
Abstract
Gallic acid (3,4,5-trihydroxybenzoic acid, GA), a phenolic acid, is ubiquitous in almost all parts of the plant. In the present study, a neuroinflammatory rat model using intranigral infusion of lipopolysaccharides (LPS, 4 μg/μL) was employed to study the neuroprotective effect of GA which was orally administered daily. Compared with the vehicle-treated rats, systemic administration of GA (100 mg/kg) significantly attenuated LPS-induced increases in glial fibrillary acidic protein (a biomarker of activated astrocytes) and ED-1 (a biomarker of activated microglia), as well as inducible nitric oxide synthase (iNOS, a proinflammatory enzyme) and interleukin-1β (a proinflammatory cytokine), in the LPS-infused substantia nigra (SN) of rat brain. At the same time, GA attenuated LPS-induced elevation in heme oxygenase-1 level (a redox-regulated protein) and α-synuclein aggregation (a hallmark of CNS neurodegeneration), suggesting that GA is capable of inhibiting LPS-induced oxidative stress and protein conjugation. Furthermore, GA prevented LPS-induced caspase 3 activation (a biomarker of programmed cell death) and LPS-induced increases in receptor-interacting protein kinase (RIPK)-1 and RIPK-3 levels (biomarkers of necroptosis), indicating that GA inhibited LPS-induced apoptosis and necroptosis in the nigrostriatal dopaminergic system of rat brain. Moreover, an in vitro study was employed to investigate the anti-inflammatory effect of GA on BV2 microglial cells which were subjected to LPS (1 μg/mL) treatment. Consistently, co-incubation of GA diminished LPS-induced increases in iNOS mRNA and iNOS protein expression in the treated BV-2 cells as well as NO production in the culture medium. The anti-oxidative activity of GA was evaluated using iron-induced lipid peroxidation of brain homogenates. After 3-h incubation at 37 °C, GA was more potent than glutathione and less potent than trolox in inhibiting iron-induced lipid peroxidation. Conclusively, the present study suggests that GA is anti-inflammatory via attenuating LPS-induced neuroinflammation, oxidative stress, and protein conjugation. Furthermore, GA prevented LPS-induced programmed cell deaths of nigrostriatal dopaminergic neurons of the rat brain, suggesting that GA may be neuroprotective by attenuating neuroinflammation in CNS neurodegenerative diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Oxidative stress, protein aggregation, and cell death has been proposed as a vicious cycle in the pathophysiology of CNS neurodegenerative diseases, including Alzheimer’s disease, Parkinsonian disease (PD), stroke, and spinal cord injury [1]; neuroinflammation appears to be the center of this vicious cycle. To support this notion, a significant body of studies has delineated a pathological role of neuroinflammation in CNS neurodegenerative diseases [2,3,4,5]. Furthermore, our previous studies employed intranigral infusion of arsenite, 1-methyl-4-phenylpyridinium, and acrolein and consistently showed neuroinflammation in the affected nigrostriatal dopaminergic system of rat brain, including activation of glial cells, increases in proinflammatory cytokines and enzymes as well as inflammasome activation, protein aggregation, and cell death [6,7,8].
To rescue the deteriorated brain function, neuroprotective strategies using anti-inflammatory treatments have been proposed for some time [9, 10]. Recently, gallic acid (3,4,5-trihydroxybenzoic acid, GA), a phenolic acid ubiquitously found in almost all parts of the plant, including seed, root, fruit, leaf, wood, and bark, has been suggested as an active ingredient for the neuroprotective action of the natural products [11, 12], including Mucuna pruriens seeds [13]. To support this finding, a pure GA preparation was used to attenuate 6-hydroxydopamine (OHDA)-induced apoptosis in the human SH-SY5Y cells [14]. Due to its permeability to the blood-brain barrier, oral administration of GA attenuated neurotoxicity induced by 6-OHDA, including a reduction in passive avoidance and thiol content as well as elevation in malondialdehyde [15]. The anti-oxidative mechanism underlying GA-induced neuroprotection was proposed for GA-induced neuroprotection in PD models [15, 16]. However, limited studies have focused on the involvement of neuroinflammation in the neuroprotective effect of GA on Parkinsonian models.
LPS has been commonly used to model neuroinflammation found in CNS neurodegenerative diseases. In vitro studies have shown that incubation of LPS activated microglia and resulted in increases in proinflammatory cytokines and proinflammatory enzymes [16]. In vivo studies using local infusion of LPS in the hippocampus [4] or substantia nigra consistently showed neuroinflammation in the defected brain regions, such as glial activation, increases in proinflammatory cytokines, leukocyte infiltration, and α-synuclein aggregation [2, 4, 17]. Moreover, systemic administration of LPS mimicked idiopathic PD in the nigrostriatal dopaminergic system of the treated mice, including dopamine neuron loss and cytokine and chemokine production [3, 17]. In the present study, the involvement of inflammation in GA-induced neuroprotection was investigated using in vivo and in vitro approaches. The in vivo study employed intranigral infusion of LPS to delineate the neuroprotective effect of GA, focusing on the LPS-induced neuroinflammation, including microglial activation, α-synuclein aggregation, and necroptosis. Furthermore, the in vitro study employed BV-2 cells as an alternative model for primary cultured microglia, to confirm the involvement of neuroinflammation in GA-induced neuroprotective effect on LPS-treated SN.
Drugs
The chemicals used were lipopolysaccharide (LPS, Sigma, St. Louis, MO, USA), GA (Sigma), trolox (Sigma), and glutathione (Calbiochem, CA, USA).
Antibodies
The primary antibodies for Western blot assay included GFAP (1:1000; #3670, Cell Signaling Tech., Beverly, MA, USA), ED-1 (1:1000, Bio-Rad Laboratories, Inc., Hercules, CA, USA), IL-1β (1:1000, #12426, Cell Signaling Tech.), iNOS (1:1000, BD, Lexington, KY, USA), HO-1 (1:1000, #ADI-SPA-895-F, Enzo Life Sciences, NY, USA), α-synuclein (1:1000, BD, Lexington), cleaved caspase 1 (1:1000, #MAB1871, Millipore, Billerica, MA, USA), and β-actin (1:15000, #MAB1501, Millipore). The secondary antibodies were horseradish peroxidase–conjugated secondary IgG (1:15000, Chemicon, Temecula, CA, USA). The primary antibodies for immunofluorescence staining included those against tyrosine hydroxylase (TH, 1:50, Cell Signaling Tech.) and α-synuclein (1:50, Abcam, Cambridge, UK). The secondary antibodies were conjugated with rhodamine and fluorescein isothiocyanate (Millipore).
Animals
Adult, male Sprague Dawley rats, weighing 250–350 g, were supplied by the National Laboratory Animal Breeding and Research Center, Taipei, Taiwan, R.O.C.. All animals (3 rats/cage) were housed in an air-conditioned room (22 ± 2 °C) on a 12-h light/dark cycle (07:00–19:00-h light) and had free access to food and water. The use of animals has been approved by the Institutional Animal Care and Use Committee of Taipei Veterans General Hospital, Taipei, Taiwan, R.O.C.. The approval number is IACUC2018-186. All experiments were performed in the accordance with relevant guidelines and regulation.
Surgery and Intranigral Infusion of Drug
Rats were anesthetized with chloral hydrate (450 mg/kg, i.p., Sigma) and placed in a stereotaxic instrument (David Kopf Instruments, Palo Alto, CA, USA). After skin incision and exposure of the parietal bone, holes were drilled above the cortical surface for intranigral infusion of drugs. One microliter of LPS (4 μg/μL) was unilaterally infused into substantia nigra (SN) stereotaxically with coordinates of 3.2 mm anterior, 2 mm above the interaural zero, 2.1 mm lateral to the midline, and 3.5 mm below the incisor bar. Drug solutions were infused at a rate of 0.2 μL/min through a 30-gauge stainless steel needle. The injection needle was held in place for an additional 3 min following drug infusion. After the surgery, rats recovered from anesthesia and were placed in home cages for the indicated times.
Oral Administration of GA
Rats were randomly divided into 3 groups. The control group received saline and two experimental groups received GA (50 and 100 mg/kg) via an intragastric needle 1 h prior to an intranigral infusion of LPS. Afterwards, daily administration of GA continued as indicated for each experiment.
Western Blot Analysis of Relevant Proteins
At the end of in vivo experiments, rats were sacrificed by decapitation. Dissected SN was homogenized with a sonicator in 40-μL ice-cold protease inhibitor cocktail (Calbiochem, CA., USA). After homogenization, the lysates were centrifuged at 15,000×g for 30 min at 4 °C; the supernatant was stored at − 80 °C. Protein samples (30 μg) were run on 8–12% sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis and then transferred onto a nitrocellulose membrane (Bio-Rad, Hercules, CA, USA) at 80 V for 120 min. Blots were probed with a monoclonal antibody against GFAP, ED-1, IL-1β, iNOS, heme oxygenase-1 (HO-1), α-synuclein, and cleaved caspase 1 at room temperature for 2 h. The immunoreaction was visualized by Amersham-enhanced chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ, USA). After this detection, the bound primary and secondary antibodies were stripped by incubating the membrane in stripping buffer (100 mM 2-mercaptoethanol, 2% SDS) at 50 °C for 45 min. The membrane was reprobed with a mouse β-actin antibody. The densities of blots were analyzed using a scanning densitometer which was operated by Scanner Control software (Molecular Dynamics, Sunnyvale, CA, USA). Results were obtained by calculating the density using Imagequant software (American Biosciences, Pittsburgh, PA, USA) and reported as relative optical density of the specific proteins.
Immunofluorescence Staining
Eight days after intranigral infusion of LPS, rats were deeply anesthetized and perfused transcardially using chilled 4% paraformaldehyde in 0.1 M PBS. Brains were dissected and placed in 30% sucrose PBS until sunken. Afterwards, the brain was coronally sectioned at 30-μm thickness using a cryostat (Leica CM 1950, Wetzlar, Germany). Brain sections were washed with 0.1 M PBS, incubated with 0.3% Triton X-100 and 1% goat serum (GS; Sigma), and blocked with 3% GS for 60 min. Then, brain sections were incubated overnight at 4 °C with primary antibodies specific for tyrosine hydroxylase and α-synuclein. Afterwards, brain sections were incubated at room temperature for 1 h with secondary antibodies conjugated with rhodamine and fluorescein isothiocyanate (Millipore). Nuclei were labeled with 4′, 6-diamidino-2-phenylindole (1 mg/mL, DAPI, Millipore) for 10 min at room temperature. Brain sections were mounted in glycerol and visualized by confocal microscopy (Olympus FV1000, Tokyo, Japan).
Cultured Cells
The immortalized BV-2 microglial cells (a gift from Dr. Young-Ji Shiao) is commonly used as an alternative model for primary cultured microglia. Dulbecco’s modified Eagle’s medium (DMEM) containing fetal bovine serum (10%), l-glutamine (1%), and penicillin-streptomycin (1%) was used to culture BV-2 cells at 37 °C under a humidified atmosphere with 5% CO2.
Cytotoxicity Assay
Cytotoxicity was determined by measuring the levels of lactate dehydrogenase (LDH) in the culture medium. Cells were seeded on a 24-well plate and treated with GA for 24 h. The LDH leaked in the culture medium was assessed by addition of reduced β-nicotinamide adenine dinucleotide and sodium pyruvate. The LDH activity was determined by measuring the absorbance at 340 nm for 6 min using an ELISA reader (TECAN, Sunrise, Männedorf, Schweiz). A positive control (100%) for normalization was done by treating BV-2 cells with 0.1% Triton X-100.
Quantitative RT-PCR Analysis
At the end of the experiment, total RNA was extracted from BV-2 cells using the Direct-zol RNA MicroPrep (Zymo Research, CA, USA). According to the manufacturer’s instruction, the cells were lysed by TRIzol reagent and then were centrifuged in 12,000×g for 1 min at 4 °C. Afterwards, the supernatant was incubated with DNase I at room temperature for 15 min. The mixture then was transferred into Zymo-Spin™ IC columns and centrifuged in 12,000×g for 1 min at 4 °C. RNA was washed with RNA wash buffer and eluted with 40 μL of RNase-free water. The PrimeScript RT Reagent Kit (Clontech, Mountain View, CA, USA) was used to reverse-transcribe total RNA (10 μg) to cDNA according to the manufacturer’s instructions. Real-time quantitative PCR was used to quantify the levels of gene expression. The PCR reaction was performed in StepOne™ Real-Time PCR system (Life Technologies, Carlsbad, CA, USA) using KAPA SYBR FAST qPCR Kits (Kapa Biosystems, Wilmington, MA, USA). The primer sequences for iNOS were 5′-TCT TGG AGC GAG TTG TGG AT-3′ (forward) and 5′-GGG TGG TAA TGT CCA GGA AGT-3′ (reverse). The primer sequences for GAPDH (internal control) were 5′-GTG TTC CTA CCC CCA ATG TGT-3′ (forward) and 5′-AGG AGA CAA CCT GGT CCT CAG T-3′ (reverse). The reaction mixture was denatured at 95 °C for 3 min. The PCR condition was 95 °C for 3 s and 60 °C for 30 s each cycle. Gene expression levels were calculated by 2−ΔΔCt method.
NO Production Using the Griess Reaction
To measure NO production in the culture medium, the culture medium was mixed with an equal volume of the Griess reagent (1% sulfanilamide, 0.1% N-(1-naphthyl)ethylenediamine in 2.5% H2PO4). After a 15-min incubation at room temperature in the dark, the concentration of nitrite was determined by measuring the absorbance at 550 nm using an ELISA plate reader (TECAN Sunrise, Männedorf, Schweiz).
In vitro Studies Using Brain Homogenates
Rat cortex was dissected and homogenized in chilled Ringer’s solution (50 mg/mL). The homogenates were incubated at 0 °C for control levels or at 37 °C with iron (1 μM) for iron-induced lipid peroxidation (LP). After a 3-h incubation, the effects of GA, trolox, and glutathione on iron-induced LP were measured using a spectrofluorometer (Hitachi F-4500, Hitachi High-Technologies Corporation, Tokyo, Japan). In brief, brain homogenates (400 μL) were transferred to an Eppendorf tube with chloroform (300 μL) and methanol (100 μL), mixed and kept on ice for 15 min. After centrifugation (8000g for 5 min), an aliquot of chloroform extract (200 μL) was transferred to another tube containing methanol (100 μL). LP was determined by measuring the levels of malondialdehyde and its dihydropyridine polymers, which emit fluorescence at 426 nm when activated by UV at 356 nm. The LP in relative fluorescence units was reported as % of control.
Statistics
Statistical comparisons were made as indicated using unpaired Student’s t test.
Results
GA Inhibited LPS-Induced Neuroinflammation and Oxidative Stress In vivo
To investigate the protection of GA against neuroinflammation, a neuroinflammation animal model was established using an intranigral infusion of LPS (4 μg/μL) in the rat brain. Eight days after intranigral infusion of LPS, significant elevation in GFAP (a biomarker of astrocyte activation) and ED-1 (a biomarker of microglia activation) was detected in the LPS-infused SN (Fig. 1a). Furthermore, LPS increased the levels of IL-1β (a proinflammatory cytokine) (Fig. 1b) and iNOS (a proinflammatory enzyme) (Fig. 1c). To study the neuroprotective effect of GA, oral administration of GA (50 mg/kg and 100 mg/kg) was performed 1 h prior to intranigral infusion of LPS and daily for 7 days. While GA did not alter the body weight of the studied animals (Fig. 3d), GA (50 mg/kg) attenuated LPS-induced increases in ED-1 and iNOS but not in GFAP and IL-1β in the infused SN. Furthermore, GA (100 mg/kg) significantly inhibited LPS-induced elevation in GFAP, ED-1, IL-1β, and iNOS (Fig. 1), suggesting that GA is capable of inhibiting LPS-induced glial cell activation and neuroinflammation.

Effects of GA on LPS-induced neuroinflammation in rat brain. LPS (4 μg/μL) was locally infused in the SN of anesthetized rats. GA (50, 100 mg/kg) was orally administered 1 h prior to the LPS infusion and daily afterwards for 7 days. At the end of the study, GFAP and ED-1 (a), IL-1β (b), and iNOS (c) levels in the LPS-infused SN were evaluated using Western blot assay. Graphs show statistical results from relative optical density of bands on the blots estimated by Imagequant software. Values are mean ± S.E.M. (n = 8/group for GFAP and ED-1; n = 4/group for IL-1β; n = 3/group for iNOS). *p < 0.05 in the LPS-infused SN compared with the intact SN in vehicle-treated rat; #p < 0.05 in the LPS-infused SN in the GA-treated rats compared with that in the vehicle-treated rats by t test. d The effect of GA on body weight of studied animals. Values are mean ± S.E.M. (n = 7/group)
At the same time, intranigral infusion of LPS significantly increased HO-1 expression (a redox-regulated chaperone protein) (Fig. 2a). Moreover, LPS slightly reduced α-synuclein monomer (17 kDa) and significantly increased α-synuclein aggregates (51 kDa, a pathological biomarker of PD) (Fig. 2b). Furthermore, our immunostaining data showed co-localized immunoreactivities of α-synuclein and tyrosine hydroxylase (a biomarker of dopaminergic neurons), suggesting the existence of α-synuclein in the nigral dopaminergic neurons (Fig. 2b). Consistently, systemic administration of GA attenuated LPS-induced increases in HO-1 and the α-synuclein aggregates (Fig. 2), indicating that GA is capable of reducing LPS-induced oxidative stress and protein aggregation.

Effects of GA on LPS-induced oxidative stress and protein aggregation in rat brain. LPS (4 μg/μL) was locally infused in the SN of anesthetized rats. GA (50, 100 mg/kg) was orally administered 1 h prior to the LPS infusion and daily afterwards for 7 days. At the end of the study, HO-1 expression levels (a) in the LPS-infused SN were evaluated using Western blot assay. b Western blot assay and immunostaining assay were used to measure α-synuclein levels as well as localization of α-synuclein and tyrosine hydroxylase in the LPS-infused SN, respectively. Graphs show statistical results from relative optical density of bands on the blots estimated by Imagequant software. Values are mean ± S.E.M. (n = 8/group). *p < 0.05 in the LPS-infused SN compared with the intact SN in vehicle-treated rat; #p < 0.05 in the LPS-infused SN in the GA-treated rats compared with that in the vehicle-treated rats by t test
GA Attenuated LPS-Induced Neurotoxicity In vivo
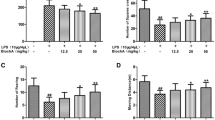
The neurotoxic mechanisms underlying the GA-induced neuroprotection of LPS-induced neurotoxicity were investigated by measuring cleaved caspase 3 (a biomarker of apoptosis) and RIPK-1 and RIPK-3 (two biomarkers of necroptosis). Intranigral infusion of LPS induced elevation in cleaved caspase 3 (Fig. 3a) as well as in RIPK-1 and RIPK-3 (Fig. 3b, c) in the infused SN. Compared with the saline-treated rats, systemic administration of GA significantly attenuated LPS-induced increases in cleaved caspase 3, RIPK-1, and RIPK-3 (Fig. 3a–c), suggesting that GA is capable of inhibiting LPS-induced apoptosis and necroptosis in the nigrostriatal dopaminergic system of rat brain.

Effects of GA on LPS-induced apoptosis and necroptosis in rat brain. LPS (4 μg/μL) was locally infused in the SN of anesthetized rats. GA (50, 100 mg/kg) was orally administered 1 h prior to the LPS infusion and daily afterwards for 7 days. At the end of the study, cleaved caspase 3 (cCas.3) (a), RIPK-1 (b), and RIPK-3 (c) levels in the LPS-infused SN were evaluated using Western blot assay. Graphs show statistical results from relative optical density of bands on the blots estimated by Imagequant software. Values are mean ± S.E.M. (n = 10/group for cCas.3; n = 8/group for RIPK-1 and RIPK-3). *p < 0.05 in the LPS-infused SN compared with the intact SN in vehicle-treated rat; #p < 0.05 in the LPS-infused SN in the GA-treated rats compared with that in the vehicle-treated rats by t test
GA Blocked LPS-Induced Neuroinflammation in BV-2 Cells
The involvement of neuroinflammation in GA-induced neuroprotection was further investigated using LPS-treated BV-2 cells. First, LDH assay was employed to investigate the potential cytotoxic effect of GA on BV-2 cells. Compared with the control group, GA (10–100 μM) was not toxic to BV-2 cells (Fig. 4a). The non-toxic concentrations of GA were employed to investigate the anti-inflammatory effect of GA. Incubation of LPS (1 μg/μL) for 24 h significantly elevated iNOS mRNA (Fig. 4b). Furthermore, LPS increased iNOS expression in the treated BV-2 cells (Fig. 4c) and NO level in the culture medium (Fig. 4d) using Western blot assay and Griess assay, respectively. Co-incubation of GA (25 μM, 50 μM) significantly attenuated LPS-induced elevation in iNOS mRNA and iNOS expression. Moreover, GA (25–100 μM) concentration-dependently reduced LPS-induced NO levels in the culture medium (Fig. 4) while GA (100 μM) alone did not affect NO production (Fig. 4d). These data indicate that GA is capable of inhibiting LPS-induced neuroinflammation in microglia.

Effects of GA on LPS-induced neuroinflammation in BV-2 cells. a BV-2 cells were incubated with GA (10–100 μM) for 24 h. LDH assay was employed for the cytotoxic effect of GA. P, a positive control by 0.1% Triton X-100. b, c BV-2 cells were incubated with LPS (1 μg/mL) plus GA (25–50 μM) for 24 h. RT-PCR and Western blot assay were employed for measuring iNOS mRNA and iNOS expression, respectively. Graphs show statistical results from iNOS mRNA and iNOS protein expression. Values are mean ± S.E.M. (n = 3/group). *p < 0.05 in the LPS-treated BV-2 cells compared with vehicle-treated BV-2 cells; #p < 0.05 in the LPS plus GA-treated BV-2 cells compared with that in LPS-treated BV-2 cells by t test. d BV-2 cells were incubated with LPS (1 μg/mL) plus GA (25–100 μM) for 24 h. The levels of NO in culture medium were measured using the Griess reaction. Values are mean ± S.E.M. (n = 3/group). *p < 0.05 in the LPS-treated BV-2 cells compared with vehicle-treated BV-2 cells; #p < 0.05 in the LPS plus GA-treated BV-2 cells compared with that in the LPS-treated BV-2 cells by t test
GA Inhibited Iron-Induced Lipid Peroxidation Using Cortical Homogenates
To study the anti-oxidative activity of GA, brain homogenates were used by incubating at 0 °C (as basal control). After a 3-h incubation at 37 °C, iron significantly increased the formation of peroxidized lipids compared with that at 0 °C (Fig. 5). Co-incubation with GA concentration-dependently attenuated iron-induced lipid peroxidation. The anti-oxidative activity of GA was compared with that of glutathione and trolox (a water-soluble analog of vitamin E). The anti-oxidative efficacy of GA was more potent than that of glutathione and less potent than that of trolox (Fig. 5).

Anti-oxidative activity of gallic acid (GA) on iron-induced lipid peroxidation. Iron-induced lipid peroxidation (LP) was performed using brain homogenates. Co-incubation of ferrous citrate (1 μM) with GA, trolox, or glutathione (GSH) at the indicated concentration at 37 °C for 3 h. The brain LP in relative fluorescence units was reported as % of control (without drug treatment). Values are mean ± S.E.M. (n = 3/group) from a representative experiment that was replicated with similar results
Discussion
In the present study, the role of neuroinflammation in the GA-induced neuroprotection was revealed using intranigral infusion of LPS in rat brain, a neuroinflammation animal model of PD. Consistent with the previous studies [2], our in vivo data demonstrated significant elevation in GFAP, ED-1, IL-1β, and iNOS in the LPS-infused SN of rat brain. Furthermore, our in vitro study showed LPS-induced increases in iNOS mRNA and iNOS protein levels in the treated BV-2 cells as well as NO production in the culture medium. These data demonstrate an LPS-induced neuroinflammation in the present study. At the same time, we found that LPS increased HO-1 protein expression (a redox-regulated chaperone protein), confirming LPS-elevated oxidative stress during neuroinflammation [2]. A pathological role of oxidative stress has been proposed for promoting protein aggregation [7, 18]. Indeed, Western blot assay showed LPS-induced production in α-synuclein trimer (51 kDa). Furthermore, immunostaining data suggested the existence of α-synuclein in dopaminergic neurons in the LPS-infused SN. Moreover, α-synuclein aggregation has been suggested as a proinflammatory factor to sensitize TLR4 in activated glial cells [18], to activate inflammasome formation [19] and to upregulate mRNA of proinflammatory cytokines [20]. Collectively, these events form a vicious cycle of neuroinflammation which in turn causes neuronal death by apoptosis and necroptosis [21].
Compared with the previous studies of GA at 100 and 200 mg/kg [22], we investigated the anti-inflammatory effect of GA at 50 and 100 mg/kg which showed no major deleterious effect on the body weight of studied animals, indicating the safety of GA. Furthermore, the present study showed dose-dependence in the GA-induced anti-inflammatory effects as follows. Low-dose GA (50 mg/kg) either had no effect or mildly inhibited LPS-induced neuroinflammation. In contrast, higher-dose GA (100 mg/kg) significantly and consistently attenuated LPS-induced neuroinflammation. Furthermore, GA prevented LPS-induced apoptosis and necroptosis in the infused SN. Due to the complexity of brain cells in the in vivo study, oral administration of GA would be expected to affect neurons and glial cells as well. The significance of neuroinflammation on activated microglia has been suggested for the necroptosis of neurons [23]. Therefore, the GA-induced attenuation of LPS-induced neuroinflammation in BV-2 microglial cells may be responsible for inhibiting neuronal necroptosis and apoptosis in the infused SN of rat brain.
Anti-oxidative activity has been reported as one major mechanism underlying GA-induced neuroprotection in cerebral damage [24] and traumatic brain injury [25]. Indeed, due to its structure with phenolic hydroxyl groups, GA may exert its protective effect via scavenging free radicals. To support this notion, GA concentration-dependently inhibited LPS-induced NO production. In contrast, we observed that 25 μM GA enhanced LPS-induced increases in HO-1 levels in the treated BV-2 cells (supplemental data). Low-dose GA potentiated LPS-induced HO-1expression in the infused SN in a few cases. One possible explanation for this discrepancy may be due to a pro-oxidant property of low-concentration GA [26]. Low GA reportedly reduced ferric iron to ferrous iron and generated hydroxyl radicals [26]. This hypothesis can support the pro-apoptotic effect of GA (25–50 μM) on cancer cells [27, 28].
In conclusion, the present study showed the involvement of inflammation in GA-induced neuroprotection in vivo and in vitro. Our data showed that oral administration of GA is capable of inhibiting LPS neuroinflammation (neuroglial activation, proinflammatory cytokines and enzyme production, α-synuclein aggregation) and programmed cell death of the nigrostriatal dopaminergic system of rat brain. In vitro data from BV-2 cells support the role of neuroinflammation in GA-induced neuroprotection in rat brain. These data indicate that GA may exert its neuroprotective action via inhibiting neuroinflammation in CNS neurodegenerative diseases.
References
Manoharan S, Guillemin GJ (2016) The role of reactive oxygen species in the pathogenesis of Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease: a mini review. 2016:8590578. https://doi.org/10.1155/2016/8590578
Flores-Martinez YM, Fernandez-Parrilla MA (2018) Acute neuroinflammatory response in the substantia nigra pars compacta of rats after a local injection of lipopolysaccharide. 2018:1838921. https://doi.org/10.1155/2018/1838921
Reinert KR, Umphlet CD, Quattlebaum A, Boger HA (2014) Short-term effects of an endotoxin on substantia nigra dopamine neurons. Brain Res 1557:164–170. https://doi.org/10.1016/j.brainres.2014.02.005
Rutherford NJ, Sacino AN, Brooks M, Ceballos-Diaz C, Ladd TB, Howard JK, Golde TE, Giasson BI (2015) Studies of lipopolysaccharide effects on the induction of alpha-synuclein pathology by exogenous fibrils in transgenic mice. Mol Neurodegener 10:32. https://doi.org/10.1186/s13024-015-0029-4
Yang QQ, Zhou JW (2018) Neuroinflammation in the central nervous system: symphony of glial cells. https://doi.org/10.1002/glia.23571
Lin AM, Fang SF, Chao PL, Yang CH (2007) Melatonin attenuates arsenite-induced apoptosis in rat brain: involvement of mitochondrial and endoplasmic reticulum pathways and aggregation of alpha-synuclein. J Pineal Res 43(2):163–171. https://doi.org/10.1111/j.1600-079X.2007.00456.x
Wang YT, Lin HC, Zhao WZ, Huang HJ, Lo YL, Wang HT, Lin AM (2017) Acrolein acts as a neurotoxin in the nigrostriatal dopaminergic system of rat: involvement of alpha-synuclein aggregation and programmed cell death. Sci Rep 7:45741. https://doi.org/10.1038/srep45741
Hung KC, Huang HJ, Wang YT, Lin AM (2016) Baicalein attenuates alpha-synuclein aggregation, inflammasome activation and autophagy in the MPP(+)-treated nigrostriatal dopaminergic system in vivo. J Ethnopharmacol 194:522–529. https://doi.org/10.1016/j.jep.2016.10.040
Fu Y, Yang J, Wang X, Yang P, Zhao Y, Li K, Chen Y (2018) Herbal compounds play a role in neuroprotection through the inhibition of microglial activation. 2018:9348046. https://doi.org/10.1155/2018/9348046
Jiang X, Ganesan P, Rengarajan T, Choi DK, Arulselvan P (2018) Cellular phenotypes as inflammatory mediators in Parkinson’s disease: interventional targets and role of natural products. Biomed Pharmacother 106:1052–1062. https://doi.org/10.1016/j.biopha.2018.06.162
Wasik A, Antkiewicz-Michaluk L (2017) The mechanism of neuroprotective action of natural compounds. Pharmacol Rep 69(5):851–860. https://doi.org/10.1016/j.pharep.2017.03.018
Daglia M, Di Lorenzo A, Nabavi SF, Talas ZS, Nabavi SM (2014) Polyphenols: well beyond the antioxidant capacity: gallic acid and related compounds as neuroprotective agents: you are what you eat! Curr Pharm Biotechnol 15(4):362–372
Rai SN, Birla H, Singh SS, Zahra W, Patil RR, Jadhav JP, Gedda MR, Singh SP (2017) Mucuna pruriens protects against MPTP intoxicated neuroinflammation in Parkinson’s disease through NF-kappaB/pAKT signaling pathways. Front Aging Neurosci 9:421. https://doi.org/10.3389/fnagi.2017.00421
Chandrasekhar Y, Phani Kumar G, Ramya EM, Anilakumar KR (2018) Gallic acid protects 6-OHDA induced neurotoxicity by attenuating oxidative stress in human dopaminergic cell line. Neurochem Res 43(6):1150–1160. https://doi.org/10.1007/s11064-018-2530-y
Mansouri MT, Farbood Y, Sameri MJ, Sarkaki A, Naghizadeh B, Rafeirad M (2013) Neuroprotective effects of oral gallic acid against oxidative stress induced by 6-hydroxydopamine in rats. Food Chem 138(2–3):1028–1033. https://doi.org/10.1016/j.foodchem.2012.11.022
Lively S, Schlichter LC (2018) Microglia responses to pro-inflammatory stimuli (LPS, IFNgamma+TNFalpha) and reprogramming by resolving cytokines (IL-4, IL-10). Front Cell Neurosci 12:215. https://doi.org/10.3389/fncel.2018.00215
Cazareth J, Guyon A, Heurteaux C, Chabry J, Petit-Paitel A (2014) Molecular and cellular neuroinflammatory status of mouse brain after systemic lipopolysaccharide challenge: importance of CCR2/CCL2 signaling. J Neuroinflammation 11:132. https://doi.org/10.1186/1742-2094-11-132
Hughes CD, Choi ML, Ryten M, Hopkins L, Drews A, Botia JA, Iljina M, Rodrigues M et al (2018) Picomolar concentrations of oligomeric alpha-synuclein sensitizes TLR4 to play an initiating role in Parkinson’s disease pathogenesis. doi:https://doi.org/10.1007/s00401-018-1907-y
Codolo G, Plotegher N, Pozzobon T, Brucale M, Tessari I, Bubacco L, de Bernard M (2013) Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS One 8(1):e55375. https://doi.org/10.1371/journal.pone.0055375
Couch Y, Alvarez-Erviti L, Sibson NR, Wood MJ, Anthony DC (2011) The acute inflammatory response to intranigral alpha-synuclein differs significantly from intranigral lipopolysaccharide and is exacerbated by peripheral inflammation. J Neuroinflammation 8:166. https://doi.org/10.1186/1742-2094-8-166
Fricker M, Vilalta A, Tolkovsky AM, Brown GC (2013) Caspase inhibitors protect neurons by enabling selective necroptosis of inflamed microglia. J Biol Chem 288(13):9145–9152. https://doi.org/10.1074/jbc.M112.427880
Maya S, Prakash T, Goli D (2018) Evaluation of neuroprotective effects of wedelolactone and gallic acid on aluminium-induced neurodegeneration: relevance to sporadic amyotrophic lateral sclerosis. Eur J Pharmacol 835:41–51. https://doi.org/10.1016/j.ejphar.2018.07.058
Qin S, Yang C, Huang W, Du S, Mai H, Xiao J, Lu T (2018) Sulforaphane attenuates microglia-mediated neuronal necroptosis through down-regulation of MAPK/NF-kappaB signaling pathways in LPS-activated BV-2 microglia. Pharmacol Res 133:218–235. https://doi.org/10.1016/j.phrs.2018.01.014
Sun J, Li YZ, Ding YH, Wang J, Geng J, Yang H, Ren J, Tang JY et al (2014) Neuroprotective effects of gallic acid against hypoxia/reoxygenation-induced mitochondrial dysfunctions in vitro and cerebral ischemia/reperfusion injury in vivo. Brain Res 1589:126–139. https://doi.org/10.1016/j.brainres.2014.09.039
Sarkaki A, Farbood Y, Gharib-Naseri MK, Badavi M, Mansouri MT, Haghparast A, Mirshekar MA (2015) Gallic acid improved behavior, brain electrophysiology, and inflammation in a rat model of traumatic brain injury. Can J Physiol Pharmacol 93(8):687–694. https://doi.org/10.1139/cjpp-2014-0546
Strlic M, Radovic T, Kolar J, Pihlar B (2002) Anti- and prooxidative properties of gallic acid in fenton-type systems. J Agric Food Chem 50(22):6313–6317
Moghtaderi H, Sepehri H, Delphi L, Attari F (2018) Gallic acid and curcumin induce cytotoxicity and apoptosis in human breast cancer cell MDA-MB-231. BioImpacts : BI 8 (3):185-194. Doi:https://doi.org/10.15171/bi.2018.21
Lee HL, Lin CS, Kao SH, Chou MC (2017) Gallic acid induces G1 phase arrest and apoptosis of triple-negative breast cancer cell MDA-MB-231 via p38 mitogen-activated protein kinase/p21/p27 axis. Anti-Cancer Drugs 28(10):1150–1156. https://doi.org/10.1097/cad.0000000000000565
Acknowledgments
The authors express their gratitude to Dr. C.Y. Chai at the Institute of Biomedical Sciences, Academia Sinica, for his encouragement and support. Special thanks are due to Dr. R.K. Freund at the Department of Pharmacology, University of Colorado, Anschutz, CO, USA, for editing this paper.
Funding
This study was supported by MOST107-2320-B-010-019-MY3 and 107DN08, Taipei, Taiwan, R.O.C.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The use of animals has been approved by the Institutional Animal Care and Use Committee of Taipei Veterans General Hospital, Taipei, Taiwan, R.O.C.. The approval number is IACUC2018. All experiments were performed in the accordance with relevant guidelines and regulation.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplemental data
Effects of GA on LPS-induced HO-1 in BV-2 cells. (A) BV-2 cells were incubated with LPS (1 μg/mL) plus GA (25-50 μM) for 24 h. Western blot assay was employed for measuring HO-1 expression. Graphs show statistical results from HO-1 protein expression. Values are the mean ± S.E.M. (n = 3/group). *, p < 0.05 in the LPS-treated BV-2 cells compared with vehicle-treated BV-2 cells; #, p < 0.05 in the LPS plus GA-treated BV-2 cells compared with LPS-treated BV-2 cells by t-test. (PNG 199 kb)
Rights and permissions
About this article

Cite this article
Liu, YL., Hsu, CC., Huang, HJ. et al. Gallic Acid Attenuated LPS-Induced Neuroinflammation: Protein Aggregation and Necroptosis. Mol Neurobiol 57, 96–104 (2020). https://doi.org/10.1007/s12035-019-01759-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-019-01759-7