
Abstract
On the basis of the evidence that rapid intracellular Zn2+ dysregulation by amyloid β1–42 (Aβ1–42) in the normal hippocampus transiently induces cognitive decline, here we report preferential neurodegeneration in the dentate gyrus by Aβ1–42-induced intracellular Zn2+ dysregulation and its defense strategy. Neurodegeneration was preferentially observed in the dentate granule cell layer in the hippocampus after a single Aβ1–42 injection into the lateral ventricle but not in the CA1 and CA3 pyramidal cell layers, while intracellular Zn2+ dysregulation was extensively observed in the hippocampus in addition to the dentate gyrus. Neurodegeneration in the dentate granule cell layer was rescued after co-injection of extracellular and intracellular Zn2+ chelators, i.e., CaEDTA and ZnAF-2DA, respectively. Aβ1–42-induced cognitive impairment was also rescued by co-injection of CaEDTA and ZnAF-2DA. Pretreatment with dexamethasone, an inducer of metalothioneins, Zn2+-binding proteins rescued neurodegeneration in the dentate granule cell layer and cognitive impairment via blocking the intracellular Zn2+ dysregulation induced by Aβ1–42. The present study indicates that intracellular Zn2+ dysregulation induced by Aβ1–42 preferentially causes neurodegeneration in the dentate gyrus, resulting in hippocampus-dependent cognitive decline. It is likely that controlling intracellular Zn2+ dysregulation, which is induced by the rapid uptake of Zn-Aβ1–42 complexes, is a defense strategy for Alzheimer’s disease pathogenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
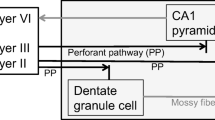
Age-related cognitive decline is believed to be initially due to changes in synaptic function rather than loss of neurons [1], while substantial synaptic and neuronal losses are observed in the early stage of Alzheimer’s disease (AD) when hippocampus-dependent memory loss becomes detectable clinically [2, 3]. The dentate gyrus and the entorhinal cortex are vulnerable to aging and AD, respectively [4]. The perforant pathway from the entorhinal cortex innervates dentate granule cells and is one of the earliest and most severely affected pathways in AD [2, 5], suggesting that the cellular disconnection between the entorhinal cortex and the dentate gyrus is involved in the AD pathogenesis. Elderly persons with mild cognitive impairment (MCI), which is a prodromal state of AD [6], have approximately 30% fewer neurons in the entorhinal cortex, which correlate with hippocampus-dependent cognitive decline [2, 5]. Therefore, dentate granule cell degeneration is a target for defending the AD pathogenesis, and its defending strategy may be useful for progressive cognitive decline.
The basal level of extracellular Zn2+ is in the range of low nanomolar concentrations (~10 nM) in the hippocampus [7] and probably increased age-dependently [8, 9]. Extracellular Zn2+ dynamics, which is often linked with Zn2+ release from zincergic terminals, a subclass of glutamatergic neurons, plays a key role for not only synaptic plasticity, a cellular mechanism of cognition but also synaptic dysfunction associated with cognitive decline [10, 11]. The basal level of intracellular Zn2+ is estimated to be less than 1 nM (~100 pM) [12, 13], and rapid intracellular Zn2+ dysregulation is critical for not only cognitive decline but also neurodegeneration. Weakened intracellular Zn2+-buffering is linked with age-related characteristic of extracellular Zn2+ influx in the aged dentate gyrus followed by cognitive decline [14, 15] and neurodegeneration [16].
Rapid intracellular Zn2+ dysregulation is due to extracellular Zn2+ influx via excess activation of glutamate receptors, glutamate excitotoxicity, while amyloid-β (Aβ), a causative candidate for the AD pathogenesis [17, 18], also induces rapid intracellular Zn2+ dysregulation. Aβ1–42 is dynamically released from synaptic vesicles [19] and rapidly captures Zn2+ at 100–500 picomolar concentration in the hippocampus extracellular fluid. Both the levels of Zn2+ and Aβ1–42 are increased in dentate granule cells 5 min after injection of Aβ1–42 into the dentate granule cell layer of rats, followed by transient Aβ1–42-induced cognitive decline that is due to increase in intracellular Zn2+ released from Aβ1–42[20, 21]. Intracellular Zn2+ dysregulation via synaptic activity-independent mechanism occurs rapidly after injection of human Aβ1–42 into rat dentate granule cell layer [22] but not after injection of human Aβ1–40 and mouse/rat Aβ1–42 because affinity of human Aβ1–42 to Zn2+ is much higher than human Aβ1–40[21] and mouse/rat Aβ1–42[23, 24], which cannot capture extracellular Zn2+ at low nanomolar concentration.
On the basis of the idea that more Zn2+ ferried by Aβ1–42 into dentate granule cells leads to degeneration in addition to cognitive decline, here, we examined whether Aβ1–42-induced intracellular Zn2+ dysregulation causes neurodegeneration in the hippocampus and also whether blocking the intracellular Zn2+ dysregulation is a useful defense strategy against Aβ1–42-induced neurodegeneration.
Materials and Methods
Animals and Chemicals
Male ddY mice (10 weeks of age) were purchased from Japan SLC (Hamamatsu, Japan). All the experiments were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals of the University of Shizuoka that refer to the American Association for Laboratory Animals Science and the guidelines laid down by the NIH (NIH Guide for the Care and Use of Laboratory Animals) in the USA. The Ethics Committee for Experimental Animals in the University of Shizuoka has approved this work.
Synthetic human Aβ1–42 was purchased from ChinaPeptides (Shanghai, China). Aβ1–42 was dissolved in saline and used immediately when the experiments were performed. SDS-PAGE showed that Aβ1–42 prepared in saline was mainly monomers with a small fraction of low order oligomers [20]. ZnAF-2DA, a membrane-permeable zinc indicator was kindly supplied from Sekisui Medical Co., LTD (Hachimantai, Japan). ZnAF-2DA is taken up into the cells through the cell membrane and is hydrolyzed by esterase in the cytosol to yield ZnAF-2 (Kd = 2.7 × 10−9 M for Zn2+), which cannot permeate the cell membrane [25, 26]. The fluorescence indicator was dissolved in dimethyl sulfoxide (DMSO) and then diluted with Ringer solution containing 119 mM NaCl, 2.5 mM KCl, 1.3 mM MgSO4, 1.0 mM NaH2PO4, 2.5 mM CaCl2, 26.2 mM NaHCO3, and 11 mM D-glucose (pH 7.3).
Intracerebroventricular (ICV) Injection of Aβ
Mice (11–13 weeks of age) were anesthetized with chloral hydrate (30 mg/kg) and placed in a stereotaxic apparatus. A microinjection canula (CXG-6, Eicom Co., Kyoto) was positioned 0.5 mm posterior to the bregma, 1.0 mm lateral, 2.2–2.4 mm inferior to the dura for ICV injection. Aβ1-42 in saline (25 μM) was injected via the microinjection canula at the rate of 0.5 μl/min for 40 min (500 pmol/mouse). Ten minutes later, the microinjection canula was slowly pulled up from the brain. The mice were individual housed for the experiments□.
Object Recognition Test
Twelve days after ICV injection of Aβ1-42, mice were placed for 10 min into an open field, which was a 56 × 67.5 cm arena surrounded by 60 cm high walls, made of a black-colored plastic. Twenty-four hours after open field exploration, mice were trained and tested in a novel object recognition task. Training in the object recognition task took place in the same area used for the open field exploration. The open field exploration was thus used as a context habituation trial for the recognition memory task. The object recognition test requires that the mice recall which of two earthenware objects they had been previously familiarized with. Twenty-four hours after arena exploration, training was conducted by placing individual mice into the field, in which two identical objects (objects A1 and A2) were positioned in two adjacent corners, 13 cm from the walls. Mice were left to explore the objects for 5 min. Mice were not used for the test when the total of the object exploration time was less than 10 s. In the test given 1 h after training, the mice explored the open field for 3 min in the presence of one familiar (A) and one novel (B) object. Behavior of mice was recorded with a video camera during the training and the test, and then two persons independently measured exploratory time and the averaged time was used. All objects presented similar textures, colors, and sizes but distinctive shapes. A recognition index calculated for each mouse was expressed by the ratio TB/(TA + TB) [TA = time spent to explore the familiar object A; TB = time spent to explore the novel object B]. Between trials the objects were washed with 70% ethanol solution. Exploration was defined as sniffing or touching the object with the nose and/or forepaws. We confirmed that there was no preference for the objects used.
Propidium Iodide (PI) Staining
Neurodegeneration was determined by PI staining after the object recognition test was finished. Fourteen days after ICV injection of Aβ1-42, the brain was quickly removed from the mice under anesthesia and immersed in ice-cold choline-Ringer containing 124 mM choline chloride, 2.5 mM KCl, 2.5 mM MgCl2, 1.25 mM NaH2PO4, 0.5 mM CaCl2, 26 mM NaHCO3, and 10 mM glucose (pH 7.3) to suppress excessive neuronal excitation. Coronal brain slices (400 μm) were prepared using a vibratome ZERO-1 (Dosaka Kyoto, Japan) in ice-cold choline-Ringer, which were continuously bubbled with 95% O2 and 5% CO2. The brain slices were bathed in PI in Ringer solution (7 μg/ml) for 30 min, bathed in Ringer solution for 30 min, and transferred to a recording chamber filled with Ringer solution. PI fluorescence (Ex/Em: 535 nm/617 nm) was captured with a confocal laser-scanning microscopic system. The region of interest was set in the dentate granule cell layer and the CA1 and CA3 pyramidal cell layers.
Fluoro-Jade B (FJB) Staining
Neurodegeneration was also determined by FJB staining after the object recognition test was finished. Fourteen days after ICV injection of Aβ1-42, the mice were anesthetized with chloral hydrate and perfused with ice-cold 4% paraformaldehyde in PBS, followed by removal of the brain and overnight fixation in 4% paraformaldehyde in PBS at 4 °C. Fixed brains were cryopreserved in 30% sucrose in PBS for 2 days and frozen in Tissue-Tek Optimal Cutting Temperature embedding medium. Coronal brain slices (30 μm) were prepared at − 20 °C in a cryostat, picked up on slides, adhered at 50 °C for 60 min, and stored at − 20 °C. The slides were first immersed in a solution containing 1% sodium hydroxide in 80% alcohol (20 ml of 5% NaOH added to 80 ml ethanol) for 5 min. This was followed by 2 min in 70% ethanol and 2 min in distilled water. The slides were then transferred to a solution of 0.06% potassium permanganate for 15 min, preferably on a shaker table to insure consistent background suppression between slices. The slides were then rinsed in distilled water for 2 min. The staining solution was prepared from a 0.01% stock solution of FJB that was made by adding 10 mg of the dye powder to 100 ml of distilled water. The stock solution and 0.1% 4′,6-Diamidino-2-phenylindole (DAPI) in distilled water were diluted with 0.1% acetic acid vehicle, resulting in a final dye concentration of 0.0004% FJB and 0.0001% DAPI in the staining solution. The staining solution was prepared within 10 min of use. The slides were bathed in the staining solution for 30 min and were rinsed for 2 min in each of three distilled water washes. Excess water was briefly removed by using a paper towel. The slides were placed at 50 °C for drying. The dry slides were twice immersed in xylene for 2 min twice before coverslipping with DPX, a non-aqueous, non-fluorescent plastic mounting media. FJB-positive cells in the unit area were measured with a confocal laser-scanning microscopic system (Ex/Em: 480 nm/525 nm). The region of interest was set in the dentate granule cell layer and the CA1 and CA3 pyramidal cell layers.
In Vivo Aβ1-42-Mediated Zn2+Dynamics
Aβ1–42 (25 μM) in saline containing 100 μM ZnAF-2DA was intracerebroventricularly injected via a microinjection canula at the rate of 0.5 μL/min for 40 min (500 pmol/mouse) into anesthetized mice as described above. One hour after the start of injection, coronal brain slices (400 μm) were prepared in ice-cold choline-Ringer solution in the same manner and transferred to a recording chamber filled with Ringer solution. The fluorescence of ZnAF-2 (laser, 488 nm; emission, 505–530 nm) was captured with a confocal laser-scanning microscopic system.
In another experiment, saline and dexamethasone sodium phosphate (10 mg/kg) in saline were intraperitoneally (i.p.) injected into mice once a day for two days. Twenty-four hours later, Aβ1–42 (25 μM) in saline containing 100 μM ZnAF-2DA was intracerebroventricularly injected, and the fluorescence of ZnAF-2 was captured in the same manner.
Data Analysis
Student’s paired t test was used for comparison of the means of paired data. For multiple comparisons, differences between treatments were assessed by one-way ANOVA followed by post hoc testing using the Tukey’s test (the statistical software, GraphPad Prism 5). A value of p < 0.05 was considered significant. Data were expressed as means ± standard error. The results of statistical analysis are described in each figure legend.
Results
Preferential Neurodegeneration in the Dentate Gyrus by Aβ1–42
To assess hippocampal neurodegeneration, we used PI, a fluorescent intercalating agent, which binds to DNA by intercalating between the bases with little or no sequence preference in dead cells, and FJB, an anionic fluorescein derivative, which is used for the histological staining of neurons undergoing degeneration [27, 28].
PI fluorescence was enhanced in the dentate granule cell layer 14 days after ICV injection of Aβ1-42 but not in the CA1 and CA3 pyramidal cell layers (Fig. 1). FJB-positive cells were also increased in the dentate granule cell layer but not in the CA1 and CA3 pyramidal cell layers (Fig. 2). On the other hand, intracellular Zn2+ level, which was determined with intracellular ZnAF-2 fluorescence, was extensively increased in the hippocampal CA1 and CA3, in addition to the dentate gyrus (Fig. 3).

A single ICV injection of Aβ1–42 induces neurodegeneration in the dentate gyrus. a Fourteen days after ICV injection of Aβ1-42, PI fluorescence was measured in the dentate granule cell layer (DGCL), the CA1 pyramidal cell layer (CA1 PCL), and the CA3 pyramidal cell layer (CA3 PCL) surrounded by the dotted line (upper). Bar, 50 μm. b Each bar and line (mean ± SEM) represent the rate (%) of PI fluorescence after Aβ1–42 injection to that after saline (vehicle) injection, which was represented as 100% (lower). ***p < 0.001 vs. saline (t test). DGCL; saline, n = 17, Aβ1–42 = 23; CA1, saline, n = 21, Aβ1–42 = 30; CA3, saline, n = 13, Aβ1–42 = 28

A single ICV injection of Aβ1–42 induces neurodegeneration in the dentate gyrus. a Fourteen days after ICV injection of Aβ1-42, FJB fluorescence was measured in the dentate granule cell layer (DGCL), the CA1 pyramidal cell layer (CA1 PCL), and the CA3 pyramidal cell layer (CA3 PCL) surrounded by the dotted line. Bar, 50 μm. b The area surrounded by the white line, which was shown in a, was magnified. c Each bar and line (mean ± SEM) represent FJB-positive cells in the unit area after injection of vehicle and Aβ1-42. ***p < 0.001 vs. saline (t test). DGCL; saline, n = 16, Aβ1–42 = 16; CA1, saline, n = 16, Aβ1–42 = 16; CA3, saline, n = 16, Aβ1–42 = 16

A single ICV injection of Aβ1–42 increases intracellular Zn2+ level in the hippocampus. a Fourteen days after ICV injection of Aβ1-42, ZnAF-2 fluorescence was measured in the dentate granule cell layer (DGCL), the CA1 pyramidal cell layer (CA1 PCL), and the CA3 pyramidal cell layer (CA3 PCL) surrounded by the dotted line (upper). Bar, 50 μm. b Each bar and line (mean ± SEM) represent the rate (%) of ZnAF-2 fluorescence after Aβ1–42 injection to that in the DGCL after saline (vehicle) injection, which was represented as 100% (lower). *p < 0.05; **p < 0.001 vs. saline (t test). DGCL; saline, n = 7, Aβ1–42 = 6; CA1, saline, n = 7, Aβ1–42 = 6; CA3, saline, n = 6, Aβ1–42 = 6
Rescue of Neurodegeneration by Reducing Aβ1–42-Induced Increase in Intracellular Zn2+ in the Dentate Gyrus
The enhanced PI fluorescence and FJB-positive cells were reduced in the dentate granule cell layer after co-injection of extracellular and intracellular Zn2+ chelators, i.e., CaEDTA and ZnAF-2DA, respectively (Figs. 4 and 5). CaEDTA competes with Aβ1–42 for binding to Zn2+ and reduces the formation of Zn-Aβ1–42 complexes, resulting in reducing both the uptake of Zn2+ and Aβ1–42[21]. Extracellular ZnAF-2DA also competes with Aβ1–42 for binding to Zn2+ and reduces the formation of Zn-Aβ1–42 complexes, while intracellular ZnAF-2 taken up into cells chelates Zn2+ released from Aβ1–42[20, 21].

Zn2+ chelators rescue Aβ1–42-induced neurodegeneration. Fourteen days after ICV injection of Aβ1-42 and Zn2+ chelators, PI fluorescence was measured in the dentate granule cell layer (DGCL) surrounded by the dotted line (upper). Bar, 50 μm. Each bar and line (mean ± SEM) represent the rate (%) of PI fluorescence after Aβ1–42 injection to that after saline (vehicle) injection, which was represented as 100% (lower). ***p < 0.001 vs. saline; #p < 0.05, ##p < 0.01 vs. Aβ1–42 (Tukey’s test). Saline, n = 17, Aβ1–42, n = 23; Aβ1–42 + CaEDTA, n = 16, Aβ1–42 + ZnAF-2DA, n = 18

Zn2+ chelators rescue Aβ1–42-induced neurodegeneration. Fourteen days after ICV injection of Aβ1-42 and Zn2+ chelators, FJB fluorescence was measured in the dentate granule cell layer (DGCL) surrounded by the dotted line (upper). Bar, 50 μm. Each bar and line (mean ± SEM) represent FJB-positive cells in the unit area after injection of vehicle and Aβ1-42 (lower). ***p < 0.001 vs. saline; ###p < 0.001 vs. Aβ1–42 (Tukey’s test). Saline, n = 16, Aβ1–42, n = 19; Aβ1–42 + CaEDTA, n = 16, Aβ1–42 + ZnAF-2DA, n = 16
Newly synthesized metalothioneins (MTs), Zn2+-binding proteins can capture Zn2+ released from intracellular Aβ1–42 without modifying intracellular Aβ1–42 level followed by rescuing Aβ1–42 neurotoxicity [29], indicating that Zn2+ released from Aβ1–42 is more neurotoxic than Aβ1–42 itself. Pretreatment with dexamethasone, an inducer of MTs reduced not only the enhanced PI fluorescence and FJB-positive cells in the dentate granule cell layer (Figs. 6 and 7) but also Aβ1–42-induced increase in intracellular Zn2+ in the dentate gyrus (Fig. 8).

Dexamethasone rescues Aβ1–42-induced neurodegeneration. Saline and dexamethasone (DEX, 10 mg/kg) in saline were i.p. injected into mice once a day for two days. Twenty-four hours later, Aβ1–42 (25 μM) in saline was intracerebroventricularly injected and PI fluorescence was measured in the dentate granule cell layer (DGCL) surrounded by the dotted line after 14 days after ICV injection (upper). Bar, 50 μm. Each bar and line (mean ± SEM) represent the rate (%) of PI fluorescence after Aβ1–42 injection to that after saline (vehicle) injection, which was represented as 100% (lower). **p < 0.01 vs. saline/saline; #p < 0.05, ##p < 0.01 vs. saline/Aβ1–42 (Tukey’s test). Saline/saline, n = 12, saline/Aβ1–42, n = 9; DEX/saline, n = 15, DEX/Aβ1–42, n = 16

Dexamethasone rescues Aβ1–42-induced neurodegeneration. Saline and dexamethasone (DEX, 10 mg/kg) in saline were i.p. injected into mice once a day for two days. Twenty-four hours later, Aβ1–42 (25 μM) in saline was intracerebroventricularly injected and FJB fluorescence was measured in the dentate granule cell layer (DGCL) surrounded by the dotted line 14 days after ICV injection (upper). Bar, 50 μm. Each bar and line (mean ± SEM) represent FJB-positive cells in the unit area after injection of vehicle and Aβ1-42 (lower). **p < 0.01 vs. saline/saline; #p < 0.05 vs. saline/Aβ1–42 (Tukey’s test). Saline/saline, n = 20, saline/Aβ1–42, n = 23, DEX/saline, n = 12, DEX/Aβ1–42, n = 14

Dexamethasone reduces Aβ1–42-induced increase in intracellular Zn2+. Saline and dexamethasone (DEX, 10 mg/kg) in saline were i.p. injected into mice once a day for two days. Twenty-four hours later, Aβ1–42 (25 μM) in saline was intracerebroventricularly injected and ZnAF-2 fluorescence was measured in the dentate granule cell layer (DGCL) surrounded by the dotted line 1 h after the start of ICV injection (upper). Bar, 50 μm. Each bar and line (mean ± SEM) represent the rate (%) of ZnAF-2 fluorescence after Aβ1–42 injection to that in the DGCL after saline (vehicle) injection, which was represented as 100% (lower). ***p < 0.001 vs. saline/saline; ###p < 0.001 vs. saline/Aβ1–42 (Tukey’s test). Saline/saline, n = 7, saline/Aβ1–42, n = 6; DEX/saline, n = 8, DEX/Aβ1–42, n = 7
Rescue of Cognitive Decline by Zn2+Chelators and Dexamethasone
Object recognition memory was impaired after ICV injection of Aβ1-42, while the impairment was rescued by co-injection of CaEDTA and ZnAF-2DA (Recognition index: saline/training, 49.1 ± 1.5%, saline/test, 64.3 ± 2.9, p < 0.01; Aβ1–42/training, 49.6 ± 3.6%, Aβ1–42/test, 48.6 ± 2.2%; Aβ1–42 + CaEDTA/training, 49.6 ± 2.4%, Aβ1–42 + CaEDTA/test, 68.3 ± 3.5%, p < 0.01; Aβ1–42 + ZnAF-2DA/training, 50.0 ± 2.5%, Aβ1–42 + ZnAF-2DA/test, 67.6 ± 3.1%, p < 0.01) (Fig. 9a). The impairment was also rescued by pretreatment with dexamethasone (Recognition index: saline/saline/training, 49.1 ± 1.3%, saline/saline/test, 64.1 ± 2.8, p < 0.01; saline/Aβ1–42/training, 49.6 ± 2.6%, saline/Aβ1–42/test, 49.6 ± 1.8%; dexamethasone (DEX)/saline/training, 45.3 ± 3.7%, DEX/saline/test, 63.9 ± 3.5%, p < 0.01; DEX/Aβ1–42/training, 50.2 ± 1.2%, DEX/Aβ1–42/test, 62.0 ± 2.2%, p < 0.05) (Fig. 9b).

Zn2+ chelators and dexamethasone rescue Aβ1–42-induced cognitive decline. a Thirteen days after ICV injection of Aβ1-42, object recognition test was performed. Each bar and line (mean ± SEM) represent the recognition index (%) in the test 1 h after training. *p < 0.05 vs. saline, ##p < 0.01 vs. Aβ1–42 (Tukey’s test). Saline, n = 6, Aβ1–42, n = 6; Aβ1–42 + CaEDTA, n = 7, Aβ1–42 + ZnAF-2DA, n = 7. b Saline and dexamethasone (DEX, 10 mg/kg) in saline were i.p. injected into mice once a day for two days. Twenty-four hours later, Aβ1–42 (25 μM) in saline was intracerebroventricularly injected and object recognition test was performed 13 days after ICV injection. **p < 0.01 vs. saline/saline; #p < 0.05, ##p < 0.01 vs. saline/Aβ1–42(Tukey’s test). Saline/saline, n = 9, saline/Aβ1–42, n = 11; DEX/saline, n = 6, DEX/Aβ1–42, n = 7
Discussion
On the basis of the rapid oligomerization of Aβ, the toxic action of soluble low-molecular-weight Aβ oligomers has been emphasized in the AD pathogenesis. Aβ1–40 and Aβ1–42 are the two most abundant isoforms, and the former is more than 10 times as abundant as the latter in the cerebrospinal fluid [30]. Importantly, Aβ1–42 far more rapidly forms aggregates and is more neurotoxic than Aβ1–40[31,32,33,34]. The neurotoxic action of low-molecular-weight Aβ oligomers has been tested in not only primary neuronal cultures or brain slices but also animal models. To determine the action of Aβ1–42 in cognitive decline and neurodegeneration in vivo, Aβ1–42 oligomers have been exclusively prepared in vitro [35, 36] and injected into experimental animals to test the neurotoxicity in vivo [37,38,39,40].
On the other hand, Zn2+ has been implicated in the AD pathogenesis because Zn2+ readily accelerates Aβ oligomerization [41, 42]. The trial for drug therapy targeting metal ions, e.g., Zn2+ has been reported for AD [43]. As a matter of fact, Zn2+ has remarkable impacts on Aβ1–42 aggregation in the range of physiological pH [44]. Zn2+ shifts the self-association of Aβ1–42 toward a non-fibrillary pathway by interfering with the aggregation process at multiple levels [45]. Judging from these findings, it is important to evaluate neurotoxicity of soluble Aβ1–42 oligomers, which is rapidly formed in the presence of extracellular Zn2+ in vivo. It is estimated that chemical structures of soluble Aβ1–42 oligomers with Zn2+ are different from those of soluble Aβ1–42 oligomers prepared without Zn2+ in vitro, which might lead to misunderstanding of Aβ1–42 neurotoxicity in vivo. On the basis of rapid intracellular Zn2+ dysregulation in dentate granule cells induced by Aβ1–42 in vivo [21], we tested the idea that rapid intracellular Zn2+ dysregulation induced by Aβ1–42 causes neurodegeneration in the hippocampus after a single injection of Aβ1–42 into the lateral ventricle.
Rapid increase in intracellular Zn2+ induced by Aβ1–42 was extensively observed in the hippocampus 20 min after finishing a single Aβ1–42 injection into the lateral ventricle. In contrast, neurodegeneration was preferentially observed in the dentate gryrus 14 days after ICV injection but not in the CA1 and CA3. When Aβ1–42 concentration reaches 100–500 pM, Aβ1–42 is taken up into dentate granule cells extracellular Zn2+-dependently. The formation of Zn-Aβ1–42 complexes in the extracellular compartment is critical for Aβ1–42 uptake [21], which is the highest in dentate gyrus neurons in hippocampal slices in vitro [22], consistent with the highest level of intracellular Zn2+ in the dentate gyrus in the present study. These data suggest that the preferential uptake of Aβ1–42 into dentate gyrus neurons causes the preferential neurodegeneration. The perforant pathway from the entorhinal cortex innervates dentate granule cells and is one of the earliest and most severely affected pathways in AD [2, 5]. Therefore, vulnerability of dentate granule cells in the AD pathogenesis is probably due to the preferential uptake of Aβ1–42, which might be linked with Aβ1–42 degradation and its elimination from the dentate gyrus [46]. However, the relationship between the preferential Aβ1–42 uptake and Aβ1–42 metabolism in the dentate gyrus remains to be clarified.
Intracellular Zn2+ level was also considerably high in CA3 and CA1 neurons, while neurodegeneration was scarcely observed in the CA3 and CA1. The high Zn2+ levels might be linked with less Aβ1–42 uptake into CA3 and CA1 neurons [22] followed by less vulnerability of CA3 and CA1 neurons to neurodegeneration. On the other hand, it is estimated that Zn2+ released from intracellular Zn-Aβ1–42 complexes taken up into dentate gyrus neurons causes neurodegeneration. This estimation was confirmed using Zn2+ chelators (Figs. 4 and 5). Extracellular Zn2+ chelators reduce the formation of Zn-Aβ1–42 complexes in the extracellular compartment and both the uptake of Aβ1–42 and Zn2+ followed by reducing Aβ1–42 toxicity[20]. Intracellular Zn2+ chelators do not reduce the formation of Zn-Aβ1–42 complexes in the extracellular compartment, while they capture Zn2+ released from intracellular Zn-Aβ1–42 complexes followed by reducing Aβ1–42 toxicity[20]. In the present study, neurodegeneration in the dentate gyrus was rescued after co-injection of extracellular and intracellular Zn2+ chelators, i.e., CaEDTA and ZnAF-2DA, respectively. Aβ1–42-induced cognitive impairment was also rescued by co-injection of CaEDTA and ZnAF-2DA. Intracellular Zn-Aβ1–42 complexes taken up into dentate gyrus neurons release toxic Zn2+, while Aβ1–42 may capture functional Zn2+ after the release, followed by neurodegeneration via dysfunction of Zn2+-requiring proteins such as a metabotropic Gq-coupled Zn2+-sensing receptor, mZnR/GPR39[47].
Intracellular MTs buffer free Zn2+ based on its varying affinities for Zn2+[48,49,50]. If intracellular Zn2+ concentration reaches approximately 10 nM, which is the estimated concentration of extracellular Zn2+[7], MTs bind up to 7 equivalents of Zn2+ and become the holo-MTs, Zn7MTs [51]. When the binding capacity of MTs is saturated by the rapid increase in intracellular Zn2+, which is induced by extracellular Aβ1–42 influx, intracellular MTs lose the Zn2+-buffering ability. Therefore, it is thought that an increase in intracellular MTs is effective for buffering the rapid increase in intracellular Zn2+ by Aβ1–42. In vivo Kd value of Zn2+ to Aβ1–42 may be in the range of ~3–30 nM [21], which is higher than that of MTs (~1 pM) [52]. Pretreatment with dexamethasone, an inducer of MT-I and MT-II, reduced neurodegeneration in the dentate granule cell layer and rescued cognitive impairment via blocking the rapid increase in intracellular Zn2+ induced by Aβ1–42. Because intracellular MTs induced by dexamethasone does not reduce the uptake of Zn-Aβ1–42, the induced MTs, which capture Zn2+ released from Aβ1–42, reduce intracellular Zn2+ levels but not modify intracellular Aβ1–42 levels [29]. In contrast, dexamethasone may reduce the increase in intracellular Zn2+ induced by Aβ1–42 via another mechanism because dexamethasone has a great many effects. In addition to the effect of Zn2+ chelators, the effect of dexamethasone indicates that neurodegeneration and cognitive decline induced by Aβ1–42 are due to intracellular Zn2+ toxicity, which is caused by release from Aβ1–42.
The present study indicates that rapid intracellular Zn2+ dysregulation induced by Aβ1–42 preferentially causes neurodegeneration in the dentate gryrus, resulting in hippocampus-dependent cognitive decline. It is likely that controlling intracellular Zn2+ dysregulation, which is induced by the rapid uptake of Zn-Aβ1–42 complexes, is a defense strategy for the AD pathogenesis.
References
Morrison JH, Hof PR (1997) Life and death of neurons in the aging brain. Science 278:412–419
Scheff SW, Price DA, Schmitt FA, Mufson EJ (2006) Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging 27:1372–1384
Crews L, Masliah E (2010) Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum Mol Genet 19:R12–R20
Small SA, Schobel SA, Buxton RB, Witter MP, Barnes CA (2011) A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat Rev Neurosci 12:585–601
Gómez-Isla T, Price JL, McKeel DW Jr, Morris JC, Growdon JH, Hyman BT (1996) Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci 16:4491–4500
Qin Y, Tian Y, Han H, Liu L, Ge X, Xue H, Wang T, Zhou L et al (2019) Risk classification for conversion frommild cognitive impairmenttoAlzheimer'sdiseasein primary care. Psychiatry Res 278:19–26
Frederickson CJ, Giblin LJ, Krezel A, McAdoo DJ, Muelle RN, Zeng Y, Balaji RV, Masalha R et al (2006) Concentrations of extracellular free zinc (pZn)e in the central nervous system during simple anesthetization, ischemia and reperfusion. Exp Neurol 198:285–293
Takeda A, Tamano H (2017) Significance of low nanomolar concentration of Zn2+ in artificial cerebrospinal fluid. Mol Neurobiol 54:2477–2482
Tamano H, Nishio R, Shakushi Y, Sasaki M, Koike Y, Osawa M, Takeda A (2017) In vitro and in vivo physiology of low nanomolar concentrations of Zn2+ in artificial cerebrospinal fluid. Sci Rep 7:42897
Takeda A, Tamano H (2017) Impact of synaptic Zn2+ dynamics on cognition and its decline. Int J Mol Sci 18:2411
Tamano H, Takeda A (2019) Age-dependent modification of intracellular Zn2+-buffering in the hippocampus and its impact. Biol Pharm Bull in press
Sensi SL, Canzoniero LM, Yu SP, Ying HS, Koh JY, Kerchner GA, Choi DW (1997) Measurement of intracellular free zinc in living cortical neurons: Routes of entry. J Neurosci 17:9554–9564
Colvin RA, Bush AI, Volitakis I, Fontaine CP, Thomas D, Kikuchi K, Holmes WR (2008) Insights into Zn2+ homeostasis in neurons from experimental and modeling studies. Am J Phys Cell Physiol 294:C726–C742
Takeda A, Koike Y, Osawa M, Tamano H (2018) Characteristic of extracellular Zn2+ influx in the middle-aged dentate gyrus and its involvement in attenuation of LTP. Mol Neurobiol 55:2185–2195
Takeda A, Tamano H, Murakami T, Nakada H, Minamino T, Koike Y (2018) Weakened intracellular Zn2+-buffering in the aged dentate gyrus and its involvement in erasure of maintained LTP. Mol Neurobiol 55:3856–3865
Tamano H, Nishio R, Morioka H, Furuhata R, Komata Y, Takeda A (2019) Paraquat as an environmental risk factor in Parkinson's disease accelerates age-related degeneration via rapid influx of extracellular Zn2+ into nigral dopaminergic neurons. Mol Neurobiol 56:7789–7799. https://doi.org/10.1007/s12035-019-01642-5
Perrin RJ, Fagan AM, Holtzman DM (2009) Multimodal techniques for diagnosis and prognosis of Alzheimer's disease. Nature 461:916–922
Kepp KP (2016) Alzheimer's disease due to loss of function: A new synthesis of the available data. Prog Neurobiol 143:36–60
Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM et al (2005) Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48:913–922
Takeda A, Nakamura M, Fujii H, Uematsu C, Minamino T, Adlard PA, Bush AI, Tamano H (2014) Amyloid β-mediated Zn2+ influx into dentate granule cells transiently induces a short-term cognitive deficit. PLoS One 9:e115923
Takeda A, Tamano H, Tempaku M, Sasaki M, Uematsu C, Sato S, Kanazawa H, Datki ZL et al (2017) Extracellular Zn2+ is essential for amyloid β1-42-induced cognitive decline in the normal brain and its rescue. J Neurosci 37:7253–7262
Tamano H, Oneta N, Shioya A, Adlard PA, Bush AI, Takeda A (2019) In vivo synaptic activity-independent co-uptakes of amyloid β1-42 and Zn2+ into dentate granule cells in the normal brain. Sci Rep 9:6498
Tamano H, Suzuki H, Kobuchi S, Adlard PA, Bush AI, Takeda A (2019) Difference in ability for extracellular Zn2+ influx between human and rat amyloid β1-42 and its significance. NeuroToxicology 72:1–5
Tamano H, Takiguchi M, Shimaya R, Adlard PA, Bush AI, Takeda A (2019) Extracellular Zn2+-independently attenuated LTP by human amyloid β1-40and rat amyloid β1-42. Biochem Bioph Res Co 514:888–892
Hirano T, Kikuchi K, Urano Y, Nagano T (2002) Improvement and biological applications of fluorescent probes for zinc, ZnAFs. J Am Chem Soc 124:6555–6562
Ueno S, Tsukamoto M, Hirano T, Kikuchi K, Yamada MK, Nishiyama N, Nagano T, Matsuki N et al (2002) Mossy fiber Zn2+ spillover modulates heterosynaptic N-methyl-D-aspartate receptor activity in hippocampal CA3 circuits. J Cell Biol 158:215–220
Schmued LC, Hopkins KJ (2000) Fluoro-jade B: A high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res 874:123–130
Piermartiri TC, Figueiredo CP, Rial D, Duarte FS, Bezerra SC, Mancini G, de Bem AF, Prediger RD et al (2010) Atorvastatin prevents hippocampal cell death, neuroinflammation and oxidative stress following amyloid-β(1-40) administration in mice: Evidence for dissociation between cognitive deficits and neuronal damage. Exp Neurol 226:274–284
Takeda A, Tamano H, Hashimoto W, Kobuchi S, Suzuki H, Murakami T, Tempaku M, Koike Y et al (2018) Novel defense by metallothionein induction against cognitive decline: From amyloid β1-42-induced excess Zn2+ to functional Zn2+ deficiency. Mol Neurobiol 55:7775–7788
Schoonenboom NS, Mulder C, Van Kamp GJ, Mehta SP, Scheltens P, Blankenstein MA, Mehta PD (2005) Amyloid beta 38, 40, and 42 species in cerebrospinal fluid: More of the same? Ann. Neurol. 58:139–142
Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I et al (1998) Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 95:6448–6453
Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G (2000) High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J Neurosci 20:4050–4058
Yan Y, Wang C (2006) Abeta42 is more rigid than Abeta40 at the C terminus: Implications for Abeta aggregation and toxicity. J Mol Biol 364:853–862
Fu L, Sun Y, Guo Y, Chen Y, Yu B, Zhang H, Wu J, Yu X et al (2017) Comparison ofneurotoxicityof different aggregated forms of Aβ40, Aβ42 and Aβ43 in cell cultures. J Pept Sci 23:245–251
Kuperstein I, Broersen K, Benilova I, Rozenski J, Jonckheere W, Debulpaep M, Vandersteen A, Segers-Nolten I et al (2010) Neurotoxicity of Alzheimer’s disease Abeta peptides is induced by small changes in the Abeta42 to Abeta40 ratio. EMBO J 29:3408–3420
Broersen K, Jonckheere W, Rozenski J, Vandersteen A, Pauwels K, Pastore A, Rousseau F, Schymkowitz J (2011) A standardized and biocompatible preparation of aggregate-free amyloid beta peptide for biophysical and biological studies of Alzheimer’s disease. Protein Eng Des Sel 24:743–750
Reed MN, Hofmeister JJ, Jungbauer L, Welzel AT, Yu C, Sherman MA, Lesné S, LaDu MJ et al (2011) Cognitive effects of cell-derived and synthetically derived Aβoligomers. Neurobiol Aging 32:1784–1794
Brouillette J, Caillierez R, Zommer N, Alves-Pires C, Benilova I, Blum D, De Strooper B, Buée L (2012) Neurotoxicity and memory deficits induced by soluble low-molecular-weight amyloid-β1-42 oligomers are revealed in vivo by using a novel animal model. J Neurosci 32:7852–7861
Forny-Germano L, Lyra e Silva NM, Batista AF, Brito-Moreira J, Gralle M, Boehnke SE, Coe BC, Lablans A et al (2014) Alzheimer's disease-like pathology induced by amyloid-β oligomers in nonhuman primates. J Neurosci 34:13629–13643
Schmid S, Jungwirth B, Gehlert V, Blobner M, Schneider G, Kratzer S, Kellermann K, Rammes G (2017) Intracerebroventricular injection of beta-amyloid in mice is associated with long-term cognitive impairment in the modified hole-board test. Behav Brain Res 324:15–20
Bush AI, Pettingell WH, Multhaup G, de Paradis M, Vonsattel JP, Gusella JF, Beyreuther K, Masters CL et al (1994) Rapid induction of Alzheimer A beta amyloid formation by zinc. Science 265:1464–1467
Bush AI (2013) The metal theory of Alzheimer's disease. J Alzheimers Dis 33:S277–S281
Adlard PA, Bush AI (2018) Metals andAlzheimer's disease: How far have we come in the clinic? J Alzheimers Dis 62:1369–1379
Guo J, Yu L, Sun Y, Dong X (2017) Kinetic insights into Zn2+-induced amyloid β-protein aggregation revealed by stopped-flow fluorescence spectroscopy. J Phys Chem B 121:3909–3917
Zhang T, Pauly T, Nagel-Steger L (2018) Stoichiometric Zn2+interferes with the self-association of Aβ42: Insights from size distribution analysis. Int J Biol Macromol 113:631–639
Yamada K, Yabuki C, Seubert P, Schenk D, Hori Y, Ohtsuki S, Terasaki T, Hashimoto T et al (2009) Abeta immunotherapy: Intracerebral sequestration of Abeta by an anti-Abeta monoclonal antibody 266 with high affinity to soluble Abeta. J Neurosci 29:11393–11398
Abramovitch-Dahan C, Asraf H, Bogdanovic M, Sekler I, Bush AI, Hershfinkel M (2016) Amyloid β attenuates metabotropic zinc sensing receptor, mZnR/GPR39, dependent Ca2+, ERK1/2 and Clusterin signaling in neurons. J Neurochem 139:221–233
Krężel A, Maret W (2006) Zinc buffering capacity of a eukaryotic cell at physiological pZn. J Biol Inorg Chem 11:1049–1062
Krężel A, Hao Q, Maret W (2007) The zinc/thiolate redox biochemistry of metallothionein and the control of zinc ion fluctuations in cell signaling. Arch Biochem Biophys 463:188–200
Krężel A, Maret W (2007) Dual nanomolar and picomolar Zn(II) binding properties of metallothionein. J Am Chem Soc 129:10911–10921
Krężel A, Maret W (2017) The functions of metamorphic Metallothioneins in zinc and copper metabolism. Int J Mol Sci 18. https://doi.org/10.3390/ijms18061237
Vasák M, Kägi JH (1983) Spectroscopic properties of metallothionein. In: Sigel H (ed) Metal Ions in Biological Systems, vol 15. Marcel Dekker, New York, pp. 213–273
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Tamano, H., Takiguchi, M., Tanaka, Y. et al. Preferential Neurodegeneration in the Dentate Gyrus by Amyloid β1–42-Induced Intracellular Zn2+Dysregulation and Its Defense Strategy. Mol Neurobiol 57, 1875–1888 (2020). https://doi.org/10.1007/s12035-019-01853-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-019-01853-w