0001) was increased after LPS/IFN stimulation. Compared with the LPS/IFN-γ group, the expression of iNOS (P = 0.0025) was lower and the expression of Arg-1 (P < 0.0001) was higher in mitochondria-treated M1 microglia for 24 h (Fig. 3a–d). We next assessed possible alterations of inflammatory factors secreted by primary microglia stimulated with LPS/IFN-γ in the absence or presence of mitochondria. The results showed that compared with the LPS/IFN-γ group, pro-inflammatory cytokines TNF-α (P < 0.0001), IL-6 (P < 0.0001), and IL-1β (P = 0.0115) were increased in the LPS/IFN-γ group, while mitochondria treatment significantly reduced the levels of TNF-α (P = 0.0034), IL-6 (P = 0.0020), and IL-1β (P = 0.0073). Anti-inflammatory cytokines were also analyzed. There were no differences in IL-10 levels after mitochondria treatment compared with the LPS/IFN-γ group. The levels of TGF-β (P = 0.0002) and IL-4 (P = 0.0005) were significantly increased in the LPS/IFN-γ + mito group compared with the LPS/IFN-γ group (Fig. 3e). Moreover, there were no differences in inflammatory cytokines between the ctrl and the ctrl + mito groups. Thus, these results indicate that microglia treated with exogenous mitochondria for 24 h after LPS/IFN-γ stimulation was adequate for skewing the microglia functional polarity from M1 to M2 phenotype and greatly easing the inflammatory storm triggered by LPS/IFN-γ.

Immunofluorescence staining and inflammatory cytokine release after primary microglia treated with exogenous mitochondrial for 24 h. a–d Representative immunofluorescence staining of iNOS and Arg-1 in LPS-/IFN-γ-stimulated primary microglia treated with or without mitochondria (n = 5). e Enzyme-linked immunosorbent assay–based comparisons of TNF-α, IL-6, IL-1β, TGF-β, IL-4, and IL-10 levels in the different treatment groups (n = 6). Data are expressed as mean ± SD. *P < 0.05, ****P < 0.0001 versus the control group; ##P < 0.01, ###P < 0.001, and ####P < 0.0001 versus the LPS/IFN-γ group, by one-way ANOVA and Tukey’s test
Mitochondrial Transplantation Reduced the Toxicity of Neuron Co-cultured with M1 Microglia In vitro
To further confirm the effects of mitochondria-mediated conversion of microglial polarization on neuronal survival, BV2 microglia were treated with LPS/IFN-γ in either the presence or the absence of mitochondria, or control, and co-cultured with HT22 neurons in a transwell co-culture system for 24 h (Fig. 4a). Results showed that LPS-/IFN-γ-stimulated microglia increased neuronal apoptosis (P < 0.0001; Fig. 4b, c), decreased cell viability (P = 0.0014; Fig. 4d), and enhanced lactate dehydrogenase release (P = 0.0021; Fig. 4e) compared with the control group, whereas when LPS-/IFN-γ-stimulated microglia were treated with mitochondria to skew its differentiation and then co-cultured with HT22 cells for 24 h, the cell apoptosis of HT22 was significantly decreased (P = 0.0022), the cell viability was enhanced (P = 0.0061), and the release of lactate dehydrogenase was reduced (P = 0.0203) compared with the LPS/IFN-γ group. These results demonstrate that exogenous mitochondria treatment modulates microglial polarization and thus enhances neuronal survival.

The effect of exogenous mitochondria–mediated microglial polarization on neuronal survival in Transwell system. a Mimetic diagram of in vitro experiment of BV2 microglia and HT22 cells transwell co-culture system. b, c The apoptosis of HT22 cells after incubation with LPS-/IFN-γ-stimulated BV2 microglia in the presence or absence of mitochondria for 24 h was analyzed by flow cytometry (n = 5). Control (ctrl) was HT22 cells co-cultured with untreated BV2 microglia. d Cell viability of HT22 cells were analyzed by the CCK8 assay (n = 6). e Neuronal death was quantified by LDH release (n = 5). Data are expressed as mean ± SD. **P < 0.01, ****P < 0.0001 versus the control group; #P < 0.05, ##P < 0.01 versus the LPS/IFN-γ group, by one-way ANOVA and Tukey’s test
Mitochondrial Transplantation Enhanced the Switch of M1 to M2 Phenotypes After Sepsis
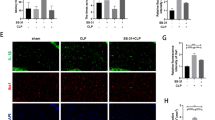
To determine whether administration of exogenous mitochondria contributed to microglial polarization through targeting of these mitochondria into the hippocampal microglia, we administered MitoTracker Red CMXRos–labeled mitochondria via ICV injection (5 μl, ~ 7.5 × 108) immediately after mice were submitted to CLP model. After 24 h, animals were perfused, and serial sections of hippocampus were processed for immunofluorescence staining. We observed that the MitoTracker Red CMXRos–labeled mitochondria were detected and well co-localized with Iba1-positive microglia in the dentate gyrus dentate (Fig. 5a), suggesting that MitoTracker Red CMXRos–labeled mitochondria internalized into microglia of the hippocampus. Furthermore, to confirm whether mitochondrial transplantation affects microglial polarization after sepsis, the expression levels of iNOS and Arg1 in the hippocampus at 24 h after CLP onset were measured. CLP + vehicle mice displayed an increase in iNOS protein level (P = 0.0003), which was reversed by the mitochondria treatment (P = 0.0120). Arg-1 protein level in the CLP + vehicle group did not differ from the sham + vehicle group (P = 0.1468), while it was significantly increased (P = 0.0077) in the CLP + mito group (Fig. 5b) compared with the CLP + vehicle group, indicating that transplanted mitochondria induced an effective shift of microglia from M1 to M2 state.

Mitochondria transplantation enhanced microglial phenotypic switch from M1 to M2 phenotype after sepsis. a After 24 h, the direction of transplanted mitochondria labeled with MitoTracker Red CMXRos was detected using the confocal microscopy to verify the presence of exogenous mitochondria internalized into microglia stained with anti-Iba1 (green) after sepsis in the hippocampus. b Western blotting showed the expressions of iNOS and Arg-1 in the hippocampus at 24 h after CLP model (n = 4). Data are expressed as mean ± SD. ***P < 0.001 versus the sham + vehicle group, #P < 0.05, ##P < 0.01 versus the CLP + vehicle group, by one-way ANOVA and Tukey’s test
Mitochondrial Transplantation Ameliorated Behavioral Deficits in Mice After Sepsis
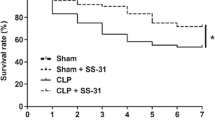
Here, the survival rates and body weight among the four groups were monitored consecutively for 10 days. The survival rates of mice from the sham + vehicle and the sham + mito groups both exhibited 100% during the 10-day follow-up. Mice from the CLP + vehicle and the CLP + mito groups presented 60.61% and 66% respectively 10 days after sepsis onset, and there were no significant differences between the two CLP groups (Fig. 6a). There were significant differences in body weight changes in sham + vehicle vs. CLP + vehicle group (P < 0.05) or sham + mito vs. CLP + mito group (P < 0.05). However, we did not observe significant differences in body weight changes between the CLP + vehicle and the CLP + mito groups on 10-day follow-up (Fig. 6b). These results demonstrated that mitochondrial transplantation did not improve the mortality and body weight of septic mice; however, it indeed ameliorated the behavioral deficits. In the open field test, there were no significant differences in the number of crossing and rearing between the CLP + vehicle group and the CLP + mito group in the training session (P = 0.5940), which demonstrated no differences in motor and exploratory activities between groups after CLP model. During the test session, after CLP, we could only observe the decreased number of crossing (P = 0.0280) and rearing (P = 0.0372) in the mitochondria treatment group as compared with those obtained in the training session, which indicated improvement in memory retention performance (Fig. 6c). In the step-down inhibitory avoidance test, mitochondrial transplantation dramatically increased the latency time when mice suffering from CLP challenged (P = 0.0286, CLP + mito vs. CLP + vehicle) (Fig. 6d). Therefore, these results indicate that mitochondrial transplantation improves cognitive impairment after sepsis.

Mitochondria transplantation ameliorated behavioral deficits after sepsis. There were 10 animals in the sham + vehicle group and the sham + mito group and 16 animals in the CLP + vehicle group and the CLP + mito group, respectively. a The survival rates were analyzed by the Kaplan–Meier method and compared by log-rank test and observed for 10 days. b Body weight changes were determined by two-way ANOVA with two-factor repeated measures ANOVA. c Open field test. The number of crossing and rearing has significant differences between the training and test sessions in the sham + vehicle group, sham + mito group, and CLP + mito group (Student’s t test, *P < 0.05). There were no differences between the training and test sessions in the CLP + vehicle group. Data are represented as mean ± SD (n = 10). d Step-down inhibitory avoidance test. Data are expressed as median and interquartile ranges (n = 10) for the mean latency in seconds and analyzed by Mann–Whitney U test. Significant differences of latency in the test sessions were found between the CLP + vehicle group and the CLP + mito group (*P < 0.05)
Discussion
The present study provides several novel insights regarding the neuroprotective effects of exogenous functional mitochondrial transplantation on modulating microglial polarization after sepsis. We demonstrated that mitochondrial content and function were restored in M2 beneficial microglial phenotype. Our in vivo and in vitro studies identified a pivotal role of mitochondrial transplantation in modulating microglial activation states. Our data also showed that M1 microglia treated with exogenous mitochondria promoted neuronal survival via suppressing multiple pro-inflammatory cytokines. Furthermore, mitochondrial transplantation improved memory retention and lessened behavioral deficits after sepsis by modulating microglial phenotype.
Mitochondrial dysfunction was observed in activated microglia [23]. However, the mitochondrial status in classically activated M1 microglia and alternatively activated M2 microglia is still unclear. In our present studies, one of the remarkable findings is that mitochondrial content increased in M2 microglia but decreased in M1 microglia. In fact, cellular mitochondrial content is not constant and could be changed under different physiological and pathological conditions [38]. Our results showed that mitochondrial biomarker COX IV expression, fluorescence intensity of MitoTracker Red CMXRos staining, and the mtDNA copy number were increased in M2 microglia. In addition, our study observed that the mitochondrial function in M2 microglia was restored as shown by increased JC-1 staining and intracellular ATP levels. These findings indicate that M2 microglia exhibit a better mitochondrial status including the mitochondrial content and function.
Given that mitochondrial dysfunction occurs in M1 pro-inflammatory phenotype, improving mitochondria status may be an efficient strategy for modulating the functional phenotype of microglia. Increasing studies have emphasized the important role of microglial phenotypic switch form M1 phenotype to M2 phenotype [16, 39]. It is argued that this switch is necessary to repair tissue damage and to promote the resolution of neuroinflammation during sepsis and thus may be a promising approach for alleviating sepsis-induced brain dysfunction. Several preclinical tests have used new drugs including antioxidants or electron transfer chain-boosting substrates to improve mitochondrial function; however, there is little effective treatment to rapidly reverse mitochondrial dysfunctions [40, 41]. Recently, the term of “mitochondrial transplantation” opens a novel horizon for many CNS diseases, such as neurodegenerative diseases, stroke, and spinal cord injury [42,43,44]. Our previous study have demonstrated that the isolated muscle-derived mitochondria were functionally intact, and the direct injection of these autologous mitochondria into the lateral ventricle after brain ischemia-reperfusion injury reduced cellular oxidative stress, apoptosis, and participated in neuroprotective effects [29]. In addition, a recent study has showed that muscle-derived mitochondria may be a good candidate for isolation and transplantation just as our previous study suggested [27]. However, skeletal muscle failure was a frequent manifestation of sepsis [45, 46]. In these cases, potential alternative viable mitochondria with normal respiratory function from allogeneic donor which derived from different individuals of the same species are necessary. Meanwhile, the usage of allogeneic mitochondrial donor sources would significantly increase the application of mitochondrial transplantation therapy. Recent research has reported that the infusion of allogeneic mitochondria does not cause alloreactivity and allorecognition reaction [47]. In our current study, we observed that exogenous mitochondria could integrate into M1 microglia in vitro. Furthermore, our results showed that exogenous mitochondria treatment sufficiently promoted the microglial shift from classically activated M1 phenotype toward protective M2 phenotype within 24 h. These results were consistent with the recent study which demonstrated that the enhancement of mitochondrial biogenesis and mitochondrial content in LPS-/IFN-γ-stimulated microglia could promote protective M2 microglial polarization [48].
Activated microglia can release a large amount of pro-inflammatory factors, including IL-1β, IL-6, and TNF-α [14]; these cytokines worsen the neurons around the activated microglia and even induce neuronal death after long-term exposure [49]. Here, we observed that the treatment of exogenous mitochondria suppressed LPS-/IFN-γ-mediated surge in pro-inflammatory cytokines. In addition, a recent study reported that injection of allogeneic mitochondria did not cause an innate inflammatory response [47]. Our results, in accordance with this evidence, showed that there was no further enhanced inflammatory response observed in the ctrl + mito group. These novel findings reveal that exogenous mitochondria play a vital role on skewing classically activated M1 phenotype toward protective M2 phenotype and, as a result, inhibit pro-inflammatory cytokine release in LPS-/IFN-γ-stimulated microglia. Microglia can directly protect neurons and promote hippocampal neurogenesis after brain injury [50]. Indeed, in our studies, LPS-/IFN-γ-stimulated microglia induced neuronal cell death, which was attenuated by exogenous mitochondria treatment.
Despite that the efficiency of mitochondrial transplantation has been observed in substantial CNS diseases, the neuroprotective effects in a clinically relevant model of sepsis have not been reported yet. To study the pathophysiology and mechanisms of sepsis, diverse animal models have been developed. Polymicrobial infection of sepsis induced by CLP is the most commonly used model because it simulated the progression and characteristics of sepsis in humans [51]. Antibiotics is one of the essential strategies for septic treatment [52]. Our present study showed that there were no differences in body weight and mortality between the mitochondria- and vehicle-treated mice after sepsis induction due to the antibiotics administration. Microglial activation and neuroinflammation have been implicated in the cognitive decline after sepsis in both animal models and humans [8, 13, 53, 54]. Memory retention or habituation to a novel environment can be measured by reduction in the number of crossing and rearing between the two sessions of open field task [37, 55]. The current study observed that mice presented a similar exploratory behavior in the CLP + vehicle group and the CLP + mito group when first exposed to the box during training session in the open field task, which indicated no difference in motor and exploratory activity between groups after CLP model. After 10 days of CLP onset, mitochondria treatment reduced the number of crossing and rearing in the test session, which indicated that the mice recognized the environment and remembered the previous exposure to this environment. However, the vehicle-treated mice did not show this pattern after sepsis, which reflected memory deficits. Our studies also observed that mitochondrial transplantation increased the latency time after sepsis in the step-down inhibitory avoidance task. These experiments reveal that mitochondria treatment has protective effects on neurologic function after sepsis as improved memory retention.
Our experiments elucidate key role for exogenous mitochondrial transplantation in mediating the microglial activation states after sepsis. Microglia could be activated at 24 h after sepsis and in a region-specific manner, with the hippocampus being more affected than cortical regions [56, 57]. Previously, we proposed that the activated microglia were increased significantly in the hippocampus early after sepsis [58]. Dentate gyrus as the input region of the hippocampus is thought to contribute to the formation of memories, the exploration of novel environments, and learning [59]. In this study, we showed that the transplanted exogenous mitochondria integrated into microglia in the dentate gyrus of the hippocampus after 24 h of CLP onset. Our study also suggested that exogenous mitochondrial transplantation caused a reduction in M1 phenotype and an increase in M2 phenotype. These in vivo studies indicated that ICV injection of exogenous functional mitochondria decreases cognitive impairment in sepsis survivors via directly promoting microglial polarization from M1 pro-inflammation phenotype to M2 anti-inflammation phenotype.
In conclusion, the current study demonstrated that mitochondrial transplantation may provide a promising therapeutic approach for sepsis-associated brain dysfunction, by modulating microglial polarization and suppressing pro-inflammatory cytokine secretion, as a consequence, improving long-term cognitive impairment after sepsis (Fig. 7). These findings not only provide new sight into the effect of transplanted mitochondria as a novel regulator of microglial phenotypes but may have potential to be translated into improved clinical outcomes in sepsis survivors.

Modulatory effects of mitochondrial transplantation on the phenotypic switch of microglia both in vivo and in vitro. Resting (M0) microglia were stimulated by LPS/IFN-γ to convert into the classical activated (M1) phenotype. The M1 microglia presented high expression of iNOS and IL-6, as well as pro-inflammatory cytokines TNF-α and IL-1β, which are cytotoxic. Additionally, the production of Arg-1 and CD206 was suppressed. Transplanted mitochondria counteracted the microglia reaction to an inflammatory challenge by converting microglia from a cytotoxic phenotype to a beneficial M2 microglia, as indicated by downregulation of iNOS and upregulation of Arg-1 and CD206. In vivo, mitochondrial transplantation had a neuroprotective effect by modulating phenotypic switch of microglia after sepsis
Data Availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Gofton TE, Young GB (2012) Sepsis-associated encephalopathy. Nat Rev Neurol 8(10):557–566. https://doi.org/10.1038/nrneurol.2012.183
Widmann CN, Heneka MT (2014) Long-term cerebral consequences of sepsis. Lancet Neurol 13(6):630–636. https://doi.org/10.1016/S1474-4422(14)70017-1
Carli F, Mayo N, Klubien K, Schricker T, Trudel J, Belliveau P (2002) Epidural analgesia enhances functional exercise capacity and health-related quality of life after colonic surgery: results of a randomized trial. Anesthesiology 97(3):540–549
Gyoneva S, Davalos D, Biswas D, Swanger SA, Garnier-Amblard E, Loth F, Akassoglou K, Traynelis SF (2014) Systemic inflammation regulates microglial responses to tissue damage in vivo. Glia 62(8):1345–1360. https://doi.org/10.1002/glia.22686
Iwashyna TJ, Ely EW, Smith DM, Langa KM (2010) Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA 304(16):1787–1794. https://doi.org/10.1001/jama.2010.1553
Annane D, Sharshar T (2015) Cognitive decline after sepsis. Lancet Respir Med 3(1):61–69. https://doi.org/10.1016/S2213-2600(14)70246-2
Ehlenbach WJ, Gilmore-Bykovskyi A, Repplinger MD, Westergaard RP, Jacobs EA, Kind AJH, Smith M (2018) Sepsis survivors admitted to skilled nursing facilities: cognitive impairment, activities of daily living dependence, and survival. Crit Care Med 46(1):37–44. https://doi.org/10.1097/CCM.0000000000002755
Michels M, Vieira AS, Vuolo F, Zapelini HG, Mendonca B, Mina F, Dominguini D, Steckert A et al (2015) The role of microglia activation in the development of sepsis-induced long-term cognitive impairment. Brain Behav Immun 43:54–59. https://doi.org/10.1016/j.bbi.2014.07.002
Dombrovskiy VY, Martin AA, Sunderram J, Paz HL (2007) Rapid increase in hospitalization and mortality rates for severe sepsis in the United States: a trend analysis from 1993 to 2003. Crit Care Med 35(5):1244–1250. https://doi.org/10.1097/01.Ccm.0000261890.41311.E9
Lemstra AW, Groen in’t Woud JC, Hoozemans JJ, van Haastert ES, Rozemuller AJ, Eikelenboom P, van Gool WA (2007) Microglia activation in sepsis: a case-control study. J Neuroinflammation 4:4. https://doi.org/10.1186/1742-2094-4-4
van Gool WA, van de Beek D, Eikelenboom P (2010) Systemic infection and delirium: when cytokines and acetylcholine collide. Lancet 375(9716):773–775. https://doi.org/10.1016/s0140-6736(09)61158-2
Sandiego CM, Gallezot J-D, Pittman B, Nabulsi N, Lim K, Lin S-F, Matuskey D, Lee J-Y et al (2015) Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc Natl Acad Sci U S A 112(40):12468–12473. https://doi.org/10.1073/pnas.1511003112
Hoogland IC, Houbolt C, van Westerloo DJ, van Gool WA, van de Beek D (2015) Systemic inflammation and microglial activation: systematic review of animal experiments. J Neuroinflammation 12:114. https://doi.org/10.1186/s12974-015-0332-6
Salter MW, Stevens B (2017) Microglia emerge as central players in brain disease. Nat Med 23(9):1018–1027. https://doi.org/10.1038/nm.4397
Eggen BJ, Raj D, Hanisch UK, Boddeke HW (2013) Microglial phenotype and adaptation. J NeuroImmune Pharmacol 8(4):807–823. https://doi.org/10.1007/s11481-013-9490-4
Cherry JD, Olschowka JA, O’Banion MK (2014) Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation 11:98. https://doi.org/10.1186/1742-2094-11-98
Mills EL, Kelly B, O'Neill LAJ (2017) Mitochondria are the powerhouses of immunity. Nat Immunol 18(5):488–498. https://doi.org/10.1038/ni.3704
d’Avila JCP, Santiago APSA, Amâncio RT, Galina A, Oliveira MF, Bozza FA (2008) Sepsis induces brain mitochondrial dysfunction. Crit Care Med 36(6):1925–1932. https://doi.org/10.1097/CCM.0b013e3181760c4b
Manfredini A, Constantino L, Pinto MC, Michels M, Burger H, Kist LW, Silva MC, Gomes LM et al (2019) Mitochondrial dysfunction is associated with long-term cognitive impairment in an animal sepsis model. Clin Sci (Lond) 133(18):1993–2004. https://doi.org/10.1042/CS20190351
Hayashi Y, Yoshida M, Yamato M, Ide T, Wu Z, Ochi-Shindou M, Kanki T, Kang D et al (2008) Reverse of age-dependent memory impairment and mitochondrial DNA damage in microglia by an overexpression of human mitochondrial transcription factor a in mice. J Neurosci 28(34):8624–8634. https://doi.org/10.1523/jneurosci.1957-08.2008
Ryu JK, Nagai A, Kim J, Lee MC, McLarnon JG, Kim SU (2003) Microglial activation and cell death induced by the mitochondrial toxin 3-nitropropionic acid: in vitro and in vivo studies. Neurobiol Dis 12(2):121–132. https://doi.org/10.1016/s0969-9961(03)00002-0
Baik SH, Kang S, Lee W, Choi H, Chung S, Kim JI, Mook-Jung I (2019) A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer’s disease. Cell Metab 30(3):493–507.e496. https://doi.org/10.1016/j.cmet.2019.06.005
Ferger AI, Campanelli L, Reimer V, Muth KN, Merdian I, Ludolph AC, Witting A (2010) Effects of mitochondrial dysfunction on the immunological properties of microglia. J Neuroinflammation 7:45. https://doi.org/10.1186/1742-2094-7-45
Hayakawa K, Bruzzese M, Chou SH, Ning M, Ji X, Lo EH (2018) Extracellular mitochondria for therapy and diagnosis in acute central nervous system injury. JAMA Neurol 75(1):119–122. https://doi.org/10.1001/jamaneurol.2017.3475
Wu S, Zhang A, Li S, Chatterjee S, Qi R, Segura-Ibarra V, Ferrari M, Gupte A et al (2018) Polymer functionalization of isolated mitochondria for cellular transplantation and metabolic phenotype alteration. Adv Sci (Weinh) 5(3):1700530–1700530. https://doi.org/10.1002/advs.201700530
Wu T-H, Sagullo E, Case D, Zheng X, Li Y, Hong JS, TeSlaa T, Patananan AN et al (2016) Mitochondrial transfer by photothermal nanoblade restores metabolite profile in mammalian cells. Cell Metab 23(5):921–929. https://doi.org/10.1016/j.cmet.2016.04.007
Nakamura Y, Park JH, Hayakawa K (2019) Therapeutic use of extracellular mitochondria in CNS injury and disease. Exp Neurol:113114. https://doi.org/10.1016/j.expneurol.2019.113114
Gollihue JL, Rabchevsky AG (2017) Prospects for therapeutic mitochondrial transplantation. Mitochondrion 35:70–79. https://doi.org/10.1016/j.mito.2017.05.007
Zhang Z, Ma Z, Yan C, Pu K, Wu M, Bai J, Li Y, Wang Q (2019) Muscle-derived autologous mitochondrial transplantation: a novel strategy for treating cerebral ischemic injury. Behav Brain Res 356:322–331. https://doi.org/10.1016/j.bbr.2018.09.005
Preble JM, Pacak CA, Kondo H, MacKay AA, Cowan DB, McCully JD (2014) Rapid isolation and purification of mitochondria for transplantation by tissue dissociation and differential filtration. J Vis Exp 91:e51682. https://doi.org/10.3791/51682
Zhai Q, Li F, Chen X, Jia J, Sun S, Zhou D, Ma L, Jiang T et al (2017) Triggering receptor expressed on myeloid cells 2, a novel regulator of immunocyte phenotypes, confers neuroprotection by relieving neuroinflammation. Anesthesiology 127(1):98–110. https://doi.org/10.1097/aln.0000000000001628
Yang X, Xu S, Qian Y, Xiao Q (2017) Resveratrol regulates microglia M1/M2 polarization via PGC-1α in conditions of neuroinflammatory injury. Brain Behav Immun 64:162–172. https://doi.org/10.1016/j.bbi.2017.03.003
Bi J, Zhang J, Ren Y, Du Z, Li Q, Wang Y, Wei S, Yang L et al (2019) Irisin alleviates liver ischemia-reperfusion injury by inhibiting excessive mitochondrial fission, promoting mitochondrial biogenesis and decreasing oxidative stress. Redox Biol 20:296–306. https://doi.org/10.1016/j.redox.2018.10.019
Xu Y, Zhi F, Peng Y, Shao N, Khiati D, Balboni G, Yang Y, Xia Y (2019) Delta-opioid receptor activation attenuates hypoxia/MPP(+)-induced downregulation of PINK1: a novel mechanism of neuroprotection against Parkinsonian injury. Mol Neurobiol 56(1):252–266. https://doi.org/10.1007/s12035-018-1043-7
Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA (2009) Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc 4(1):31–36. https://doi.org/10.1038/nprot.2008.214
Barichello T, Martins MR, Reinke A, Feier G, Ritter C, Quevedo J, Dal-Pizzol F (2005) Cognitive impairment in sepsis survivors from cecal ligation and perforation. Crit Care Med 33(1):221–223; discussion 262-223. https://doi.org/10.1097/01.ccm.0000150741.12906.bd
Tuon L, Comim CM, Petronilho F, Barichello T, Izquierdo I, Quevedo J, Dal-Pizzol F (2008) Time-dependent behavioral recovery after sepsis in rats. Intensive Care Med 34(9):1724–1731. https://doi.org/10.1007/s00134-008-1129-1
Navarro E, Gonzalez-Lafuente L, Perez-Liebana I, Buendia I, Lopez-Bernardo E, Sanchez-Ramos C, Prieto I, Cuadrado A et al (2017) Heme-oxygenase I and PCG-1alpha regulate mitochondrial biogenesis via microglial activation of Alpha7 nicotinic acetylcholine receptors using PNU282987. Antioxid Redox Signal 27(2):93–105. https://doi.org/10.1089/ars.2016.6698
Franco R, Fernández-Suárez D (2015) Alternatively activated microglia and macrophages in the central nervous system. Prog Neurobiol 131:65–86. https://doi.org/10.1016/j.pneurobio.2015.05.003
Wang W, Karamanlidis G, Tian R (2016) Novel targets for mitochondrial medicine. Sci Transl Med 8(326):326rv323. https://doi.org/10.1126/scitranslmed.aac7410
El-Hattab AW, Zarante AM, Almannai M, Scaglia F (2017) Therapies for mitochondrial diseases and current clinical trials. Mol Genet Metab 122(3):1–9. https://doi.org/10.1016/j.ymgme.2017.09.009
Chang JC, Wu SL, Liu KH, Chen YH, Chuang CS, Cheng FC, Su HL, Wei YH et al (2016) Allogeneic/xenogeneic transplantation of peptide-labeled mitochondria in Parkinson’s disease: restoration of mitochondria functions and attenuation of 6-hydroxydopamine-induced neurotoxicity. Transl Res 170:40–56.e43. https://doi.org/10.1016/j.trsl.2015.12.003
Huang PJ, Kuo CC, Lee HC, Shen CI, Cheng FC, Wu SF, Chang JC, Pan HC et al (2016) Transferring xenogenic mitochondria provides neural protection against ischemic stress in ischemic rat brains. Cell Transplant 25(5):913–927. https://doi.org/10.3727/096368915x689785
Gollihue JL, Patel SP, Eldahan KC, Cox DH, Donahue RR, Taylor BK, Sullivan PG, Rabchevsky AG (2018) Effects of mitochondrial transplantation on bioenergetics, cellular incorporation, and functional recovery after spinal cord injury. J Neurotrauma 35(15):1800–1818. https://doi.org/10.1089/neu.2017.5605
Callahan LA, Supinski GS (2009) Sepsis-induced myopathy. Crit Care Med 37(10 Suppl):S354–S367. https://doi.org/10.1097/CCM.0b013e3181b6e439
Owen AM, Patel SP, Smith JD, Balasuriya BK, Mori SF, Hawk GS, Stromberg AJ, Kuriyama N et al (2019) Chronic muscle weakness and mitochondrial dysfunction in the absence of sustained atrophy in a preclinical sepsis model. Elife 8:e49920. https://doi.org/10.7554/eLife.49920
Ramirez-Barbieri G, Moskowitzova K, Shin B, Blitzer D, Orfany A, Guariento A, Iken K, Friehs I et al (2019) Alloreactivity and allorecognition of syngeneic and allogeneic mitochondria. Mitochondrion 46:103–115. https://doi.org/10.1016/j.mito.2018.03.002
Ma L, Niu W, Lv J, Jia J, Zhu M, Yang S (2018) PGC-1α-mediated mitochondrial biogenesis is involved in cannabinoid receptor 2 agonist AM1241-induced microglial phenotype amelioration. Cell Mol Neurobiol 38:1529–1537. https://doi.org/10.1007/s10571-018-0628-z
Chu X, Zhou S, Sun R, Wang L, Xing C, Liang R, Kong Q (2018) Chrysophanol relieves cognition deficits and neuronal loss through inhibition of inflammation in diabetic mice. Neurochem Res 43(4):972–983. https://doi.org/10.1007/s11064-018-2503-1
Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, Tsirka SE, Maletic-Savatic M (2010) Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 7(4):483–495. https://doi.org/10.1016/j.stem.2010.08.014
Dejager L, Pinheiro I, Dejonckheere E, Libert C (2011) Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends Microbiol 19(4):198–208. https://doi.org/10.1016/j.tim.2011.01.001
Gotts JE, Matthay MA (2016) Sepsis: pathophysiology and clinical management. BMJ 353:i1585–i1585. https://doi.org/10.1136/bmj.i1585
Michels M, Danielski LG, Dal-Pizzol F, Petronilho F (2014) Neuroinflammation: microglial activation during sepsis. Curr Neurovasc Res 11(3):262–270. https://doi.org/10.2174/1567202611666140520122744
Andonegui G, Zelinski EL, Schubert CL, Knight D, Craig LA, Winston BW, Spanswick SC, Petri B et al (2018) Targeting inflammatory monocytes in sepsis-associated encephalopathy and long-term cognitive impairment. JCI Insight 3(9):e99364. https://doi.org/10.1172/jci.insight.99364
Viegas CM, Busanello ENB, Tonin AM, Grings M, Moura AP, Ritter L, Zanatta A, Knebel LA et al (2012) Chronic postnatal ornithine administration to rats provokes learning deficit in the open field task. Metab Brain Dis 27(4):479–486. https://doi.org/10.1007/s11011-012-9322-x
Moraes CA, Santos G, de Sampaio e Spohr TC, D’Avila JC, Lima FR, Benjamim CF, Bozza FA, Gomes FC (2015) Activated microglia-induced deficits in excitatory synapses through IL-1beta: implications for cognitive impairment in sepsis. Mol Neurobiol 52(1):653–663. https://doi.org/10.1007/s12035-014-8868-5
Hernandes MS, D’Avila JC, Trevelin SC, Reis PA, Kinjo ER, Lopes LR, Castro-Faria-Neto HC, Cunha FQ et al (2014) The role of Nox2-derived ROS in the development of cognitive impairment after sepsis. J Neuroinflammation 11:36. https://doi.org/10.1186/1742-2094-11-36
Zhang Z, Lei Y, Yan C, Mei X, Jiang T, Ma Z, Wang Q (2019) Probenecid relieves cerebral dysfunction of sepsis by inhibiting pannexin 1-dependent ATP release. Inflammation 42(3):1082–1092. https://doi.org/10.1007/s10753-019-00969-4
Jonas P, Lisman J (2014) Structure, function, and plasticity of hippocampal dentate gyrus microcircuits. Front Neural Circuits 8:107. https://doi.org/10.3389/fncir.2014.00107
Acknowledgments
We thank the National Local Joint Engineering Research Center for Precision Surgery & Regenerative Medicine, Shaanxi Provincial Center for Regenerative Medicine and Surgery Engineering Research, Xi’an Jiaotong University, 76 Yanta West Road, Xi’an 710061, Shaanxi, China, for providing the platform of some biochemistry experiments.
Funding
This study was supported by the National Natural Science Foundation of China (Grants 81774113, 81974540, 81801958), the Natural Science Basic Research Program of Shaanxi (Grant 2017JZ029), and the Fundamental Research Funds for the Central Universities (Grant xzy012019100).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. The study was conceived and coordinated by Qiang Wang and Chaoying Yan. Material preparation was performed by Zhi Ma and Hongli Ma. Data collection and analysis were performed by Chaoying Yan, Tao Jiang, and Qian Zhai. The first draft of the manuscript was written by Chaoying Yan and Zhanqin Zhang. The manuscript was reviewed and edited by Qiang Wang, Zhanqin Zhang, and Qing Li. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethics Approval
All procedures performed in studies involving animals were approved by the ethical standards of Institutional Animal Care and Use Committees of Xi’an Jiaotong University, Xi’an, China. Mice received humane care in accordance with the Guidelines for the Care and Use of Laboratory Animals of the National Institutes of Health.
Code Availability (Software Application and Custom Code)
GraphPad Prism Version 8.0; SPSS Version 20.0; ImageJ Version 1.51J.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yan, C., Ma, Z., Ma, H. et al. Mitochondrial Transplantation Attenuates Brain Dysfunction in Sepsis by Driving Microglial M2 Polarization. Mol Neurobiol 57, 3875–3890 (2020). https://doi.org/10.1007/s12035-020-01994-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-01994-3