
Abstract
Microglia-mediated neuroinflammatory responses are well known to inhibit neurogenesis in the dentate gyrus (DG) of the adult hippocampus, and growing evidence indicates that therapeutic intervention to suppress microglial activation could be an effective strategy for restoring the impaired neurogenesis and memory performance. In the present study, we investigated the effects of water-soluble arginyl–diosgenin analog (Arg-DG) on the adult hippocampal neurogenesis using a central LPS-induced inflammatory mice model, along with the fundamental mechanisms in vivo and in vitro using LPS-stimulated microglial BV2 cells. Arg-DG (0.6 mg/kg) attenuates LPS-impaired neurogenesis by ameliorating the proliferation and differentiation of neural stem cells (NSCs), and prolonging their survival. The impaired neurogenesis in the hippocampal DG triggered the cognitive function, and that treatment of Arg-DG led to the recovery of cognitive decline. Arg-DG also suppressed the production of LPS-induced pro-inflammatory cytokines in hippocampal DG by blocking microglial activation. In in vitro study, Arg-DG inhibited the production of nitric oxide (NO), nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2) expression, and prostaglandin D2 production (PGD2), as well as the pro-inflammatory cytokines, such as interleukin (IL)-6, IL-1β, and tumor necrosis factor alpha (TNF-α). The anti-inflammatory effect of Arg-DG was regulated by NF-κB and MAPK JNK signaling both in vivo, and in LPS-stimulated microglial BV2 cells. Taken together, these results suggest that Arg-DG might have the potential to treat various neurodegenerative disorders resulting from microglia-mediated neuroinflammation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Accumulated evidence has demonstrated that adult neurogenesis spontaneously and continuously occurs at two distinct neurogenic niches throughout adulthood, at least in the subgranular zone (SGZ) of hippocampal dentate gyrus (DG), and the subventricular zone of the lateral ventricle (SVZ), in several species [1, 2]. Though it is still a matter of extensive debate, the functions of regenerating neurons in the adult brain remain largely unexplored. Many rodent studies indicated that adult neurogenesis is necessary for hippocampus-dependent learning and memory tasks, and the population of neurogenesis has a positive correlation with behavioral tasks [3, 4]. Dysregulation in adult hippocampal neurogenesis appears to be a common feature in various neurodegenerative diseases, such as Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, amyotrophic lateral sclerosis, ischemia, and major depression [5, 6].
Adult neurogenesis is dynamically modulated by many physiological and pathological stimuli, such as learning, physical exercise [7], environmental enrichment [8], aging, seizure, ischemia, stroke, and acute or chronic stress [9]. It has been reported that adult hippocampal neurogenesis is suppressed by chronic neuroinflammation in normal aging, chronic stress, injuries, and neurodegenerative diseases [10]. Microglia are brain resident immune cells of the CNS, and have emerged as the primary mediators in brain injury and inflammatory responses. Microglial activation induced by the central or peripheral LPS administration has been shown to inhibit hippocampal neurogenesis [11, 12]. In addition, pro-inflammatory cytokines, such as IL-1β, IL-6, and TNF-α released from over-activated microglia after certain brain injuries, can specifically suppress adult neurogenesis by regulating the proliferation, differentiation, survival, and integration of newborn neurons into the existing neural networks [13,14,15], further suggesting that neuroinflammatory responses in activated microglia play a crucial role in regulating adult neurogenesis.
Toll-like receptors (TLR) play a crucial role in the innate immune response to various stimuli, including pathogens [16, 17]. These receptors are widely expressed on the surface of immune cells, such as monocytes, macrophages, and microglia. TLR4, as one type of receptor of the TLR family, serves as the primary modulator of innate immune responses to stimuli, by triggering a cascade of pro-inflammatory events in microglia [18]. It was reported that the Gram-negative bacterial lipopolysaccharide (LPS) from Escherichia coli could dramatically enhance the expression of TLR4 in both protein and mRNA level, leading to the activation of downstream intracellular signaling factors, such as nuclear factor-ĸB (NF-ĸB) and MAPKs, and ultimately initiating the expression of differential inflammation target genes [19]. LPS specifically binds to TLR4, thereby promoting the activation of downstream intracellular singling pathways in microglia [20]. It has been shown that NF-κB and MAPK signaling are involved in LPS-induced microglial activation and adult hippocampal neurogenesis. They also play key roles in the pathogenesis of microglia-mediated neurodegenerative diseases. NF-κB is a major transcription factor that plays an important role in the regulation of numerous pro-inflammatory genes. It is also important in the regulation of LPS-stimulated expression of iNOS, COX-2, and pro-inflammatory cytokines in microglial BV2 cells [21], and also involved in adult neurogenesis through the TLR4/NF-κB signaling pathway in mice [19]. In addition, MAPK-signaling pathways, such as p38, c-Jun N-terminal kinase (JNK), and extracellular-signal-regulated kinase (ERK1/2), are associated with the regulation of the production of inflammatory mediators in activated microglia, as well as neurogenesis in the adult hippocampus [22]. Therefore, regulation of NF-κB and MAPK activity in activated microglia has been considered a potentially useful strategy for balancing adult hippocampal neurogenesis dysfunction of various neurodegenerative diseases induced by neuroinflammation.
Several approaches to developing therapeutic strategies for impaired hippocampal neurogenesis have been attempted, namely exogenous stem cell transplantation, stimulation of endogenous neurogenesis, neuroprotection, and anti-inflammatory strategies [12]. Pharmacological interventions, consisting of the administration of free radical scavengers, apoptosis inhibitors, neurotrophic agents, and anti-inflammatory drugs to control microglia-mediated inflammatory response, seem to be promising therapeutic strategies for countering neuroinflammatory and neurodegenerative diseases [19, 23,24,25].
Diosgenin (DG), a naturally occurring steroidal saponin, has been reported to offer a variety of pharmacological activities, including anti-diabetic and anti-tumor activity, anti-inflammatory character, and the ability to improve severe memory dysfunction in transgenic model mice of Alzheimer’s disease (AD) [26], potential neuroprotection against transient focal cerebral ischemia–reperfusion injury in rats [27], and multiple sclerosis [28, 29]. Caprospinol, a lipophilic diosgenin derivative, reduced amyloid deposits in an Aβ1–42-infused AD rat model, thereby improving memory deficits [30]. However, the clinical application of DG is limited by its extremely low solubility and poor pharmacokinetic profile. In observations by our group and others [21, 31], a series of DG derivatives with balanced hydrophobicity exhibited improved anti-inflammatory activity against LPS-induced microglial BV2 cells. We have reported a water-soluble arginyl–diosgenin analog (Arg-DG) for osteogenesis [32]. Moreover, similar to that found in our previous study [21], in the pretest, Arg-DG also showed significant anti-inflammatory effect in LPS-stimulated microglial BV2 cells. Based on these studies, we selected Arg-DG for systemic investigation of its role in LPS-impaired hippocampal neurogenesis and the underlying mechanisms in vitro and in vivo.
Materials and Methods
Reagents
Arg-DG was previously reported, and Fig. S1A of the Supplementary Information (SI) shows the structure [32]. LPS (E. coli, serotype 055:B5, St. Louis, MO, USA), dimethylsulfoxide (DMSO), Tween-20, Griess reagent kit (Invitrogen, Eugene, Oregon, USA), Triton X-100, 2′,7′-dichlorofluorescein diacetate (DCF-DA), and phosphate buffered saline (PBS) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Cell culture medium and fetal bovine serum (FBS) were obtained from Life Technologies (Frederick, MD, USA). Primary antibodies against IL-1β/6, phosphorylated IκB, IκB, JNK, phosphorylated p38, p38, and phosphorylated ERK1/2 and ERK1/2 were purchased from Cell Signaling Technology (Beverly, MA, USA). TLR4, TNF-α, COX-2, iNOS, Histone H1, phosphorylated RelA, and β-actin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). NF-κB p65 was purchased from Abcam (Cambridge, UK).
Animals and Treatments
The 7-week-old male C57BL/6 mice from DAMUL Science (Daejeon, Korea) were housed under standard conditions (23 ± 1 °C, 55% humidity, 12 h:12 h of light/dark cycle) and were allowed free access to food and water. Mice were divided into four groups: saline-treated group (sham control), Arg-DG-treated group with intraperitoneal (i.p.) injection, saline-treated group with a single dose LPS (3 μg) by intracerebroventricular (ICV) injection, and Arg-DG-treated with LPS ICV injection group (n = 5, mice/group). Arg-DG solution (0.6 mg/kg) dissolved in saline was administrated by i.p. injection three times at 8-h intervals after recovering from anesthesia. BrdU labeling reagent (50 mg/kg, Invitrogen) was administrated by i.p. injection at 3 h before sacrifice, to examine the proliferation of NSCs in the hippocampal DG. The mice treated by Arg-DG and BrdU were sacrificed after 3 days for immature differentiation of NSCs. Mice receiving BrdU administration once daily for five consecutive days were used to evaluate the survival rate of newborn cells and mature differentiation of NSCs. The corresponding animals were sacrificed at the times indicated in the experimental schedule (Fig. S1B and C of the SI), and all animal experiments were carried out according to the guidelines of the Animal Care and Use Committee of Chonnam National University.
Tissue Preparation and Fluorescent Immunohistochemical Staining (IHC)
Mice were anesthetized with a combination of Zoletil 50 and Rompun, and transcardially perfused with sterile normal saline solution, then with fresh and ice-cold 4% paraformaldehyde (PFA) in PBS. Brains were isolated, placed in 4% PFA in PBS at 4 °C overnight, and transferred into a 30% sucrose solution for 3 days. The dehydrated mouse brains were embedded using Tissue-Tek OCT Compound (Sakura Finetek, Alphen and den Rijn, Netherlands) and stored at − 80 °C. They were then sectioned coronally at 40 μm using a freezing microtome (Model CM3050; Leica Microsystems, Bannockburn, IL, USA). Sections with hippocampal formation were collected and stored in a buffer solution containing 0.1 M PB (250 mL), glycerin (125 mL), and ethylene glycol (125 mL) at − 20 °C.
Sections were placed in slide glasses and washed by PBS solution containing 0.1% Triton X-100 (PBST) after 1 h air drying. The sections were incubated in 4% PFA solution and activated in 2 N HCl for 30 min at 37 °C, and then neutralized in PBS for BrdU immunostaining. The sections were blocked with 5% normal horse serum (Thermo Fisher Scientific, MA, USA) in PBST for 30 min. The sections were incubated overnight at 4 °C in PBST with the following primary antibodies: anti-BrdU, -DCX, -NeuN, -Iba1, and -cleaved caspase-3 (Abcam, Cambridge, UK). Secondary antibodies conjugated with FITC or Cy3 from Santa Cruz Biotechnology (Santa Cruz, CA, USA) were incubated for 2 h at room temperature (RT). They were then washed with PBST for 1 h. The nuclei were stained using propidium iodide (PI; Thermo Fisher Scientific, MA, USA) or 4′,6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific, MA, USA) and then mounted with mounting medium (Vector Laboratories Inc., Burlingame, CA).
A confocal microscope equipped with an argon/krypton laser and two helium/neon lasers (LSM5, Zeiss, Thornwood, NY, USA) were used to acquire IHC images. ImageJ (version 1.4; NIH, USA) was used to analyze the obtained images, and the cells were counted in a defined frame size of (20–50) with (35 μm × 35 μm) in each section [33, 34]. LSM software was used to visualize the double-labeled samples by z-stack acquisitions and three-dimensional reconstructions.
Open Field Test
Mice were placed into an “open field arena” of (40 cm × 40 cm), with 36-cm-high walls. The amount of locomotor activity was analyzed using ANY-maze software (Stoelting, Wood Dale, USA) system equipped with a video camera.
Novel Object Recognition Test
Each animal was habituated to open field arena of (40 cm × 40 cm), with 36-cm-high walls, for 20 min. Mice were then further exposed to two equal objects (A and A) placed in opposing sides of the arena for 8 min on the next day. After 24 h, object A was exchanged with object B (A and B) to observe memory assessment. Preference index was presented as [object (B)/both objects (A and B)] × 100. The mice were tracked and analyzed using ANY-maze software (Stoelting, Wood Dale, USA).
Morris Water Maze Test
Spatial learning and memory of mice was assessed by Morris water maze. A circular water pool (110 cm diameter) was used, and the surrounding was attached with visual cues. A clear escape platform of (10.5 cm × 8 cm), 6.5 cm high, was placed 1 cm below the water surface. Hidden platform assay was performed as learning trials. Mice were trained for four consecutive days with four trials (each trial for 40 s) per day. The time taken by each mouse to arrive on the hidden platform refers to escape latency. On the fifth day, probe test was performed to evaluate the memory retention with no platform for 80 s. After removing the platform, each mouse was placed in the opposite quadrant to the target zone. The time in the target zone and the numbers of platform crossing were calculated. On the seventh day, reversal test was performed. For two consecutive days, tests were performed with the platform placed in the opposite quadrant to the original one. Data were recorded using a video camera and analyzed by ANY-maze software (Stoelting, Wood Dale, USA).
Western Blot Analysis
The DG region of brains was isolated and homogenized in an RIPA buffer (Biosesang, South Korea) containing 1% Xpert Protein Inhibitor Cocktail Solution and 1% Xpert Phosphatase Inhibitor Cocktail Solution (GenDEPOT, Barker, TX, USA). BCA protein assay kit (Thermo Scientific, Rockford, IL, USA) was used to measure protein concentration. Protein was separated in 10–15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) and transferred to a nitrocellulose membrane for 2 h with 10% methanol in Tris–glycine buffer. After skim-milk blocking, the membranes were incubated with the indicated primary antibodies (1:1000). The immunoreactive proteins were developed with Immobilon Western Chemiluminescent HRP Substrate (Millipore, Darmstadt, Germany) in Fusion FX (Vilber Lourmat, Eberhardzell, Germany), and densitometry analysis was used to quantify the relative protein expression levels.
In in vitro study, microglial BV2 cells were employed to study the anti-inflammatory effects and underlying mechanism of Arg-DG under LPS stimulation, as previously reported by our group. Briefly, BV2 cells seeded at a density of 1 × 106 cells/mL overnight were pretreated with or without Arg-DG (1–5 μM) for 1 h and stimulated with 100 ng/mL LPS for 24 h or 30 min. The protein was extracted with a RIPA buffer consisting of 1% Xpert Protein Inhibitor Cocktail Solution and 1% Xpert Phosphatase Inhibitor Cocktail Solution (GenDEPOT, Barker, TX, USA). The Western blot analysis described above was used to analyze the sample.
Measurement of pro-inflammatory cytokines
A Western blot and quantitative real-time polymerase chain reaction (PCR) (Corbett Life Science, Sydney, Australia) were used to examine the expression levels of pro-inflammatory cytokine, including IL-1β, IL-6, and TNF-α, in the DG (Table 1). The sample was homogenized, and the total RNA was extracted by TRIzol reagent (TaKaRa, Shiga, Japan), according to the manufacturer’s instruction. Total RNA (1 μg) was reversely transcribed using a cDNA synthesis kit (Enzynomics, Daejeon, Korea). One microliter of cDNA, QuantiNova SYBR Green PCR Kit (500), and Rotor-Gene 3000 System (Corbett Research, Morklake, Australia) were used for quantitative real-time PCR studies. A single fluorescence reading was recorded at each extension step. The relative mRNA expression level was calculated by using mean cycle threshold (△Ct) values from quadruplicate measurements, after normalizing to the level of GADPH as an internal control, using Corbett Robotics Rotor-Gene software (Rotor-Gene 6, version 6.1, build 90). The pro-inflammatory cytokine mRNA expression level in LPS-stimulated microglia was tested according to the same method reported [21]. For the in vitro study, the production of pro-inflammatory cytokines at mRNA and protein levels was determined using the same conditions as recently reported [21].
Measurement of Nitrite (NO) Production Cell Viability
NO production and cell viability of Arg-DG were measured as per the previously reported method by our group [21]. Briefly, cells seeded into a 96-well plate at a density of 2 × 104 cells/well were pretreated with a series of Arg-DG concentration for 1 h, and then incubated in the presence or absence of LPS for 24 h. Cell viability was determined by incubation of WST solution (DoGen, South Korea) for 2 h. This was followed by an absorbance measurement at a wavelength of 450 nm and a reference wavelength of 630 nm on a microplate reader. The supernatant was subjected to quantify the amount of nitrite released in cultured media using a Griess reagent kit (Thermo Fisher Scientific, MA, USA), according to the manufacturer’s protocol. In detail, equal volume of supernatants (50 μL) and fresh mixture solution of Griess reagent were incubated for 30 min under light protection at RT. The absorbance was read at 548 nm wavelength on a microplate reader, and absorbance readings were converted to nitrite concentrations according to the standard curve.
Measurement of the PEG2 Level and IL-6 and TNF-α
The production of PEG2 released in cultured media was determined using an ELISA kit (Minneapolis, MN, USA). In brief, cells seeded into a 96-well plate at a density of 2 × 104 cells/well were pretreated with a series of Arg-DG concentrations for 1 h, and then incubated in the presence or absence of LPS for 24 h. The detailed procedures followed the manufacturer’s instructions. In addition, the secretion of IL-6 and TNF-α in the supernatant was determined by DuoSet ELISA kit (R&D system, Minneapolis, MN, USA), as previously reported by our group [21].
Measurement of Intracellular ROS Level
Intracellular RS production was quantified using a fluorescent spectrometer (Hitachi F-4500) (Hitachi, Inc., Tokyo, Japan) with the ROS fluorescent probe, 2′, 7′-dichlorofluorescein diacetate (H2DCF-DA). Cells seeded into a 96-well black microplate at 6 × 104 cells/mL were pretreated with the indicated concentrations of Arg-DG for 1 h. This was followed by LPS stimulation for 30 min. Cells were treated with 10 μM H2DCF-DA for 20 min in a dark place, and washed twice with 1× HBSS buffer solution. ROS levels were analyzed with a fluorescent spectrometer and an argon laser scanning confocal microscope (Zeiss Thornwood, NY) (Leica Microsystems, Wetzlar, Germany). N-Acetylcysteine (NAC, 5 mM) as ROS scavenger was used as a positive control.
Analysis of p65/DNA Binding
Cells seeded at 6 × 105 cells/mL were cultured overnight, pretreated with an Arg-DG, and incubated further with or without LPS for 30 min. The nuclear protein was isolated using the NE-PER Nuclear and Cytoplasmic Extraction Reagents kit (Thermo Fisher Scientific, MA, USA), according to the manufacturer’s protocol. The NF-κBp65/DNA binding activity was determined by the use of a commercial ELISA-based NF-κB p65 transcription factor assay kit (Thermo Fisher Scientific, MA, USA), according to the manufacturer’s instruction. Briefly, the streptavidin-coated 96-well plates with bound NF-κB biotinylated-consensus sequence were equilibrated with working binding buffer, and 2 mL of TNF-α and 5 μg nuclear protein extract were added to the appropriate wells, and incubated for 1 h at RT after sealing. After washing, the active transcription factor bound to the consensus sequence was further incubated with NF-κB p65 primary antibody and then with a secondary HRP-conjugated antibody. The chemiluminescent substrate working solution was added to each well, and the resulting signal was measured using a luminometer.
Statistical Analysis
All data are expressed as means ± SEM. Statistical analysis of the differences in the cytokine levels were performed by Mann–Whitney U tests and differences were considered significant if the p value was < 0.05. Statistical significance was analyzed by one-way or two-way analysis of variance (ANOVA), followed by a Student–Newman–Keuls test for multiple comparisons or Student’s t test using GraphPad Prism software, version 6 (GraphPad, San Diego, CA, USA). Statistical significance was considered at p < 0.05.
Results
Arg-DG Enhanced the Proliferation of Adult NSCs in the LPS-Injured Hippocampal DG
To investigate the effect of Arg-DG on the hippocampal neurogenesis impaired by LPS, 7-week-old male C57BL/6 mice received a single I.C.V. injection of LPS (3 μg/mouse), following administration of either saline or Arg-DG solution every 8 h for three times by i.p. injection, after waking from anesthesia. A single injection of BrdU (i.p. 100 μL/mouse) was then administered to label proliferating cells within the hippocampus, before sacrifice according to the experiment schedule (SI 1). As previously reported, LPS administration resulted in a significant decrease in the number of BrdU-positive cells in the hippocampal DG relative to the sham control with a 70.18% reduction at day 1, and a 72.85% reduction at day 3, respectively. However, Arg-DG treatment led to significant increases in the number of BrdU-positive cells in the hippocampal DG of LPS-challenged mice by 2.36 times at day 1 and 3.15 times at day 3, respectively, compared to LPS treatment only (Fig. 1a, b). These results indicate that the reduced proliferation of NSCs treated by LPS was reversed by the treatment with Arg-DG.

Arg-DG recovered the proliferation of adult NSCs in the LPS-challenged hippocampal DG. IHC of mouse brain tissue sections were harvested at day 1 and day 3 after LPS with Arg-DG treatment. (a) The coronal sections were immunohistochemically labeled for BrdU (green). (b) Graphs reflect the number of BrdU-positive cells in the hippocampal DG at day 1 and day 3 after LPS challenge, respectively. ***p < 0.001 versus control; ##p < 0.01, ###p < 0.001 versus LPS plus Arg-DG group. Data are presented as means ± SEM (n = 5 each). Scale bar in (a), 100 μm
Arg-DG Prevented the Decrease of the Immature Neuronal Differentiation of Adult NSCs in the LPS-Challenged Hippocampal DG
To assess the role of Arg-DG in the neuronal differentiation of the surviving NSCs, we next examined immature neurons in the subgranular zone using immunostaining against BrdU, along with an immature neuronal marker Doublecortin (DCX), which is a cytoskeleton-associated protein expressed transiently in the early stage of new neurons during adult neurogenesis. As presented in Fig. 2, LPS caused a significant reduction (50.41%) in the number of DCX/BrdU co-labeled neurons, compared to the sham control, at day 3. The numbers of BrdU/DCX co-labeled cells were significantly recovered in the combination treatment of LPS and Arg-DG, in comparison with LPS treatment alone. In addition, there was a significant difference in the proportion of BrdU/DCX-positive cells to the BrdU retaining cells between LPS-only treatment and LPS plus Arg-DG treatment together, indicating that Arg-DG also had an effect on the neuronal lineage commitment (Fig. 2c).

Arg-DG prevented the decrease in the immature neuronal differentiation of adult NSCs in the LPS-challenged hippocampal DG. (a) Representative photomicrograph shows the BrdU and DCX double labeling cells in the hippocampal DG. Brains were harvested at day 3 after LPS and Arg-DG treatment, and the coronal sections were successively immunostained for DCX (red) and BrdU (green), respectively, and the newborn immature neurons are marked by arrows. BrdU-DCX double positive cells (yellow) represent the differentiated immature neurons. (b) Graph displays the number of BrdU- and DCX-positive cells in the hippocampal DG at day 3 after LPS challenge, respectively. (c) The ratio of BrdU-positive/DCX-positive to total BrdU-positive cells in the DG. **p < 0.01, ***p < 0.001 versus control; ##p < 0.01, ###p < 0.001 versus LPS plus Arg-DG group. Data are presented as means ± SEM (n = 5 each). Scale bar in (a), 100 μm
Arg-DG Ameliorated the Mature Neuronal Differentiation of Adult NSCs in the LPS-Challenged Hippocampal DG
Since Arg-DG ameliorated the immature differentiation of NSCs impaired by LPS treatment, this prompted us to further investigate whether or not Arg-DG affects the differentiation of immature neuronal cells into mature neurons in LPS-impaired hippocampal DG. Immunostaining analysis was performed using double labeling with BrdU and mature neuronal marker, NeuN, to assess the mature neuronal differentiation. As shown in Fig. 3a–c, the number of BrdU/NeuN double-labeling mature neurons was more significantly decreased in the LPS-impaired (50.64%) mice than those in the sham control group; however, after 28 days, a significant increase of BrdU/NeuN co-labeling cells (1.84 times increase) was observed in the treatment of Arg-DG after LPS injury, compared to the LPS only. Additionally, the increased ratio of NeuN/BrdU double-labeling cells to the overall BrdU-positive cells was also changed in the treatment of the LPS only and Arg-DG combined LPS groups, indicating that Arg-DG enhanced neurogenesis by promoting the proliferation and differentiation of NSCs in LPS-impaired mice (Fig. 3c).

Arg-DG ameliorated the mature neuronal differentiation of adult NSCs in the LPS-challenged hippocampal DG. (a) Representative images show the BrdU and NeuN double labeling cells in the hippocampal DG. Brains were harvested at day 28 after LPS and Arg-DG treatment, and the coronal sections were successively immunostained for NeuN (red) and BrdU (green), respectively. (b) Graph displays the number of BrdU+/NeuN+ cells in the hippocampal DG at day 28 after LPS challenge. BrdU and NeuN double-positive cells represented mature neurons from adult NSCs. (c) The ratio of BrdU-positive/NeuN-positive to total BrdU-positive cells in the DG. **p < 0.01, ***p < 0.001 versus control; ##p < 0.01 versus LPS plus Arg-DG group. Data are presented as means ± SEM (n = 5 each). Scale bar in (a), 100 μm
Arg-DG Increased Adult NSC Survival in the LPS-Challenged Hippocampal DG
Neuronal population in the subgranular zone of hippocampal DG might be crucial for learning and memory functions, and LPS exposure significantly decreased the survival of newly generated NSCs in the DG of adult mice [35]. Therefore, BrdU staining was used to quantify the role of Arg-DG on the survival of newly generated NSCs in LPS-injured hippocampal DG at 5 and 28 days. Mice received a single LPS administration by I.C.V. injection and treatment of Arg-DG by i.p. injection three times at 8-h intervals, followed by consecutive administration of BrdU for 5 days, and the mice were sacrificed at the indicated time. As shown in Fig. 4a and b, LPS treatment led to a significant decrease in the total number of BrdU-positive cells in hippocampal DG, with a 55.21% reduction at day 5, and a 61.64% reduction at day 28, compared to sham control. However, treatment with Arg-DG significantly improved the total number of BrdU-positive cells in LPS-impaired proliferation of NSCs at day 5 (2.16 times increase verses LPS) and survived NSCs at day 28 (2.44 times increase versus LPS). These results demonstrated that Arg-DG could improve the survival rate of NSCs in LPS-induced neuroinflammation.

Arg-DG increased adult NSC survival in the LPS-challenged hippocampal DG. (a) Representative images show the BrdU labeling cells (green) in the DG at 5 or 28 days after consecutive BrdU injection for 5 days, following LPS. Brains were harvested at day 5 and day 28 after LPS and Arg-DG treatment, and the coronal sections were immunostained for BrdU (green). (b) Graph displays the number of BrdU-positive cells in the hippocampal DG at day 28 after LPS challenge, respectively. (c) Quantitative analysis of survival rate, represented as the ratio of BrdU-positive cells at 28 days. *p < 0.05, ***p < 0.001 versus control; #p < 0.05, ###p < 0.001 versus LPS plus Arg-DG group. Data are presented as means ± SEM (n = 5). Scale bar in (a), 100 μm
Arg-DG Ameliorated the Memory Impairment in the LPS-Challenged Hippocampal DG
To examine the restorative effect of Arg-DG on the impairment of memory function, a Morris water maze test, a novel object recognition test, and an open field test were used to assess the recovery of cognitive decline induced by the impaired neurogenesis in the hippocampus of adult mice. The open field test was used to observe the basal locomotion, and we noticed no obvious difference between each group, indicating that Arg-DG did not affect locomotor activity (Fig. 5a). However, compared to wild-type mice, LPS-treated mice showed less preference to the new objects reflecting the memory loss, and Arg-DG treatment significantly restored the LPS-induced the memory impairment (Fig. 5b). In addition, LPS-treated mice were slow to reach the location of hidden platform, demonstrating memory impairment compared to wild-type mice, whereas Arg-DG treatment significantly ameliorated the memory impairment of LPS-injected mice on escape latencies close to the wild-type group. A similar trend was found during the learning and reversal test phase (Fig. 5c, d).

Arg-DG1 ameliorated LPS-challenged memory deficits in mice. (a) Basal locomotor activity was assessed by an open field test. (b) Memory loss was observed by a novel object recognition test. (c) Representative swim paths of the last trial test on the training day 4. (d) Escape latencies during the training and reversal trials were assayed with Morris water maze test. (e) Time spent by mice in target quadrant in probe test. (f) Number of platform crossings in probe test. Data are presented as mean ± SEM (n = 10 each). *p < 0.05, **p < 0.01, ***p < 0.001 versus control; #p < 0.05, ##p < 0.01, ###p < 0.001 versus LPS plus Arg-DG treated groups; ns, not significant
To determine the maintenance of memory function, we performed a probe test, by removing the platform 1 day after water maze training day 4. Compared to wild-type mice, the average time spent in the target quadrant was obviously decreased in the LPS-treated mice, but mice in the treatment of LPS and Arg-group spent longer time in searching the platform quadrant than the LPS-injected mice (Fig. 5e). Moreover, the number of platform crossings was also decreased in LPS-treated mice compared to wild-type mice, while Arg-DG treatment showed a significant increase in the number of platform crossings relative to LPS-injected mice, suggesting the possible memory improvements in LPS-injected mice (Fig. 5e, f). Overall, these results suggest that Arg-DG could alleviate the memory impairment in LPS-stimulated mice.
Arg-DG Alleviated Microglial Activation and the Expression of Pro-inflammatory Cytokines in the Mice Hippocampal DG Injured by LPS
Following LPS administration, microglial activation always changed the morphology from ramified to a reactive amoeboid form. Previous studies reported by our group and others showed that the inhibition of microglial activation with anti-inflammatory molecules, such as EGCG, minocycline, or TGF-β, could restore adult neurogenesis suppressed by neuroinflammation [19, 36, 37]. Thus, we asked whether the restorative effect of Arg-DG on adult neurogenesis impaired by LPS exposure is mediated through modulation of the microglial state and production of inflammatory mediators. First, immunostaining with microglial marker Iba-1 was used to morphologically assay microglial activation. As previously reported, Iba-1 labeling microglial cells in LPS treatment did indeed cause a greater morphology change from ramified to reactive amoeboid than in the sham control group (Fig. 6a). However, Arg-DG treatment made microglial cell bodies become smaller, with a brushy appearance, from an amoeboid, unlike the results in LPS-treated mice, especially after 3 days of treatment. Furthermore, the administration of LPS resulted in a significant increase in the number of activated microglia at day 1 (11.6 times reduction versus sham control) and day 3 (18.8 times reduction versus sham control), respectively, whereas the number of activated microglia in LPS-treated mice with Arg-DG was reduced at day 1 and day 3 more significantly than with LPS treatment only (Fig. 6b, c).

Arg-DG suppressed microglial activation and the production of pro-inflammatory cytokines in the hippocampal DG injured by LPS. (a) The representative gels showed the Iba-1-positive cells (red) in the hippocampal DG at day 1 and day 3 after LPS challenge. (b, c) Graphs display the suppressive effect of Arg-DG on the number of activated microglia induced by LPS. Brains were harvested at day 1 and day 3 after the treatment of LPS and Arg-DG, and the coronal sections were subjected to microglial activation in SGZ using the microglia-specific marker Iba-1. Data are presented as means ± SEM (n = 5 each). ***p < 0.001 versus control; ##p < 0.01, ###p < 0.001 versus LPS plus Arg-DG group. Data are presented as means ± SEM (n = 5). Scale bar in (a), 100 μm
Microglial activation is usually accompanied by the increased expression of pro-inflammatory cytokines, which have been reported to have negative impacts on adult hippocampal neurogenesis in mice. Therefore, the pro-inflammatory gene expression of TNF-α, IL-1β, and IL-6 in the hippocampus of the mice was measured by Western blot analysis. Prior to biological assay, the effective concentration of Arg-DG ranging from 0.3 to 1.2 mg/kg on the inhibition of pro-inflammatory cytokines in hippocampal DG of LPS-injected mice was investigated. Arg-DG exhibited strong inhibition on the expression of pro-inflammatory cytokines at the concentration of 0.6 mg/kg in LPS-treated mice, whereas Arg-DG alone had no effect at the concentration range of 0.3 to 1.2 mg/kg (data not shown). Thus, 0.6 mg/kg of Arg-DG was used in all the studies above. As shown in Fig. 7, it was used to examine the effects of Arg-DG on the expression of pro-inflammatory cytokines in the hippocampal DG of LPS-treated mice using Western blot analysis. As presented in Fig. 7, LPS induced a more significant increase in the expression of pro-inflammatory mediators after 1 day than did the sham control. However, Arg-DG significantly reduced the expression of IL-6, IL-1β, and TNF-α in LPS-treated mice after 1 day. Arg-DG also significantly reduced the expression of pro-inflammatory cytokines, which had been increased by LPS after 3 days (Fig. 7c, d). The administration of only Arg-DG had no effect on the production of pro-inflammatory cytokines compared to the sham control in all tests. These results suggest that Arg-DG acts as an anti-inflammatory molecule to restore impaired neurogenesis through the inhibition of microglial activation induced by LPS.

Arg-DG inhibited the production of pro-inflammatory cytokines in the hippocampal DG injured by LPS. (a, b) The representative gels and graph show the inhibitory effect of Arg-DG on the expression levels of pro-inflammatory cytokines at protein level after 1 day in LPS-injured hippocampal DG of adult mice. (c, d) The representative gels and graph show the inhibitory effect of Arg-DG on the expression levels of pro-inflammatory cytokines at protein level after 3 days in LPS-injured hippocampal DG of adult mice. Brains were harvested at day 1 and day 3 after LPS and Arg-DG treatment, the hippocampus was isolated, and the protein was extracted for the analysis of pro-inflammatory cytokines IL-6, IL-1β, TNF-α, and β-actin using Western blot. Data are presented as means ± SEM (n = 5). **p < 0.01, ***p < 0.001 versus control; #p < 0.05, ##p < 0.01 versus LPS plus Arg-DG group. Data are presented as means ± SEM (n = 5). Scale bar in (a), 100 μm
Effect of Arg-DG on the NF-κB and MAPK Signaling in LPS-Challenged Mice
Because of their ability to up-regulate many genes influencing a broad range of biological processes, NF-κB and MAPK signaling have been characterized as major mediators of microglial activation under different endotoxin stimuli, including LPS. They also play key roles in the pathogenesis of microglia-mediated neurodegenerative diseases. The role of Arg-DG in the regulation of both pathways in the hippocampal DG of LPS-injected mice was examined in order to clarify its mechanism of action. As shown in Fig. 8a and b, LPS treatment after 1 day resulted in increases in the expression of TLR4, phosphorylated IκB, and phosphorylated RelA that were more significant than in the sham control, indicating that NF-κB signaling was activated after LPS exposure in the hippocampus of mice, whereas treating LPS with Arg-DG significantly decreased their expression levels, compared to only LPS treatment. In addition, we found that there was a dramatic increase in the ratios of p-ERK1/2/ERK1/2, p-JNK/JNK, and p-p38/p38 compared to sham control, whereas combination treatment of LPS and Arg-DG significantly reduced the ratios of p-JNK/JNK in comparison with LPS treatment alone. However, it had no effect on reducing the ratios of p-ERK1/2/ERK1/2 and ratios of p-p38/p38 (Fig. 8e, f). In a manner similar to those of the 1-day results, Arg-DG also inhibited the activation of NF-κB and MAPK signaling through inhibition of the abovementioned protein in the LPS-treated mice compared to LPS only at day 3 (Fig. 8c, d, g, h).

Arg-DG blocked activation of NF-κB and MAPK signaling induced by LPS in the hippocampal DG. (a–d) The representative gels and graphs showed the inhibitory effect of Arg-DG on the expression levels of TLR4, phosphorylated RelA, phosphorylated IκB, in the hippocampal DG at day 1 and day 3 after LPS challenge. (e–h) The representative gels and graphs showed the effect of Arg-DG on the phosphorylation of ERK1/2, JNK, and p38 in the hippocampal DG at day 1 and day 3 after LPS challenge. Brains were harvested at day 1 and day 3 after LPS and Arg-DG treatment, and the hippocampus was isolated, and the protein was extracted for analyzing the expression levels of TLR4, phosphorylated RelA, RelA, phosphorylated IκB, an IκB for NF-κB signaling, and the phosphorylation of ERK1/2, JNK, and p38 for MAPK signaling using Western blot analysis. Data are presented as means ± SEM (n = 5 each). **p < 0.01, ***p < 0.001 versus control; #p < 0.05, ##p < 0.01, ###p < 0.01 versus LPS plus Arg-DG groups
Effect of Arg-DG on the LPS-Induced Pro-inflammatory Cytokines and Reactive Oxidative Species in Cultured Microglial BV2 Cells
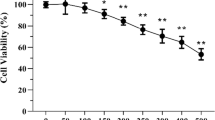
Microglia in the hippocampal DG are very abundant, and accumulating evidence indicates that the inflammation-induced reduction in neurogenesis is mediated by microglia. Therefore, we used the cultured microglial BV2 cells to further confirm the inhibitory effect of Arg-DG on LPS-induced neuroinflammation. Prior to biological assay, the toxic effects of Arg-DG in microglial BV2 cells were measured using cell viability assay, and the results showed that Arg-DG was non-cytotoxic at a concentration range of 5 to 20 μM. Pretreatment with Arg-DG significantly reduced the cytotoxicity induced by LPS in a dose-dependent manner (data not shown).
It is well known that the main reason for neuroinflammation is the overactivation of microglia. Accumulating evidence suggests that inflammatory stimuli, such as iNOS, COX-2, TNF-α, IL-1β/6, nitrite, and ROS, decrease hippocampal neurogenesis by modulation of the proliferation, differentiation, cell survival, and integration of newborn neurons into the existing neural networks, and inhibition of the inflammatory cytokines slows down the progression of neurodegenerative disorders [15, 34]. With this in mind, we investigated whether or not Arg-DG inhibits the LPS-induced production of pro-inflammatory cytokines and ROS. We found that LPS treatment significantly augmented NO production and the expression of iNOS and COX-2 at mRNA and protein levels in microglial BV2 cells. However, pretreatment with Arg-DG could significantly inhibit the production of NO, PEG2, and the levels of iNOS and COX-2 expression in LPS-induced microglial BV2 cells at the concentration range of 1 to 5 μM (Fig. 9a–e). The expression levels of pro-inflammatory cytokines, such as IL-1β, IL-6, and TNF-α, at mRNA level in LPS-induced BV2 cells were also increased more significantly than with untreated cells, whereas pretreatment with Arg-DG significantly suppressed them (Fig. 9b). Arg-DG also reduced the release of IL-6 and TNF-α at protein levels, but not IL-1β (data not shown) in LPS-induced microglial BV2 cells (Fig. 9f, g).

Arg-DG inhibited the expression of pro-inflammatory cytokines and ROS in LPS-stimulated BV2 cells. Cells were pretreated with the indicated concentrations of Arg-DG for 1 h, and then incubated in the presence or absence of LPS for 24 h. (a, b) mRNA levels of iNOS, COX-2, IL-6, IL-1β, TNF-α, and β-actin were examined by RT-PCR and quantitative real-time PCR. (c) iNOS, COX-2, and β-actin levels were determined by Western blot analysis. (d) NO production was measured by Griess reaction. (e) Production of PEG2 was determined by ELISA kit. (f, g) The production of IL-6 and TNF-α released from microglia was determined by ELISA kit. (h, i) ROS production was measured using a fluorescent dye H2DCFDA. Cells were pretreated with Arg-DG at the indicated concentrations, and 5 mM NAC for 1 h, followed by LPS stimulation for 30 min. After incubation with 5 μM H2DCFDA for 20 min, cells were examined under a fluorescent microplate reader and an argon laser scanning confocal microscope. Scale bar in (i), 20 μm. Unless otherwise stated, each experiment was performed in triplicate, and repeated at least three times. Data are presented as means ± SEM. The term “ns” means no significant difference compared to the group treated with LPS. *p < 0.05, **p < 0.01, ***p < 0.001 versus control; #p < 0.05, ##p < 0.01, ###p < 0.001 versus LPS plus Arg-DG treated groups
Microglia activation always accompanies the overproduction of ROS. ROS plays a critical role in inflammatory responses and cytotoxic damage to surrounding neurons. It also triggers signaling activation in neurodegenerative disorders. To determine the effect of Arg-DG on the production of ROS, a fluorescent probe DC-FDA was used in LPS-induced microglial BV2 cells. Cells were pretreated with a series of Arg-DG solutions (1, 2, and 5 μM) and ROS scavenger NAC (5 mM) for 1 h. This was followed by exposure to LPS for 30 min. Pretreatment with Arg-DG significantly suppressed LPS-induced intracellular ROS levels in microglia. This was similar to the effect of NAC treatment (Fig. 9h). A microscope showed that LPS treatment increased the fluorescent intensity of DCF, which was reduced by pretreatment with Arg-DG (5 μM) or NAC, indicating that Arg-DG has ROS scavenging activity (Fig. 9i).
Effect of Arg-DG on the LPS-Induced NF-κB and JNK Signaling
Since Arg-DG inhibited the activation of TLR4-NF-κB and JNK signaling in the hippocampal DG of LPS-induced mice, we further examined the role of Arg-DG on the activation of TLR-NF-κB and MAPK signaling by LPS in cultured microglial BV2 cells. As expected, LPS caused significant augment in the TLR4 and phosphorylation of IκB, the nuclear translocation of RelA, and the transcriptional activity of NF-κB. Pretreatment with Arg-DG could markedly decrease the activation of TLR4 and phosphorylation of IκB in LPS-induced microglial BV2 cells. Pretreatment with Arg-DG also inhibited the nuclear translocation of RelA protein in a dose-dependent manner (Fig. 10a–c). Moreover, LPS treatment significantly increased the DNA binding activity of NF-κB, which was dramatically reduced by pretreatment with Arg-DG at 2 and 5 μM for 1 h (Fig. 9d). In summary, these findings suggest that Arg-DG inhibits the activation of TLR4-NF-κB signaling in LPS-induced microglia by inhibiting the phosphorylation of IκBα, nuclear translocation, and the DNA binding activity of NF-κB.

Arg-DG inhibited the activation of NF-κB and MAPK signaling in LPS-stimulated BV2 cells. Cells were pretreated with Arg-DG at the indicated concentrations for 1 h before exposure to 100 ng/mL LPS for 30 min. (a, b) The expression of TLR4, p-IκB, IκB, and translocation of NF-κB p65 nucleus were measured by Western blot analysis. The expression levels of NF-κB p65 were normalized to Histone H1 and presented as relative change compared to the group treated with LPS. (c) Representative images show the NF-κB p65 labeling cells in microglial BV2 cells. (d) DNA binding activity was measured by using NF-κB p65 transcription factor assay kit. (e, f) Phosphorylation levels of ERK1/2, JNK, and p38 in whole cell lysates were determined by Western blot analysis. Unless otherwise stated, all the experiments were performed in triplicate and repeated at least three times. Data are presented as means ± SEM. The term “ns” means no significant difference compared to the group treated with LPS. *p < 0.05, **p < 0.01, ***p < 0.001 versus control; #p < 0.05, ##p < 0.01 versus LPS plus Arg-DG treated groups
MAPK signaling plays a key role in the pathogenesis of microglia-mediated neuroinflammation. To further clarify its mechanism of action, we examined the phosphorylation of ERK1/2, JNK, and p38 kinases. Our results showed that LPS treatment for 30 min significantly augmented the phosphorylation of ERK1/2, JNK, and p38, although their total forms for ERK1/2, JNK, and p38 were unchanged. Pretreatment with Arg-DG for 1 h prior to LPS stimulation selectively decreased the phosphorylation of JNK in a dose-dependent manner. However, pretreatment with Arg-DG did not affect the phosphorylation levels of ERK1/2 or p38 (Fig. 10e, f).
Discussion
The main findings of the present study were that Arg-DG administration promotes hippocampal neurogenesis impaired by LPS in the DG of adult mice. The observed beneficial effects may be attributed to an increase in the proliferation, differentiation, and cell survival of the newly generated cells. This is done by suppressing the production of pro-inflammatory cytokines underlying TLR4/NF-κB and MAPK/JNK signaling in activated microglia. Moreover, Arg-DG displayed strong anti-inflammatory activity in LPS-stimulated microglial BV2 cells via the inhibition of NF-κB and JNK signaling [21].
As part of the brain system critically involved in learning and memory, the hippocampus has been extensively studied, and alterations in adult hippocampal neurogenesis have been considered a hallmark in many neurodegenerative diseases [38, 39]. As seen in Fig. 1, our results showed that LPS exposure decreased the proliferation and survival rate of adult NSCs. LPS also suppressed the number of immature and mature neurons more than did the sham control. These observations were consistent with previous reports showing that in the hippocampal DG of adult mice, LPS administration decreased cell proliferation, survival, and the differentiation of NSCs [11, 19]. Arg-DG treatment boosted the proliferation of NSCs impaired by LPS. Furthermore, we found that Arg-DG significantly increased the number of DCX/BrdU and NeuN/BrdU double-labeling cells relative to LPS treatment. DCX and NeuN are expressed specifically in newly generated neurons and have been frequently used as markers for the differentiation of NSCs [40]. This study also indicated that Arg-DG prolonged the cell survival rate of newly generated cells more than LPS treatment. Previous studies showed that long-term survival cells in the DG of hippocampal circuitry were associated with processing spatial information [33]. To the best of our knowledge, we are the first to report that Arg-DG rescued the LPS-impaired neurogenesis by increasing cell proliferation and differentiation, and by prolonging the survival of NSCs in the hippocampal DG of adult mice.
Many studies have demonstrated that the newborn neurons in hippocampus of adult animals are functionally integrated and contribute to cognitive function [41], especially learning and memory processes [42]. It was reported that the system administration of LPS resulted in an increase in the intracellular accumulation of amyloid-β and memory deficiency, with concomitantly increased neuroinflammation [43]. In our study, we found that LPS treatment by I.C.V. injection led to the impairment of memory function in adult mice. But mice treated with Arg-DG showed enhanced memory and learning with increased number of neurons, compared to the LPS treated only (Fig. 5). Therefore, the present study provided evidence that hippocampal neurogenesis ablation in LPS-induced mice had memory dysfunction, and Arg-DG is a promising candidate to ameliorate the reduced neurogenesis-related loss of memory and learning.
There are several possible reasons for the observed beneficial effects of Arg-DG. One of the most plausible is that Arg-DG blocks microglial activation and the production of pro-inflammatory cytokines. Immune cells, including microglia, play a crucial role in the maintenance of neurogenesis and memory function in adulthood. Following neuroinflammation, microglial pathology has been reported to negatively impact hippocampal neurogenesis [11, 12]. Microglia usually undergo dramatic changes in their morphology and in the expression pattern of various pro-inflammatory cytokines in LPS stimulation [44]. We observed that LPS treatment resulted in a morphological change from ramified to reactive amoeboid, and Arg-DG reversed that effect. Moreover, LPS resulted in a significant increase in the number of activated microglia at both day 1 and day 3, which were reduced after treatment with Arg-DG, indicating that Arg-DG downregulates the microglial activation. Similar to its effects on day 1, Arg-DG suppressed the overactivation of microglia at day 3 (Fig. 6). The overactivated microglia usually contribute to aberrant expression of various cytokines, including NO, iNOS, COX-2, ROS, TNF-α, and IL-1β/6, which have been considered to be the key modulators in neurogenesis [36, 45]. For example, overproduction of TNF-α, IL-1β/6 in the hippocampus hampered neurogenesis, which could be restored by the inhibition of microglial activation with anti-inflammatory agents, such as minocycline and indomethacin [46, 47]. Furthermore, microglia activated by both LPS and IFN-γ produce high levels of NO, resulting in neuronal cell injury in co-culture in vitro [36]. In the present study, Arg-DG reduced the expression levels of the inflammatory cytokines mentioned above more significantly than LPS treatment (Fig. 9). Similarly, Arg-DG inhibited LPS-induced overexpression of the pro-inflammatory mediators in cultured microglial BV2 cells (Fig. 8). These findings suggest that one of the major mechanisms of Arg-DG on hippocampal neurogenesis is due, at least in part, to the inhibition of neuroinflammation induced by LPS, indicating that the anti-inflammatory effect of Arg-DG on microglia may contribute to restoring neurogenesis impaired by LPS [48].
TLR4, part of the toll-like receptor (TLR) family, is predominately expressed on microglia, and it has been reported that LPS specifically binds to TLR4, thereby promoting activation of downstream intracellular singling pathways in microglia [49]. NF-κB and MAPKs signaling have been involved in the LPS-induced microglial activation and adult hippocampal neurogenesis. They also play key roles in the pathogenesis of microglia-mediated neurodegenerative diseases [50]. In line with previous studies, LPS exposure dramatically augmented the expression level of TLR4, leading to the NF-κB and MAPKs downstream signaling in the hippocampus of mice after 1 day and 3 days [19, 51], whereas Arg-DG significantly inhibited the TLR4 expression after 1-day and 3-day treatment. We also found that Arg-DG obviously decreased the phosphorylated IκB, RelA, and phosphorylation of JNK in LPS-treated mice, and that it had no effects on the phosphorylated ERK1/2 and p38 (Fig. 8). This observation is in line with our recent report that diosgenin derivative incorporated with primary amine decreases the inflammatory activity through the regulation of NF-κB and JNK MAPK signaling in LPS-stimulated BV2 cells, indicating that JNK is the principal MAPK involved in neuroinflammation after modification [21]. A previous study demonstrated that the inhibition of NF-κB and p38 activation caused diosgenin to attenuate the LPS-induced acute lung injury in mice [52]. Another study showed that diosgenin suppressed the inflammation induced by conditioned medium through interfering with IκB degradation and the phosphorylation of JNK signaling in RAW 264 macrophages [53]. Interestingly, a recent study showed that the anti-inflammatory effect of diosgenin glycoside acts through suppression of ERK1/2 and p38 activation, instead of JNK, in LPS-stimulated microglial BV2 cells, suggesting that diosgenin is responsible for the anti-inflammatory activity [31]. Our results clearly showed that Arg-DG inhibited LPS-induced inflammation via NF-κB and JNK signaling in hippocampal DG of adult mice, which might be attributed to their structural difference, or targeting of different tissues. Similarly, Arg-DG inhibited LPS-activated NF-κB pathway in microglial BV2 cells via the inhibition of phosphorylation of IκB, nuclear translocation, and DNA binding activity of RelA. However, Arg-DG did not inhibit the expression of TLR4 in LPS-stimulated BV2 cells, which was different from the observed results in LPS-induced mice. Moreover, Arg-DG specifically decreased the expression level of the phosphorylated JNK/MAPK, but not p38 or ERK1/2 in cultured microglial BV2 cells (Fig. 10). These results were similar to those in our recent report, which stated that a diosgenin derivative with polyamine pretreatment also suppressed the activation of the NF-κB and JNK pathways in LPS-stimulated microglial BV2 cells [21]. Taken together, the present findings show that the administration of LPS induced excessive activation of microglia, leading through the activation of NF-κB and JNK signaling to the overexpression of various pro-inflammatory cytokines in the hippocampal DG of mice. Arg-DG acted as an anti-inflammatory agent, blocking microglial activation and the production of pro-inflammatory cytokines via NF-κB and JNK signaling in LPS-induced mice, which would have led to the restoration of LPS-impaired neurogenesis. These findings imply that the possible mechanism of Arg-DG on the improvement of impaired neurogenesis may at least partially involve the inhibition of microglial activation, and the production of pro-inflammatory cytokines induced by LPS, through blocking NF-κB and MAPK signaling.
Conclusion
In summary, a significant decrease in the proliferation and differentiation of neuronal stem cells (NSCs) was caused by the administration of LPS, which was restored by Arg-DG treatment. LPS treatment also resulted in memory dysfunction. LPS also led to microglial activation and an increase in the production of pro-inflammatory cytokines in the dentate gyrus. Arg-DG, a water-soluble arginyl–diosgenin analog, exhibited improved neurogenesis by lessening the proliferation and differentiation of NSCs, suppressing the apoptosis of NSCs, as well as enhanced memory and learning ability in LPS-injured mice. Arg-DG exerted an inhibitory action on microglial activation and the production of LPS-induced pro-inflammatory cytokines in the dentate gyrus of the hippocampus of mice. Arg-DG apparently acts by suppressing pro-inflammatory cytokines and microglial activation through the inhibition of NF-κB and JNK MAPK signaling in mice. Similar results were further observed in LPS-stimulated microglial BV2 cells. Taken together, these results suggest that Arg-DG might have the potential to treat various neurodegenerative disorders resulting from microglia-mediated neuroinflammation.
References
Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH (1998) Neurogenesis in the adult human hippocampus. Nat Med 4(11):1313–1317. https://doi.org/10.1038/3305
Altman J, Das GD (1965) Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol 124(3):319–335
Shors TJ, Miesegaes G, Beylin A, Zhao M, Rydel T, Gould E (2001) Neurogenesis in the adult is involved in the formation of trace memories. Nature 410(6826):372–376. https://doi.org/10.1038/35066584
Bassani TB, Bonato JM, Machado MMF, Coppola-Segovia V, Moura ELR, Zanata SM, Oliveira R, Vital M (2018) Decrease in adult neurogenesis and neuroinflammation are involved in spatial memory impairment in the streptozotocin-induced model of sporadic Alzheimer's disease in rats. Mol Neurobiol 55(5):4280–4296. https://doi.org/10.1007/s12035-017-0645-9
Winner B, Winkler J (2015) Adult neurogenesis in neurodegenerative diseases. Cold Spring Harb Perspect Biol 7(4):a021287. https://doi.org/10.1101/cshperspect.a021287
Singh S, Mishra A, Bharti S, Tiwari V, Singh J, Parul SS (2018) Glycogen synthase kinase-3beta regulates equilibrium between neurogenesis and gliogenesis in rat model of Parkinson's disease: a crosstalk with Wnt and notch signaling. Mol Neurobiol 55(8):6500–6517. https://doi.org/10.1007/s12035-017-0860-4
van Praag H, Kempermann G, Gage FH (1999) Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat Neurosci 2(3):266–270. https://doi.org/10.1038/6368
Kempermann G, Kuhn HG, Gage FH (1997) More hippocampal neurons in adult mice living in an enriched environment. Nature 386(6624):493–495. https://doi.org/10.1038/386493a0
Ming GL, Song H (2011) Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 70(4):687–702. https://doi.org/10.1016/j.neuron.2011.05.001
Carpentier PA, Palmer TD (2009) Immune influence on adult neural stem cell regulation and function. Neuron 64(1):79–92. https://doi.org/10.1016/j.neuron.2009.08.038
Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O (2003) Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci U S A 100(23):13632–13637. https://doi.org/10.1073/pnas.2234031100
Monje ML, Toda H, Palmer TD (2003) Inflammatory blockade restores adult hippocampal neurogenesis. Science (New York, NY) 302(5651):1760–1765. https://doi.org/10.1126/science.1088417
Wu MD, Montgomery SL, Rivera-Escalera F, Olschowka JA, O'Banion MK (2013) Sustained IL-1beta expression impairs adult hippocampal neurogenesis independent of IL-1 signaling in nestin+ neural precursor cells. Brain Behav Immun 32:9–18. https://doi.org/10.1016/j.bbi.2013.03.003
Cacci E, Claasen JH, Kokaia Z (2005) Microglia-derived tumor necrosis factor-alpha exaggerates death of newborn hippocampal progenitor cells in vitro. J Neurosci Res 80(6):789–797. https://doi.org/10.1002/jnr.20531
Vallieres L, Campbell IL, Gage FH, Sawchenko PE (2002) Reduced hippocampal neurogenesis in adult transgenic mice with chronic astrocytic production of interleukin-6. J Neurosci 22(2):486–492
Rosadini CV, Kagan JC (2017) Early innate immune responses to bacterial LPS. Curr Opin Immunol 44:14–19. https://doi.org/10.1016/j.coi.2016.10.005
Rehman SU, Ali T, Alam SI, Ullah R, Zeb A, Lee KW, Rutten BPF, Kim MO (2018) Ferulic acid rescues LPS-induced neurotoxicity via modulation of the TLR4 receptor in the mouse hippocampus. Mol Neurobiol. https://doi.org/10.1007/s12035-018-1280-9
Younger D, Murugan M, Rama Rao KV, Wu LJ, Chandra N (2018) Microglia receptors in animal models of traumatic brain injury. Mol Neurobiol. https://doi.org/10.1007/s12035-018-1428-7
Seong KJ, Lee HG, Kook MS, Ko HM, Jung JY, Kim WJ (2016) Epigallocatechin-3-gallate rescues LPS-impaired adult hippocampal neurogenesis through suppressing the TLR4-NF-kappaB signaling pathway in mice. Korean J Physiol Pharmacol 20(1):41–51. https://doi.org/10.4196/kjpp.2016.20.1.41
Doring C, Regen T, Gertig U, van Rossum D, Winkler A, Saiepour N, Bruck W, Hanisch UK et al (2017) A presumed antagonistic LPS identifies distinct functional organization of TLR4 in mouse microglia. Glia 65(7):1176–1185. https://doi.org/10.1002/glia.23151
Cai B, Seong KJ, Bae SW, Chun C, Kim WJ, Jung JY (2018) A synthetic diosgenin primary amine derivative attenuates LPS-stimulated inflammation via inhibition of NF-kappaB and JNK MAPK signaling in microglial BV2 cells. Int Immunopharmacol 61:204–214. https://doi.org/10.1016/j.intimp.2018.05.021
Kim SJ, Son TG, Park HR, Park M, Kim MS, Kim HS, Chung HY, Mattson MP et al (2008) Curcumin stimulates proliferation of embryonic neural progenitor cells and neurogenesis in the adult hippocampus. J Biol Chem 283(21):14497–14505. https://doi.org/10.1074/jbc.M708373200
Ajmone-Cat MA, Cacci E, Minghetti L (2008) Non steroidal anti-inflammatory drugs and neurogenesis in the adult mammalian brain. Curr Pharm Des 14(14):1435–1442
Kimura T, Hong Nguyen PT, Ho SA, Tran AH, Ono T, Nishijo H (2009) T-817MA, a neurotrophic agent, ameliorates the deficits in adult neurogenesis and spatial memory in rats infused I.C.V. with amyloid-beta peptide. Br J Pharmacol 157(3):451–463. https://doi.org/10.1111/j.1476-5381.2009.00141.x
Ryu JR, Hong CJ, Kim JY, Kim EK, Sun W, Yu SW (2016) Control of adult neurogenesis by programmed cell death in the mammalian brain. Mol Brain 9:43. https://doi.org/10.1186/s13041-016-0224-4
Tohda C, Urano T, Umezaki M, Nemere I, Kuboyama T (2012) Diosgenin is an exogenous activator of 1,25D(3)-MARRS/Pdia3/ERp57 and improves Alzheimer's disease pathologies in 5XFAD mice. Sci Rep 2:535. https://doi.org/10.1038/srep00535
Zhang X, Xue X, Zhao J, Qian C, Guo Z, Ito Y, Sun W (2016) Diosgenin attenuates the brain injury induced by transient focal cerebral ischemia–reperfusion in rats. Steroids 113:103–112. https://doi.org/10.1016/j.steroids.2016.07.006
Liu W, Zhu M, Yu Z, Yin D, Lu F, Pu Y, Zhao C, He C et al (2017) Therapeutic effects of diosgenin in experimental autoimmune encephalomyelitis. J Neuroimmunol 313:152–160. https://doi.org/10.1016/j.jneuroim.2017.10.018
Xiao L, Guo D, Hu C, Shen W, Shan L, Li C, Liu X, Yang W et al (2012) Diosgenin promotes oligodendrocyte progenitor cell differentiation through estrogen receptor-mediated ERK1/2 activation to accelerate remyelination. Glia 60(7):1037–1052. https://doi.org/10.1002/glia.22333
Lecanu L, Tillement L, Rammouz G, Tillement JP, Greeson J, Papadopoulos V (2009) Caprospinol: moving from a neuroactive steroid to a neurotropic drug. Expert Opin Investig Drugs 18(3):265–276. https://doi.org/10.1517/13543780902762827
Wang S, Wang F, Yang H, Li R, Guo H, Hu L (2017) Diosgenin glucoside provides neuroprotection by regulating microglial M1 polarization. Int Immunopharmacol 50:22–29. https://doi.org/10.1016/j.intimp.2017.06.008
Liao AM, Jung H, Yu JW, Lee DH, Park SS, Cai B, Chun C (2018) Synthesis and biological evaluation of arginyl–diosgenin conjugate as a potential bone tissue engineering agent. Chem Biol Drug Des 91(1):17–28. https://doi.org/10.1111/cbdd.13050
Liu L, Hoang-Gia T, Wu H, Lee MR, Gu L, Wang C, Yun BS, Wang Q et al (2011) Ginsenoside Rb1 improves spatial learning and memory by regulation of cell genesis in the hippocampal subregions of rats. Brain Res 1382:147–154. https://doi.org/10.1016/j.brainres.2011.01.051
Nam SM, Kim JW, Yoo DY, Jung HY, Chung JY, Kim DW, Hwang IK, Yoon YS (2018) Hypothyroidism increases cyclooxygenase-2 levels and pro-inflammatory response and decreases cell proliferation and neuroblast differentiation in the hippocampus. Mol Med Rep 17(4):5782–5788. https://doi.org/10.3892/mmr.2018.8605
Jarlestedt K, Naylor AS, Dean J, Hagberg H, Mallard C (2013) Decreased survival of newborn neurons in the dorsal hippocampus after neonatal LPS exposure in mice. Neuroscience 253:21–28. https://doi.org/10.1016/j.neuroscience.2013.08.040
Mattei D, Djodari-Irani A, Hadar R, Pelz A, de Cossio LF, Goetz T, Matyash M, Kettenmann H et al (2014) Minocycline rescues decrease in neurogenesis, increase in microglia cytokines and deficits in sensorimotor gating in an animal model of schizophrenia. Brain Behav Immun 38:175–184. https://doi.org/10.1016/j.bbi.2014.01.019
Battista D, Ferrari CC, Gage FH, Pitossi FJ (2006) Neurogenic niche modulation by activated microglia: transforming growth factor beta increases neurogenesis in the adult dentate gyrus. Eur J Neurosci 23(1):83–93. https://doi.org/10.1111/j.1460-9568.2005.04539.x
Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N, Cohen H, Kipnis J et al (2006) Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci 9(2):268–275. https://doi.org/10.1038/nn1629
Zheng JY, Liang KS, Wang XJ, Zhou XY, Sun J, Zhou SN (2017) Chronic estradiol administration during the early stage of Alzheimer’s disease pathology rescues adult hippocampal neurogenesis and ameliorates cognitive deficits in Abeta1–42 mice. Mol Neurobiol 54(10):7656–7669. https://doi.org/10.1007/s12035-016-0181-z
Farioli-Vecchioli S, Saraulli D, Costanzi M, Pacioni S, Cina I, Aceti M, Micheli L, Bacci A et al (2008) The timing of differentiation of adult hippocampal neurons is crucial for spatial memory. PLoS Biol 6(10):e246. https://doi.org/10.1371/journal.pbio.0060246
Zhang QJ, Li J, Zhang SY (2018) Effects of TRPM7/miR-34a gene silencing on spatial cognitive function and hippocampal neurogenesis in mice with type 1 diabetes mellitus. Mol Neurobiol 55(2):1568–1579. https://doi.org/10.1007/s12035-017-0398-5
Juliandi B, Tanemura K, Igarashi K, Tominaga T, Furukawa Y, Otsuka M, Moriyama N, Ikegami D et al (2015) Reduced adult hippocampal neurogenesis and cognitive impairments following prenatal treatment of the antiepileptic drug valproic acid. Stem Cell Rep 5(6):996–1009. https://doi.org/10.1016/j.stemcr.2015.10.012
Kim YE, Hwang CJ, Lee HP, Kim CS, Son DJ, Ham YW, Hellstrom M, Han SB et al (2017) Inhibitory effect of punicalagin on lipopolysaccharide-induced neuroinflammation, oxidative stress and memory impairment via inhibition of nuclear factor-kappaB. Neuropharmacology 117:21–32. https://doi.org/10.1016/j.neuropharm.2017.01.025
Hoogland IC, Houbolt C, van Westerloo DJ, van Gool WA, van de Beek D (2015) Systemic inflammation and microglial activation: systematic review of animal experiments. J Neuroinflammation 12:114. https://doi.org/10.1186/s12974-015-0332-6
Borsini A, Zunszain PA, Thuret S, Pariante CM (2015) The role of inflammatory cytokines as key modulators of neurogenesis. Trends Neurosci 38(3):145–157. https://doi.org/10.1016/j.tins.2014.12.006
Lopes RS, Cardoso MM, Sampaio AO, Barbosa MS Jr, Souza CC, MC DAS, Ferreira EM, Freire MA et al (2016) Indomethacin treatment reduces microglia activation and increases numbers of neuroblasts in the subventricular zone and ischaemic striatum after focal ischaemia. J Biosci 41(3):381–394
Inta D, Lang UE, Borgwardt S, Meyer-Lindenberg A, Gass P (2017) Microglia activation and schizophrenia: lessons from the effects of minocycline on postnatal neurogenesis, Neuronal Survival and Synaptic Pruning. Schizophr Bull 43(3):493–496. https://doi.org/10.1093/schbul/sbw088
Sellner S, Paricio-Montesinos R, Spiess A, Masuch A, Erny D, Harsan LA, Elverfeldt DV, Schwabenland M et al (2016) Microglial CX3CR1 promotes adult neurogenesis by inhibiting Sirt 1/p65 signaling independent of CX3CL1. Acta Neuropathol Commun 4(1):102. https://doi.org/10.1186/s40478-016-0374-8
Zhou H, Lapointe BM, Clark SR, Zbytnuik L, Kubes P (2006) A requirement for microglial TLR4 in leukocyte recruitment into brain in response to lipopolysaccharide. J Immunol 177(11):8103–8110
Zhang G, Li J, Purkayastha S, Tang Y, Zhang H, Yin Y, Li B, Liu G et al (2013) Hypothalamic programming of systemic ageing involving IKK-beta, NF-kappaB and GnRH. Nature 497(7448):211–216. https://doi.org/10.1038/nature12143
Yang R, Chen W, Lu Y, Li Y, Du H, Gao S, Dong X, Yuan H (2017) Dioscin relieves endotoxemia induced acute neuro-inflammation and protect neurogenesis via improving 5-HT metabolism. Sci Rep 7:40035. https://doi.org/10.1038/srep40035
Gao M, Chen L, Yu H, Sun Q, Kou J, Yu B (2013) Diosgenin down-regulates NF-kappaB p65/p50 and p38MAPK pathways and attenuates acute lung injury induced by lipopolysaccharide in mice. Int Immunopharmacol 15(2):240–245. https://doi.org/10.1016/j.intimp.2012.11.019
Hirai S, Uemura T, Mizoguchi N, Lee JY, Taketani K, Nakano Y, Hoshino S, Tsuge N et al (2010) Diosgenin attenuates inflammatory changes in the interaction between adipocytes and macrophages. Mol Nutr Food Res 54(6):797–804. https://doi.org/10.1002/mnfr.200900208
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (2018R1D1A3B07051424, 2018R1A6A3A11040439, 2018R1D1A1B07049876) and by a grant from the Chonnam National University Hospital Research Institute of Clinical Medicine (CRI 12052-22).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Fig. S1
(A) Chemical structures of Arg-DG and (B) schematic of the experimental design. Mice were randomly divided into three groups (1, sham control; 2, Arg-DG treatment, LPS intracerebroventricular (I.C.V) injection; and LPS I.C.V. injection with Arg-DG co-treatment) and received 3 μg of LPS with I.C.V. injection. Arg-DG solution (0.6 mg/kg) dissolved in saline was intraperitoneally administrated (IP) three times at 8-h intervals after recovering from anesthesia. 5-Bromo-2-deoxyuridine (BrdU) was intraperitoneally injected at the indicated time points. Animals were sacrificed at the indicated date presented in the experimental schedule. (C) The proliferation of adult NSCs in the hippocampal DG was not affected by Arg-DG or PBS I.C.V. injection compared to sham control, as determined by the numbers of BrdU-positive cells using immunohistochemical analysis. (PNG 736 kb)
ESM 1
(TIF 20504 kb)
Rights and permissions
About this article

Cite this article
Cai, B., Seong, KJ., Bae, SW. et al. Water-Soluble Arginyl–Diosgenin Analog Attenuates Hippocampal Neurogenesis Impairment Through Blocking Microglial Activation Underlying NF-κB and JNK MAPK Signaling in Adult Mice Challenged by LPS. Mol Neurobiol 56, 6218–6238 (2019). https://doi.org/10.1007/s12035-019-1496-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-019-1496-3