
Abstract
Specific variations in the amino acid sequence of prion protein (PrP) are key determinants of susceptibility to prion diseases. We previously showed that an amino acid substitution specific to canids confers resistance to prion diseases when expressed in mice and demonstrated its dominant-negative protective effect against a variety of infectious prion strains of different origins and characteristics. Here, we show that expression of this single amino acid change significantly increases survival time in transgenic mice expressing bank vole cellular prion protein (PrPC), which is inherently prone to misfolding, following inoculation with two distinct prion strains (the CWD-vole strain and an atypical strain of spontaneous origin). This amino acid substitution hinders the propagation of both prion strains, even when expressed in the context of a PrPC uniquely susceptible to a wide range of prion isolates. Non-inoculated mice expressing this substitution experience spontaneous prion formation, but showing an increase in survival time comparable to that observed in mutant mice inoculated with the atypical strain. Our results underscore the importance of this PrP variant in the search for molecules with therapeutic potential against prion diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Prions are self-propagating infectious proteins that cause fatal neurodegenerative disorders known as transmissible spongiform encephalopathies (TSE) or prion diseases. Characterized by spongiform changes, gliosis, and neuronal degeneration in the central nervous system (CNS), these diseases include scrapie in sheep and goats, bovine spongiform encephalopathy (BSE) in cattle, chronic wasting disease (CWD) in cervids, and Creutzfeldt-Jakob disease (CJD) in humans [1, 2]. While the underlying trigger can be sporadic, genetic, or infectious in origin [3], the characteristic event in the pathogenesis of these diseases is misfolding of normal cellular prion protein (PrPC), giving rise to a protease-resistant, β-sheet-rich isoform known as PrPSc, which accumulates in the CNS leading to neurodegeneration [4].
The presence of aspartic acid (D) at codon 163 of PrPC, a polymorphism exclusive to the Canidae family [5], may account for the unusual resistance of canid species to prion diseases [6]. Studies of recombinant proteins exposed to denaturing agents and in vitro and in vivo prion propagation studies assessing the susceptibility of species historically considered prion-resistant (leporids, equids, and canids) have demonstrated that canid PrPC shows the greatest resistance to misfolding [7,8,9,10,11]. Bank voles (Myodes glareolus), by contrast, are highly susceptible to prion infection and have been used widely in prion research owing to their ability to efficiently propagate a broad spectrum of prion strains [12,13,14,15,16]. Bank vole PrPC is polymorphic, and either methionine (M) or isoleucine (I) can be expressed at codon 109 [17]. The adaptation of CWD in voles expressing isoleucine at codon 109 led to the isolation of the fastest prion strain (survival time, ∼ 35 days) identified to date [16]. Interestingly, overexpression of bank vole I109 PrP in transgenic mice leads to the development of spontaneous TSE, providing a very useful model for the study of sporadic prion diseases [18]. Given that sporadic forms are the most common prion diseases in humans, and their origin remains unknown, the generation of these models is essential for research. It has been suggested that sporadic prion diseases may be caused by random and stochastic misfolding of PrPC, resulting in the accumulation of PrPSc and consequent clinical and neuropathological features associated with prion disorders [19].
The canid D163 PrPC polymorphism is likely the main determinant of Canidae resistance to prion diseases. Indeed, the presence of this single substitution in mouse PrPC (N158D substitution in mouse PrPC numbering) prevents prion propagation both in vitro and in vivo [6]. We previously demonstrated that in vivo coexpression of the N158D PrPC variant and wild-type mouse PrPC significantly delays disease onset in transgenic mice inoculated with several prion strains of different origins and characteristics [20], suggesting that N158D PrPC is a promising candidate in the search for proteins with dominant-negative effects against a broad spectrum of prion strains. In the present study, we investigated whether this substitution could also prevent or delay the onset of prion disease in a highly susceptible model. Transgenic mice overexpressing bank vole I109 PrP and carrying this specific residue (TgVole-N159D mice) were inoculated with two prion isolates, and the resulting survival times compared with those of transgenic mice expressing comparable levels of bank vole PrPC (TgVole mice) but lacking this PrPC residue. To corroborate the ability of this amino acid substitution to confer protection against prion propagation, we used two distinct strains with very different neuropathological and biochemical features. For both inoculated prion strains, survival periods in TgVole-N159D mice were 52–108% longer than those of TgVole mice. These results are in good agreement with our previous findings demonstrating that expression of this specific amino acid substitution, even when expressed in a PrPC highly susceptible to misfolding, interferes with prion propagation and delays the onset of disease caused by multiple, distinct prion strains.
Materials and Methods
Ethics Statement
All procedures involving animals were approved by the University of Zaragoza’s Ethics Committee for Animal Experiments (permit number PI32/13) and were performed in accordance with recommendations for the care and use of experimental animals and with Spanish law (R.D. 1201/05).
Characterization of Transgenic Mouse Lines
Two different transgenic mouse models were used in the present study: transgenic mice expressing ∼ 3–4× the I109 polymorphic variant of bank vole PrP and carrying the critical dog amino acid substitution (I109-N159D PrPC), hereafter referred to as TgVole-N159D mice, and mice overexpressing ∼ 3–4× bank vole I109 PrPC, hereafter referred to as TgVole mice, which were used as controls. The murine PRNP promoter was used for both I109-N159D and I109 PrPC expression, in a murine Prnp0/0 background. Mice were generated and characterized as previously described [21].
Histological Analysis of PrP Distribution
The histological localization and distribution of cellular PrP in the brains of both TgVole-N159D and TgVole mice were analyzed by immunohistochemistry as previously described [20]. Briefly, paraffin-embedded brain sections were incubated with a 1% peroxidase solution for 20 min followed by hydrated autoclaving at 100 °C in citrate buffer for 30 min. PrP immunodetection was performed overnight at 4 °C using SAF84 (1:1000; Cayman Chemical) antibody. The anti-mouse Envision polymer (Dako) was used as the visualization system and DAB (diaminobenzidine, Dako) as the chromogen.
The cellular localization of PrP was further analyzed in the brains of TgVole-N159D and TgVole mice using immunofluorescence and confocal imaging. The immunofluorescence staining was performed as described previously [20]. Paraffin-embedded tissue sections from TgVole-N159D and TgVole mice were pre-treated with 1% peroxidase for 30 min. Sections were subsequently permeabilized with 0.1% Triton X-100 for 3 h at room temperature and subjected to hydrated autoclaving. Immunodetection was carried out with SAF84 antibody (1:1000) followed by a goat anti-mouse IgG biotin conjugate (1:100; Invitrogen) and an Alexa fluor 594 streptavidin conjugate (1:1000; Invitrogen). Sections were observed under a Zeiss laser-scanning confocal microscope LSM 510 (Carl Zeiss MicroImaging).
Analysis of PrPC Expression Levels of the Transgenic Lines
PrP expression levels from 10% brain homogenates TgVole and TgVoleN159D mice were analyzed by Western blot using SAF83 (1:400) monoclonal antibody (Bertin bioreagents) and compared with those from bank vole brain homogenates. Serial 1:2 dilutions of the brain homogenates were run and compared to the same dilutions of bank vole brain homogenates.
Inoculation of Transgenic Mice and Sample Processing
Mice were anesthetized with isoflurane and intracerebrally inoculated (right cerebral hemisphere) with 20 μl of a 1% brain homogenate. Both TgVole-N159D and TgVole mice were inoculated with one of two different isolates: CWD-vole, a CWD strain adapted to bank voles that contains I109 PrP and is characterized by very short survival times [16]; and an atypical prion isolate (Sp-TgVole isolate) of spontaneous origin. Sp-TgVole inoculum was obtained from brain homogenates from TgVole mice [22] that were sacrificed at 182 ± 5 days of age after developing a spontaneous neurodegenerative disorder linked to the overexpression of the I109 variant of bank vole PrP [18] (Supplementary Fig. 1). Intracerebral injections were performed using a 50-μl precision syringe and a 25-G needle. After inoculation, mice received a subcutaneous injection of buprenorphine (0.3 mg/kg) to induce analgesia.
Mice were monitored for the onset of neurologic signs after inoculation, sacrificed by cervical dislocation upon detection of clinical signs of terminal disease (i.e., severe locomotor disorders, poor body condition, and any signs of impaired feeding ability), and their brains collected. Coronal sections were cut at the level of the frontal cortex and medulla oblongata and immediately stored at − 80 °C for biochemical analyses. The remaining brain tissue was stored in 10% formalin fixative for histological analyses.
Histopathological Evaluation
Formalin-fixed brains were sectioned transversely at four standard levels for neuropathological analyses of the following brain areas: frontal cortex (Fc), septal area (Sa), thalamic cortex (Tc), hippocampus (Hc), thalamus (T), hypothalamus (Ht), mesencephalon (Mes), cerebellar cortex (Cbl), and medulla oblongata (Mo) [23]. Formalin-fixed brain tissues were embedded in paraffin wax, and 4-μm-thick tissue sections were mounted on microscope slides for hematoxylin-eosin staining. The intensity and distribution of spongiform changes were blindly evaluated using an optical microscope (Zeiss Axioskop 40) and semiquantitatively scored on a scale of 0 (absence of lesions) to 5 (high intensity lesions) in each of the aforementioned brain regions.
Analysis of PrPSc Deposition
The detection of PrPSc deposition in paraffin-embedded brains was performed using the paraffin-embedded tissue (PET) blot technique, as previously described [24, 25]. PrPSc was detected by incubation with the Sha31 primary monoclonal antibody (1:8000; SPI-Bio), followed by an alkaline phosphatase-coupled goat anti-mouse antibody (1:500; Dako). Immunolabeling was visualized using the NBT/BCIP substrate chromogen (nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl-phosphate; Sigma-Aldrich). The presence, intensity, and distribution of PrPSc aggregates were evaluated using a Zeiss Stemi DV4 stereomicroscope and semiquantitatively scored as described for spongiform lesions.
The distribution of PrPSc deposition was also analyzed by immunohistochemistry using a previously described protocol [26], with some modifications. Paraffin-embedded sections were pre-incubated with 98% formic acid for 5 min and underwent hydrated autoclaving in citrate buffer for 20 min at 96 °C. Peroxidase activity was blocked for 5 min using a peroxidase blocking reagent (Dako). Immunodetection of PrPSc was achieved by incubation with 6H4 monoclonal antibody (1:100, Prionics) followed by an anti-mouse Envision polymer (Dako). Sections were subsequently incubated with DAB (diaminobenzidine, Dako) and counterstained with hematoxylin.
Biochemical Analysis of Inoculated Strains and of PrPres in Spontaneously Affected and Inoculated Animals
Proteinase K (PK)-resistant PrPSc was detected and characterized in both the CWD-vole and Sp-TgVole inocula before inoculation and also in the brains of the diseased animals after culling. To this end, 10% brain homogenates from CWD-vole I109 inoculated animals (CWD-vole strain) were incubated with 85 μg/ml PK for 1 h at 42 °C with constant agitation (450 rpm), as previously described [27]. Biochemical characterization of spontaneously generated TgVole I109 prions (Sp-TgVole strain) and of those animals inoculated with this Sp-TgVole strain was also carried out as reported previously [28]. Briefly, brain homogenates (20% w/v) from clinically diseased animals were mixed with an equal volume of 100 mM Tris-HCl + 4% sarkosyl and incubated for 30 min at 37 °C. Homogenates were then digested with 200 μg/ml PK (Sigma-Aldrich) for 1 h at 55 °C with gentle agitation. Aliquots of samples were mixed with an equal volume of isopropanol/butanol (1:1 v/v) and centrifuged for 5 min at 20,000×g. Supernatants were discarded and pellets were re-suspended in denaturing sample buffer (NuPage). Both CWD-vole and Sp-TgVole inocula and also the brains of animals spontaneously affected and inoculated with the previous two inocula were analyzed by Western blotting using the 12B2 antibody (1:2500), which recognizes the 89–93 epitopes of bank vole PrP and with D18 antibody (1:5000), which recognizes the 143–149 epitopes.
Data Analysis
Survival times were analyzed using the Kaplan-Meier method and the survival curves for mice carrying the N159D substitution were compared with those of controls using the log rank test (α = 0.05). Differences in histopathological and PrPSc deposition profiles between transgenic mouse models were analyzed using the nonparametric Mann-Whitney U test, with p values < 0.05 considered significant. GraphPad Prism version 6.0 (GraphPad Software, La Jolla, CA, USA) was used for data analysis and to generate Kaplan-Meier curves and histopathology graphs.
Results
Expression of the N159D PrPC Substitution Markedly Increases Survival Time in Mice Challenged with the CWD-vole and Sp-TgVole Strains
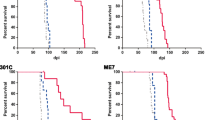
Transgenic mice expressing bank vole I109 PrPC and carrying the prion resistance-associated N159D amino acid substitution (TgVole-N159D mice) were intracerebrally inoculated with either the classical CWD-vole strain or the atypical Sp-TgVole strain (Supplementary Fig. 1). As controls, TgVole mice expressing comparable levels of bank vole I109 PrPC (∼ 3–4×) were challenged with the same isolates. Both transgenic lines present a normal cellular distribution of PrP through the brain and show a good expression of PrP in the cellular membrane that is about ~ 3–4× of that in bank vole brain homogenates (Supplementary Fig. 2). After challenge with the CWD-vole strain, survival time in TgVole-N159D mice was 108% longer than that of control TgVole mice, in which the mean survival time was 61 ± 4 days post-inoculation (dpi). In TgVole-N159D mice inoculated with the Sp-TgVole isolate, survival time was 52% longer than that of control TgVole mice (Table 1).
In both the TgVole and TgVole-N159D transgenic mice, overexpression of bank vole I109 PrPC (∼ 3–4×) leads to the development of a spontaneous neurodegenerative disorder. However, TgVole-N159D mice develop the spontaneous form of the disease showing an increase in survival time of 60% relative to non-inoculated TgVole animals (Table 1). This relative increase in survival is comparable to that observed in TgVole-N159D mice inoculated with the Sp-TgVole strain. For both inoculated strains, survival time was significantly longer in TgVole-N159D versus TgVole mice (Fig. 1). Significant differences in survival were obtained between TgVole-N159D and TgVole mice for both strains inoculated and between non-inoculated, spontaneously sick TgVole-N159D and TgVole mice (Fig. 1).

Survival curves for TgVole and TgVole-N159D mice (non-inoculated or after challenge with CWD-vole or Sp-TgVole isolates). Analysis of survival curves using the log rank test (α = 0.050) revealed significant differences between TgVole and TgVole-N159D mice after inoculation with either CWD-vole (p = 0.0007) or Sp-TgVole (p = 0.0011) isolates and between non-inoculated TgVole and TgVole-N159D mice (p < 0.0001) that developed spontaneous forms of the disease. Survival periods are expressed as days post-inoculation (dpi) for inoculated groups and as days of age at which mice succumbed to the spontaneous TSE for non-inoculated groups
Despite the significant delay in disease onset, TgVole-N159D mice exhibited clinical signs of neurodegeneration identical to those seen in TgVole mice. Animals inoculated with the CWD-vole strain exhibited dorsal kyphosis, circling behavior, cachexia, and tremor. By contrast, those inoculated with the Sp-TgVole isolate showed mild kyphosis and rapidly progressing ataxia.
Expression of the N159D Substitution Does Not Alter the Neuropathological Features of the Disease nor the Biochemical Features of the Different Strains
Expression of the PrPC N159D substitution, a key amino acid substitution associated with prion disease resistance in dogs [6], considerably delayed the onset of clinical signs, but did not significantly alter the neuropathological features exhibited by TgVole-N159D mice. Similar results were observed in our previous study, in which the effects of this dog-specific substitution were studied in the mouse PrP backbone [20].
Semi-quantitative analysis of spongiosis and prion protein deposition patterns in nine brain areas revealed no significant differences between TgVole-N159D and TgVole mice inoculated with the same isolate (Fig. 2). All mice inoculated with the CWD-vole strain showed moderate spongiform changes and discretely distributed PrPSc deposits, which were particularly conspicuous in the thalamus. In other brain regions, such as the hippocampus, spongiosis was minimal and PrPSc deposition scores were very low (Figs. 2 and 3). These neuropathological features coincide with those described in I109 bank voles infected with the same strain, in which prominent involvement of the thalamus has been reported [16]. Moreover, both TgVole-N159D and TgVole mice inoculated with the atypical Sp-TgVole isolate exhibited severe vacuolar changes in the cortex (Fc and Tc) and in the hippocampus, in which we observed the most intense PrPSc deposition for this isolate (Figs. 2 and 3). To verify that the Sp-TgVole strain maintained its neuropathological characteristics after experimental transmission to TgVole and TgVole-N159D mice, we performed a histopathological assessment of brain samples from TgVole and TgVole-N159D animals that spontaneously developed the disease at ∼ 182 and ∼ 292 days of age, respectively. The lesion profiles and PrPSc deposition patterns in these animals were almost identical to one another and to those of mice inoculated with the Sp-TgVole isolate (Fig. 2).

Spongiosis and PrPSc deposition profiles in the brains of TgVole and TgVole-N159D mice inoculated with CWD-vole or Sp-TgVole prion isolates, or non-inoculated. Spongiosis and PrPSc deposition were evaluated semiquantitatively on a scale of 0 (absence of lesions/deposits) to 5 (high intensity lesions/deposition) in the following nine brain areas: frontal cortex (Fc), septal area (Sa), thalamic cortex (Tc), hippocampus (Hc), thalamus (T), hypothalamus (Ht), mesencephalon (Mes), cerebellum (Cbl), and medulla oblongata (Mo). Graphs represent the mean with SEM of at least 5 mice per group. Comparison of the lesion and PrPSc deposition profiles of TgVole and TgVole-N159D mice revealed no significant differences between groups for any parameter (α = 0.05, Mann-Whitney U test)

a PET blot images of coronal brain sections from TgVole and TgVole-N159D mice inoculated with CWD-vole or Sp-TgVole isolates. The distribution pattern of PrPSc deposition (dark purple) in mice expressing the N159D substitution is very similar to that of non-inoculated TgVole controls. Animals inoculated with the Sp-TgVole strain show marked PrPSc deposition in the hippocampus. By contrast, in CWD-vole-inoculated animals immunolabeling with the Sha31 antibody is much weaker in the hippocampus, and strongest in the thalamus. b Immunohistochemical analysis of a TgVole mouse with the spontaneous form of the disease and of TgVole-N159D and TgVole mice inoculated with the Sp-TgVole strain. Note that the morphology and distribution of PrPSc deposits is almost identical in the three mice, all of which show abundant granular PrPSc deposition in the dentate gyrus and in Ammon’s horn of the hippocampus. Immunodetection was performed using the 6H4 monoclonal antibody (1:100)
The fact that similar neuropathological features were observed in TgVole and TgVole-N159D mice inoculated with the Sp-TgVole strain, and in non-inoculated TgVole mice, suggests the development of the same spontaneous disease in these inoculated mice in an accelerated or induced manner. This is also supported by the conservation of the biochemical features of the PrPres in spontaneously affected TgVole and TgVoleN159D animals and the animals inoculated with CWD-vole and Sp-TgVole strains (Fig. 4). The characteristic electrophoretic migration pattern after PK digestion of atypical prion disease with a 7 kDa band was detected on the spontaneously affected and Sp-TgVole inoculated animals, identical for both TgVole and TgVoleN159D models, whereas CWD-vole inoculated animals show a classical three banded pattern as expected with no observable differences between the two models.

PrPres detection from spontaneously affected, CWD-vole and Sp-TgVole inoculated TgVole and TgVoleN159D mouse brains. Ten percent brain homogenates from spontaneously affected, CWD-vole and Sp-TgVole inoculated TgVole and TgVoleN159D mice, selected for showing neurological disease onset at different days post-inoculation (dpi) were digested following two different protocols: spontaneously affected and Sp-TgVole-inoculated animal brains were treated according the atypical prion digestion protocol (as described in “Materials and methods” section) and the CWD-vole inoculated ones were digested with 85 μg/ml of Protease-K (PK). Digested samples were analyzed by Western blot using 12B2 (1:2500) and Saf83 (1:400). As expected from previous studies with spontaneously affected TgVole animals, both TgVole and TgVoleN159D animals showed the same ~ 7 kDa band, characteristic of atypical prion disorders and showing no difference between the electrophoretic migration pattern of the two distinct models. Similarly, Sp-TgVole inoculated animals show also the low molecular weight band associated with atypical prion diseases in higher amounts than the spontaneously affected animals, as expected for inoculated ones. CWD-vole inoculated animals present the classical three-banded pattern as the strain of origin. Overall, the results suggest that there is no alteration of the biochemical features of each strain upon infection of TgVoleN159D, all being comparable to the TgVole counterparts and the originally inoculated prion strains. Control: undigested bank vole brain homogenate. Mw: molecular weight
Discussion
The role in prion disease resistance of certain naturally occurring variants in the amino acid sequence of PrPC has been intensively studied. The presence of at least one arginine at codon 171 in sheep PrPC appears to confer low susceptibility to classical scrapie infection [29, 30]. Studies have demonstrated that this single amino acid substitution exerts a dominant-negative inhibitory effect on prion replication both in vitro and in vivo [31,32,33,34]. Heterozygosity for specific human PrPC polymorphisms also protects against acquired, sporadic, and some familial prion diseases [35, 36]. The resistance-associated human polymorphisms E219K and G127V not only confer strong protection against human prion diseases, but also exert a dominant-negative inhibitory effect on prion propagation when coexpressed with wild-type prion protein [33, 36, 37]. Introduction of these naturally occurring single mutations into an exogenous PrPC therefore represents a potential therapeutic strategy [33, 37]. However, when searching for dominant-negative PrPs, it is important to bear in mind that susceptibility to TSE depends on both the PRNP genotype and the infectious strain [38, 39]. Although strong, the resistance to prion propagation conferred by most of these polymorphisms is strain-specific [40,41,42]. In this study, we investigated the potential protective effect of the D163 (D159 in bank vole numbering) PrPC residue. This amino acid is almost exclusive to canid species [5, 6], in which no naturally occurring TSE have been described. Moreover, the PrPC form expressed by these species is highly resistant to misfolding in vitro [7] and appears not to undergo misfolding in vivo [6, 8].
We previously demonstrated that mice overexpressing a mutated prion protein carrying the N158D amino acid substitution are completely resistant to prion infection when inoculated with a variety of mouse-adapted prion strains [6]. Moreover, we have shown that coexpression of wild-type mouse PrPC and a mutant PrPC variant carrying this specific dog amino acid substitution has a dominant-negative effect on the in vivo propagation of mouse-adapted prion strains of scrapie and BSE origins [20]. However, could this amino acid substitution, characteristic of the most resistant mammalian species, prevent the misfolding of a PrPC characterized by an extraordinary promiscuity to propagate numerous prion strains [15]?
Here, we show that in mice overexpressing bank vole PrPC I109, whose misfolding ability is such that its single overexpression leads to the development of a spontaneous prion disease [18], the presence of the N159D substitution in PrPC significantly delays the onset of clinical signs. These findings suggest that the protective effect of this amino acid change characteristic of canids is stronger when expressed in mouse PrPC [6]. Although non-inoculated TgVole-N159D mice present a delay in the onset of clinical signs, we have observed that the expression of this resistance-associated substitution does not prevent spontaneous prion formation in these animals. However, it is important to remember that bank vole PrPC is greatly prone to conversion and allows the propagation of prion strains which are refractory to be transmitted in wild-type and transgenic mice [12, 14, 43]. Bank voles expressing the I109 polymorphic variant of PrPC are more susceptible to certain familial prion disorders than transgenic mice overexpressing homologous PrP carrying the corresponding mutation that causes the disease in humans [14, 44]. However, we show that the N159D substitution can increase survival time in a model that overexpresses a form of PrPC that is highly susceptible to misfolding induced by almost all prion strains. The prolongation of survival time was especially striking in TgVole-N159D mice after inoculation with the classical CWD-vole prion strain, which is the prion strain causing the shortest survival times described to date [16]: survival time in these mice was 108% longer than that of TgVole mice. While a significant increase in survival time was also observed in TgVole-N159D mice inoculated with Sp-TgVole isolate, an atypical strain of spontaneous origin, this effect was less marked than that observed for the CWD-vole strain (Table 1). Thus, the protective effect of the N159D substitution, although considerable in all cases, appears not to be homogeneous across strains. This observation is in good agreement with our previous findings in mice coexpressing the protein carrying the dog-specific substitution, in which the mutated protein inhibited prion propagation in a strain-specific manner [20]. It could be discussed, however, that comparing a single transgenic line carrying the mutation (TgVole-N159D mice) with a single control line (TgVole mice) is a too straightforward approach to evaluate the protective effect of the substitution. We generated other mutated lines, but their PrP expression levels were not comparable with those of their corresponding controls. Nevertheless, the goal of the present study is to analyze the transmission barrier to TgVole-N159D mice, and we consider that the most important parameters determining that transmission are the PrP expression level and the distribution of PrP. TgVole-N159D and TgVole transgenic lines have a normal cellular distribution of PrP and they show no differences between PrP expression levels or electrophoretic migration patterns (Supplementary Fig. 2). Therefore, we believe that the comparison between these lines is a suitable way to evaluate the effect of the N-to-D substitution.
Analysis of neuropathological features in TgVole-N159D and TgVole mice revealed no significant differences in lesional or PrPSc deposition profiles (Fig. 2). All mice inoculated with the Sp-TgVole isolate, generated by spontaneous misfolding of I109 PrPC, showed near identical neuropathological profiles. This profile was clearly distinguishable from that of mice inoculated with the CWD-vole strain (Figs. 2 and 3). The distribution and morphology of PrPSc deposition in mice inoculated with the Sp-TgVole isolate was comparable to that of TgVole-N159D and TgVole mice that developed spontaneous forms of the disease (Figs. 2 and 3b). However, it should be borne in mind that non-inoculated TgVole-N159D and TgVole mice develop spontaneous TSE. Therefore, once they have exceeded the age at which this spontaneous disorder develops, it cannot be determined with certainty whether the observed neuropathology is a result of this phenomenon or a consequence of inoculation. Comparison of age at disease onset revealed that mice inoculated with the Sp-TgVole isolate succumbed to disease at a younger age than non-inoculated TgVole-N159D and TgVole mice (Table 1). Inoculation of the Sp-TgVole isolate thus appears to cause a seeding acceleration phenomenon, i.e., the spontaneous pathological process characteristic of the model is accelerated by exogenous inoculation of the isolate [45]. Similar findings were reported in a study using transgenic mice overexpressing bank vole I109 prion protein [18]; inoculation of brain extracts from spontaneously diseased mice accelerated disease onset, reproducing the neuropathological hallmarks seen in transgenic mice expressing the same transgene. Moreover, as mentioned, expression of the substitution did not impede the spontaneous generation of the prion, and TgVole-N159D mice experimented an increase in survival similar to that of Sp-TgVole inoculated animals. These results agree with the suggestion that the effects of the N159D substitution are strain-dependent, and that both non-inoculated and Sp-TgVole inoculated mice propagate the same strain. In addition, it could be discussed that the delay in the onset of clinical signs observed in Sp-TgVole inoculated TgVole-N159D mice could be associated with the fact that this isolate was obtained from spontaneously sick TgVole animals. Thus, it could be possible that Sp-TgVole isolate is more adapted to the TgVole model. However, we should consider that non-inoculated TgVole-N159D mice show an almost identical delay in the onset of the disease. Moreover, in a parallel experiment, we inoculated brain homogenates from spontaneously sick TgVole-N159D mice in TgVole and TgVole-N159D mice. We observed again that TgVole-N159D inoculated mice presented a survival period ~ 50% longer than TgVole animals (data not shown), although the isolate was obtained from TgVole-N159D mice and could therefore be better adapted to this model. We can conclude that while expression of the resistance-associated amino acid change significantly increased survival times, it did not alter the pathological features of the inoculated strains. These results are in agreement with our previous findings [20] and suggest that the delayed appearance of clinical signs in TgVole-N159D mice is not caused by strain modifications resulting from the N159D substitution.
The precise molecular mechanisms by which the N159D substitution prolongs survival time in TgVole-N159D mice remain unclear. However, this residue can extend the survival period in both inoculated mice and those that develop spontaneous disease, as evidenced by the significant differences in survival time between TgVole (182 ± 5 days) and TgVole-N159D mice (292 ± 10 days) with spontaneous disease (Table 1 and Fig. 1). Several theories have been proposed to explain the molecular basis of the anti-prion propagation effect of certain single amino acid changes in PrPC. We previously described the fully protective effect of the N-to-D substitution in mouse PrP against prion inoculation [6], and the dominant negative effect of this protective mutation [20]. It has been suggested that coexpression with wild-type PrP of certain heterologous PrPs carrying putative protective mutations may interfere with the interaction between similar PrP monomers [46,47,48]. This interference could potentially disrupt the mechanism by which PrPC is converted into PrPSc [48], since allelic variants can be structurally incompatible [49]. In fact, this disruptive effect has been tested as a potential anti-prion therapy both in vitro [50] and in vivo [51], with successful results. Furthermore, the introduction of single point mutations, and the heterologous interference they cause, has also been proposed to account for the long survival periods observed when a prion strain is transmitted to a new host [48]. The N159D substitution may exert a protective effect by inducing protein alterations that attenuate the rate of fibril formation and the stability of newly formed fibrils [6]. This molecular mechanism has been previously proposed for other PrPC amino acid substitutions [37, 52]. In fact, it has been shown that the protective human PrPC variant G127V, which also acts as a dominant-negative protein [37], hinders the formation of dimers and stable fibrils, thereby protecting against the development of prion diseases [53]. Given that the N-to-D substitution at this specific position significantly alters the surface charge of PrP [6], the N159D substitution may have a similar effect in the context of bank vole PrPC.
The results presented here indicate that the introduction of a specific canid N-to-D amino acid substitution into exogenously administered PrPC not only confers complete resistance to TSE in certain mouse models but also significantly increases survival times in models overexpressing PrPs that are highly susceptible to misfolding. Moreover, the N159D substitution delays the onset of clinical signs of both infectious and spontaneous forms of prion diseases. Together with our previous findings demonstrating a dominant-negative effect of prion protein carrying the N-to-D substitution against a variety of prion strains of different origins [20], we can conclude that this mutation could represent a useful tool to control the propagation of different prion strains when present in the correct PrP background.
References
Colby DW, Prusiner SB (2011) Prions. Cold Spring Harb Perspect Biol 3(1):a006833. https://doi.org/10.1101/cshperspect.a006833
Collins SJ, Lawson VA, Masters CL (2004) Transmissible spongiform encephalopathies. Lancet 363(9402):51–61
Prusiner SB (1998) The prion diseases. Brain Pathol 8(3):499–513
Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216(4542):136–144
Stewart P, Campbell L, Skogtvedt S, Griffin KA, Arnemo JM, Tryland M, Girling S, Miller MW et al (2012) Genetic predictions of prion disease susceptibility in carnivore species based on variability of the prion gene coding region. PLoS One 7(12):e50623. https://doi.org/10.1371/journal.pone.0050623
Fernandez-Borges N, Parra B, Vidal E, Erana H, Sanchez-Martin MA, de Castro J, Elezgarai SR, Pumarola M et al (2017) Unraveling the key to the resistance of canids to prion diseases. PLoS Pathog 13(11):e1006716. https://doi.org/10.1371/journal.ppat.1006716
Vidal E, Fernandez-Borges N, Pintado B, Ordonez M, Marquez M, Fondevila D, Torres JM, Pumarola M et al (2013) Bovine spongiform encephalopathy induces misfolding of alleged prion-resistant species cellular prion protein without altering its pathobiological features. J Neurosci 33(18):7778–7786. https://doi.org/10.1523/JNEUROSCI.0244-13.2013
Polymenidou M, Trusheim H, Stallmach L, Moos R, Julius C, Miele G, Lenz-Bauer C, Aguzzi A (2008) Canine MDCK cell lines are refractory to infection with human and mouse prions. Vaccine 26(21):2601–2614. https://doi.org/10.1016/j.vaccine.2008.03.035
Khan MQ, Sweeting B, Mulligan VK, Arslan PE, Cashman NR, Pai EF, Chakrabartty A (2010) Prion disease susceptibility is affected by beta-structure folding propensity and local side-chain interactions in PrP. Proc Natl Acad Sci U S A 107(46):19808–19813. https://doi.org/10.1073/pnas.1005267107
Chianini F, Fernandez-Borges N, Vidal E, Gibbard L, Pintado B, de Castro J, Priola SA, Hamilton S et al (2012) Rabbits are not resistant to prion infection. Proc Natl Acad Sci U S A 109(13):5080–5085. https://doi.org/10.1073/pnas.1120076109
Bian J, Khaychuk V, Angers RC, Fernandez-Borges N, Vidal E, Meyerett-Reid C, Kim S, Calvi CL et al (2017) Prion replication without host adaptation during interspecies transmissions. Proc Natl Acad Sci U S A 114(5):1141–1146. https://doi.org/10.1073/pnas.1611891114
Nonno R, Di Bari MA, Cardone F, Vaccari G, Fazzi P, Dell'Omo G, Cartoni C, Ingrosso L et al (2006) Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog 2(2):e12. https://doi.org/10.1371/journal.ppat.0020012
Agrimi U, Nonno R, Dell'Omo G, Di Bari MA, Conte M, Chiappini B, Esposito E, Di Guardo G et al (2008) Prion protein amino acid determinants of differential susceptibility and molecular feature of prion strains in mice and voles. PLoS Pathog 4(7):e1000113. https://doi.org/10.1371/journal.ppat.1000113
Pirisinu L, Di Bari MA, D'Agostino C, Marcon S, Riccardi G, Poleggi A, Cohen ML, Appleby BS et al (2016) Gerstmann-Straussler-Scheinker disease subtypes efficiently transmit in bank voles as genuine prion diseases. Sci Rep 6:20443. https://doi.org/10.1038/srep20443
Watts JC, Giles K, Patel S, Oehler A, DeArmond SJ, Prusiner SB (2014) Evidence that bank vole PrP is a universal acceptor for prions. PLoS Pathog 10(4):e1003990. https://doi.org/10.1371/journal.ppat.1003990
Di Bari MA, Nonno R, Castilla J, D'Agostino C, Pirisinu L, Riccardi G, Conte M, Richt J et al (2013) Chronic wasting disease in bank voles: Characterisation of the shortest incubation time model for prion diseases. PLoS Pathog 9(3):e1003219. https://doi.org/10.1371/journal.ppat.1003219
Cartoni C, Schinina ME, Maras B, Nonno R, Vaccari G, Di Baria MA, Conte M, Liu QG et al (2005) Identification of the pathological prion protein allotypes in scrapie-infected heterozygous bank voles (Clethrionomys glareolus) by high-performance liquid chromatography-mass spectrometry. J Chromatogr A 1081(1):122–126
Watts JC, Giles K, Stohr J, Oehler A, Bhardwaj S, Grillo SK, Patel S, DeArmond SJ et al (2012) Spontaneous generation of rapidly transmissible prions in transgenic mice expressing wild-type bank vole prion protein. Proc Natl Acad Sci U S A 109(9):3498–3503. https://doi.org/10.1073/pnas.1121556109
Will RG, Ironside JW (2017) Sporadic and infectious human prion diseases. Cold Spring Harb Perspect Med 7(1). https://doi.org/10.1101/cshperspect.a024364
Otero A, Bolea R, Hedman C, Fernández-Borges N, Marín B, López-Pérez O, Barrio T, Eraña H et al (2017) An amino acid substitution found in animals with low susceptibility to prion diseases confers a protective dominant-negative effect in prion-infected transgenic mice. Mol Neurobiol 55:6182–6192. https://doi.org/10.1007/s12035-017-0832-8
Castilla J, Gutierrez-Adan A, Brun A, Doyle D, Pintado B, Ramirez MA, Salguero FJ, Parra B et al (2004) Subclinical bovine spongiform encephalopathy infection in transgenic mice expressing porcine prion protein. J Neurosci 24(21):5063–5069. https://doi.org/10.1523/Jneurosci.5400-03.2004
Fernandez-Borges N, Di Bari MA, Erana H, Sanchez-Martin M, Pirisinu L, Parra B, Elezgarai SR, Vanni I et al (2018) Cofactors influence the biological properties of infectious recombinant prions. Acta Neuropathol 135(2):179–199. https://doi.org/10.1007/s00401-017-1782-y
Fraser H, Dickinson AG (1968) The sequential development of the brain lesion of scrapie in three strains of mice. J Comp Pathol 78(3):301–311
Schulz-Schaeffer WJ, Tschoke S, Kranefuss N, Drose W, Hause-Reitner D, Giese A, Groschup MH, Kretzschmar HA (2000) The paraffin-embedded tissue blot detects PrP(Sc) early in the incubation time in prion diseases. Am J Pathol 156(1):51–56. https://doi.org/10.1016/S0002-9440(10)64705-0
Andreoletti O, Simon S, Lacroux C, Morel N, Tabouret G, Chabert A, Lugan S, Corbiere F et al (2004) PrPSc accumulation in myocytes from sheep incubating natural scrapie. Nat Med 10(6):591–593. https://doi.org/10.1038/nm1055
Monleon E, Monzon M, Hortells P, Bolea R, Acin C, Vargas F, Badiola JJ (2005) Approaches to scrapie diagnosis by applying immunohistochemistry and rapid tests on central nervous and lymphoreticular systems. J Virol Methods 125(2):165–171. https://doi.org/10.1016/j.jviromet.2005.01.013
Castilla J, Saa P, Hetz C, Soto C (2005) In vitro generation of infectious scrapie prions. Cell 121(2):195–206. https://doi.org/10.1016/j.cell.2005.02.011
Pirisinu L, Marcon S, Di Bari MA, D'Agostino C, Agrimi U, Nonno R (2013) Biochemical characterization of prion strains in bank voles. Pathogens 2(3):446–456. https://doi.org/10.3390/pathogens2030446
Westaway D, Zuliani V, Cooper CM, Da Costa M, Neuman S, Jenny AL, Detwiler L, Prusiner SB (1994) Homozygosity for prion protein alleles encoding glutamine-171 renders sheep susceptible to natural scrapie. Genes Dev 8(8):959–969
Clouscard C, Beaudry P, Elsen JM, Milan D, Dussaucy M, Bounneau C, Schelcher F, Chatelain J et al (1995) Different allelic effects of the codons 136 and 171 of the prion protein gene in sheep with natural scrapie. J Gen Virol 76(Pt 8):2097–2101
Kaneko K, Zulianello L, Scott M, Cooper CM, Wallace AC, James TL, Cohen FE, Prusiner SB (1997) Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc Natl Acad Sci U S A 94(19):10069–10074
Zulianello L, Kaneko K, Scott M, Erpel S, Han D, Cohen FE, Prusiner SB (2000) Dominant-negative inhibition of prion formation diminished by deletion mutagenesis of the prion protein. J Virol 74(9):4351–4360
Perrier V, Kaneko K, Safar J, Vergara J, Tremblay P, DeArmond SJ, Cohen FE, Prusiner SB et al (2002) Dominant-negative inhibition of prion replication in transgenic mice. Proc Natl Acad Sci U S A 99(20):13079–13084. https://doi.org/10.1073/pnas.182425299
Geoghegan JC, Miller MB, Kwak AH, Harris BT, Supattapone S (2009) Trans-dominant inhibition of prion propagation in vitro is not mediated by an accessory cofactor. PLoS Pathog 5(7):e1000535. https://doi.org/10.1371/journal.ppat.1000535
Palmer MS, Dryden AJ, Hughes JT, Collinge J (1991) Homozygous prion protein genotype predisposes to sporadic Creutzfeldt-Jakob disease. Nature 352(6333):340–342. https://doi.org/10.1038/352340a0
Shibuya S, Higuchi J, Shin RW, Tateishi J, Kitamoto T (1998) Codon 219 Lys allele of PRNP is not found in sporadic Creutzfeldt-Jakob disease. Ann Neurol 43(6):826–828. https://doi.org/10.1002/ana.410430618
Asante EA, Smidak M, Grimshaw A, Houghton R, Tomlinson A, Jeelani A, Jakubcova T, Hamdan S et al (2015) A naturally occurring variant of the human prion protein completely prevents prion disease. Nature 522(7557):478–481. https://doi.org/10.1038/nature14510
Hill AF, Joiner S, Linehan J, Desbruslais M, Lantos PL, Collinge J (2000) Species-barrier-independent prion replication in apparently resistant species. Proc Natl Acad Sci U S A 97(18):10248–10253
Collinge J (2001) Prion diseases of humans and animals: Their causes and molecular basis. Annu Rev Neurosci 24:519–550. https://doi.org/10.1146/annurev.neuro.24.1.519
Atarashi R, Sim VL, Nishida N, Caughey B, Katamine S (2006) Prion strain-dependent differences in conversion of mutant prion proteins in cell culture. J Virol 80(16):7854–7862. https://doi.org/10.1128/JVI.00424-06
Striebel JF, Race B, Meade-White KD, LaCasse R, Chesebro B (2011) Strain specific resistance to murine scrapie associated with a naturally occurring human prion protein polymorphism at residue 171. PLoS Pathog 7(9):e1002275. https://doi.org/10.1371/journal.ppat.1002275
Houston F, Goldmann W, Chong A, Jeffrey M, Gonzalez L, Foster J, Parnham D, Hunter N (2003) Prion diseases: BSE in sheep bred for resistance to infection. Nature 423(6939):498. https://doi.org/10.1038/423498a
Di Bari MA, Chianini F, Vaccari G, Esposito E, Conte M, Eaton SL, Hamilton S, Finlayson J et al (2008) The bank vole (Myodes glareolus) as a sensitive bioassay for sheep scrapie. J Gen Virol 89(Pt 12):2975–2985. https://doi.org/10.1099/vir.0.2008/005520-0
Asante EA, Linehan JM, Smidak M, Tomlinson A, Grimshaw A, Jeelani A, Jakubcova T, Hamdan S et al (2013) Inherited prion disease A117V is not simply a proteinopathy but produces prions transmissible to transgenic mice expressing homologous prion protein. PLoS Pathog 9(9):e1003643. https://doi.org/10.1371/journal.ppat.1003643
Fernandez-Borges N, Erana H, Elezgarai SR, Harrathi C, Gayosso M, Castilla J (2013) Infectivity versus seeding in neurodegenerative diseases sharing a prion-like mechanism. Int J Cell Biol 2013:583498. https://doi.org/10.1155/2013/583498
Prusiner SB, Scott M, Foster D, Pan KM, Groth D, Mirenda C, Torchia M, Yang SL et al (1990) Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 63(4):673–686
Hope J, Morton LJ, Farquhar CF, Multhaup G, Beyreuther K, Kimberlin RH (1986) The major polypeptide of scrapie-associated fibrils (SAF) has the same size, charge distribution and N-terminal protein sequence as predicted for the normal brain protein (PrP). EMBO J 5(10):2591–2597
Priola SA, Caughey B, Race RE, Chesebro B (1994) Heterologous PrP molecules interfere with accumulation of protease-resistant PrP in scrapie-infected murine neuroblastoma cells. J Virol 68(8):4873–4878
Jahandideh S, Jamalan M, Faridounnia M (2015) Molecular dynamics study of the dominant-negative E219K polymorphism in human prion protein. J Biomol Struct Dyn 33(6):1315–1325. https://doi.org/10.1080/07391102.2014.945486
Horiuchi M, Priola SA, Chabry J, Caughey B (2000) Interactions between heterologous forms of prion protein: Binding, inhibition of conversion, and species barriers. Proc Natl Acad Sci U S A 97(11):5836–5841. https://doi.org/10.1073/pnas.110523897
Seelig DM, Goodman PA, Skinner PJ (2016) Potential approaches for heterologous prion protein treatment of prion diseases. Prion 10(1):18–24. https://doi.org/10.1080/19336896.2015.1123372
Lee CI, Yang Q, Perrier V, Baskakov IV (2007) The dominant-negative effect of the Q218K variant of the prion protein does not require protein X. Protein Sci 16(10):2166–2173. https://doi.org/10.1110/ps.072954607
Zhou S, Shi D, Liu X, Liu H, Yao X (2016) Protective V127 prion variant prevents prion disease by interrupting the formation of dimer and fibril from molecular dynamics simulations. Sci Rep 6:21804. https://doi.org/10.1038/srep21804
Acknowledgments
We thank MINECO for the Severo Ochoa Excellence Accreditation (SEV-2016-0644). The authors would like to thank the following for their support: the IKERBasque Foundation, the staff at the CIC bioGUNE animal facility and Patricia Piñeiro and Dr. Jan Langeveld from Central Veterinary Institute, Wageningen for kindly providing the 12B2 antibody.
Funding
This work was supported financially by the Spanish (AGL2015-65046-C2-1-R, AGL2015-65560-R) (MINECO/FEDER) and Interreg (POCTEFA EFA148/16) grants. Alicia Otero was supported by a research grant from the Government of Aragón (C020/2014) co-financed by the European Social Fund.
Author information
Authors and Affiliations
Contributions
JC and RB conceived the study; AO, CH, NFB, HE, BM, and MM performed most of the experiments; MASM and RN collaborated in the creation of the transgenic lines used; AO, HE, JJB, RB, and JC evaluated the results; AO, RB, JJB, HE, and JC wrote and reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethics Statement
All procedures involving animals were approved by the University of Zaragoza’s Ethics Committee for Animal Experiments (permit number PI32/13) and were performed in accordance with recommendations for the care and use of experimental animals and with Spanish law (R.D. 1201/05).
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 1080 kb)
Rights and permissions
About this article

Cite this article
Otero, A., Hedman, C., Fernández-Borges, N. et al. A Single Amino Acid Substitution, Found in Mammals with Low Susceptibility to Prion Diseases, Delays Propagation of Two Prion Strains in Highly Susceptible Transgenic Mouse Models. Mol Neurobiol 56, 6501–6511 (2019). https://doi.org/10.1007/s12035-019-1535-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-019-1535-0