
Abstract
Neuronal apoptosis is a common and critical pathology following subarachnoid hemorrhage (SAH). We investigated the anti-apoptotic property of fibroblast growth factor (FGF)-2 after SAH in rats. A total of 289 rats underwent endovascular perforation to induce SAH or sham operation. Three dosages (3, 9, or 27 μg) of recombinant FGF-2 (rFGF-2) or vehicle was administered intranasally to rats 30 min after SAH induction. The pan-FGF receptor (FGFR) inhibitor PD173074 or vehicle was administered intracerebroventricularly (i.c.v.) 1 h before modeling, in addition to rFGF-2 treatment. Small interfering ribonucleic acid (siRNA) for FGFR1 and FGFR3 or scrambled siRNA was administered i.c.v. 48 h before SAH induction in addition to rFGF-2 treatment. Anti-FGF-2 neutralizing antibody or normal mouse immunoglobulin G (IgG) was administered i.c.v. 1 h before SAH model. Neurobehavioral tests, SAH severity, brain water content, immunofluorescence, Fluoro-Jade C, TUNEL staining, and western blot were evaluated. The expression of FGF-2, FGFR1, and FGFR3 increased after SAH. FGFR1 and FGFR3 were expressed in the neurons. Nine micrograms of FGF-2 alleviated neurological impairments, brain edema, and neuronal apoptosis following SAH. A rFGF-2 treatment improved motor skill learning and spatial memory and increased the number of surviving neurons postinjury to 28 days after SAH. PD173074 abolished the anti-apoptotic effects of rFGF-2 via suppression of the expression of PI3k, phosphorylated Akt (p-Akt), and Bcl-2 leading to enhancement of the expression of Bax. FGFR3 siRNA worsened neurobehavioral function and suppressed the expression of PI3k, p-Akt, and Bcl-2 rather than FGFR1 siRNA in SAH rats treated with rFGF-2. Anti-FGF-2 neutralizing antibody suppressed the expression of PI3k and p-Akt after SAH. FGF-2 may be a promising therapy to reduce post-SAH neuronal apoptosis via activation of the FGFR3/PI3k/Akt signaling pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Subarachnoid hemorrhage (SAH) is a cerebrovascular disease with devastating consequences. Early brain injury (EBI) is a type of acute pathophysiological change that occurs in the brain within the first 72 h following SAH and has been recognized as the primary factor for morbidity and mortality after SAH [1]. Neuronal apoptosis is a common and critical pathology of EBI, which causes cerebral cortex damage and exacerbates neurobehavioral impairments [2]. Neuronal apoptosis is a possible therapeutic target after SAH. Previous studies suggest that the PI3k/Akt signaling pathway plays an essential regulatory role in apoptosis after SAH. Phosphorylated Akt (p-Akt) enhances cell survival and inhibits neuronal apoptosis by inactivating its downstream targets [3].
Fibroblast growth factor (FGF)-2 is a member of the fibroblast growth factor family and is expressed much higher in the central nervous system (CNS) [4]. FGF-2 regulates a variety of cell functions including proliferation, morphogenesis, and suppression of apoptosis [5]. FGF-2 is secreted from damaged neurons, although the function of endogenous FGF-2 has never been investigated after SAH. In contrast, previous studies have demonstrated that exogenous FGF-2 exerts neuroprotective effects after trauma, intracerebral hemorrhage, and ischemic injury via activation of the PI3k/Akt pathway [5,6,7,8]. However, previous studies have not demonstrated post-SAH neuroprotective effects of exogenous FGF-2 as well as endogenous FGF-2. The FGF receptor (FGFR) is composed of five subtypes: FGF-2 activates FGFR1, FGFR2, and FGFR3 and has high affinities with FGFR1 and FGFR3 [9, 10], which are expressed diffusely throughout the brain after traumatic injury, while FGFR2 is localized primarily to the white matter in the cerebral hemispheres and cerebellum [9]. However, previous studies have never been able to confirm which FGFR subtype was responsible for neuroprotective effects of FGF-2 after CNS injury in vivo.
FGF-2 may prevent EBI including anti-neuronal apoptosis via activation of the FGFR/PI3k/Akt pathway. Thus, this study aimed to thoroughly investigate whether FGF-2 attenuates neuronal apoptosis via activation of the FGFR/PI3k/Akt signaling pathway after SAH.
Materials and Methods
The Institutional Animal Care and Use Committee (IACUC) of Loma Linda University approved the study protocol, and all experiments were conducted in accordance with the NIH Guidelines for the Use of Animals in Neuroscience Research. All authors have read and approved the submitted manuscript.
Animal Model and Study Protocol
Ten-week-old Sprague–Dawley male rats (weight, 300–320 g) were used to produce SAH models by endovascular perforation as previously described [11]. Briefly, each rat was placed under deep anesthesia (4.5% for 2 min) and maintained by 3% isoflurane for the duration of the surgery. After exposing the left common, external, and internal carotid arteries, a sharpened 4-0 monofilament nylon suture was advanced rostrally into the left internal carotid artery from the external carotid artery stump to perforate the bifurcation of the anterior and the middle cerebral arteries. We monitored peak inspiration pressure and respiratory rate by a pressure-controlled ventilator, heart rate, skin pigmentation, and pedal reflex (firm toe pinch) during operation. We monitored these vitals every 5 min to ensure that the animal(s) were not in distress and were responding to the anesthesia and procedure accordingly. Body temperature was kept constant 37 °C by a heating pad until the animals recovered. Sham-operated rats underwent identical procedures except that the suture was withdrawn without puncturing an artery. All rats were randomly assigned to the following experiments as described (Additional File 1: Fig. S1).
Experiment 1
To determine the time course and cellular localization of endogenous FGF-2, FGFR1, and FGFR3 expression, 44 rats were randomly divided and assigned to six groups: sham and SAH 3, 6, 12, 24, and 72 h groups. After mortality and SAH severity were evaluated, western blot (WB) in the left hemisphere sample was analyzed (n = 6 per group). Also, to assess the colocalization of FGFR1 and FGFR3 expression in neuron, double immunohistochemistry staining of FGFR1 and FGFR3 in conjunction with neurons was performed in the sham and SAH groups at 24 h after modeling in accordance with further experiments (n = 4 per group).
Experiment 2.1
To evaluate short-term effects of recombinant rat FGF-2 (rFGF-2) on post-SAH neuronal apoptosis, 42 rats were randomly divided and assigned to five groups: sham+phosphate-buffered saline (PBS; n = 10), SAH+PBS (n = 10), SAH+rFGF-2 3 μg (n = 6), SAH+rFGF-2 9 μg (n = 10), and SAH+rFGF-2 27 μg (n = 6) groups. PBS or rFGF-2 (4039, BioVision Inc., Milpitas, CA) was administered intranasally 30 min after modeling. Mortality, neurological scores, SAH severity, and brain water content were evaluated at 24 h after modeling (n = 6 per group). Based on the results of neurological scores and brain water content, 9 μg of rFGF-2 was used in the staining and mechanistic studies. Sham+PBS, SAH+PBS, and SAH+rFGF-2 (n = 4 per group) were used for Fluoro-Jade C (FJC) staining for neuronal degeneration and apoptotic marker, cleaved caspase-3 staining to evaluate neuronal apoptosis in the left temporal cortex 24 h after modeling.
Experiment 2.2
To evaluate the long-term effects of rFGF-2 on post-SAH neuronal loss, 24 rats were randomly divided and assigned into three groups: sham, SAH+PBS, and SAH+rFGF-2 (9 μg) groups (n = 8 per group). PBS and rFGF-2 were administered intranasally 30 min after SAH induction. Following SAH induction, mortality was evaluated, and rotarod test was performed on day 7, day 14, and day 21 after modeling. In addition, the Morris water maze was performed on days 22–27 after modeling. The rats were euthanized on day 28 and Nissl staining was performed to determine the neuronal loss of the hippocampal region.
Experiment 3.1
To explore the mechanism of post-SAH neuroprotective function following rFGF-2 administration, 46 rats were randomly divided and assigned to five groups: sham+PBS+dimethyl sulfoxide (DMSO; n = 6), sham+rFGF-2 (9 μg)+PD173074 (80 μg; n = 10), SAH+PBS+DMSO (n = 10), SAH+rFGF-2+DMSO (n = 10), and SAH+rFGF-2+PD173074 (n = 10) groups. The pan-FGFR inhibitor PD173074 (P2499, Sigma-Aldrich Inc., St Louis, MO) and DMSO were administered i.c.v. 1 h before modeling, while rFGF-2 and PBS were administered intranasally 30 min after modeling. After mortality, neurobehavioral scores, and SAH severity 24 h after modeling were evaluated, WB in the left hemisphere samples (n = 6 per group), terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), and cleaved caspase-3 staining (n = 4 per group) were performed.
Experiment 3.2
To determine the efficacy in each FGFR subtype, 36 rats were randomly divided and assigned to six groups: sham, SAH, SAH+rFGF-2 (9 μg), SAH+rFGF-2+scrambled small interfering ribonucleic acid (Scr siRNA; 500 pmol), SAH+rFGF-2+FGFR1 siRNA (500 pmol), and SAH+rFGF-2+FGFR3 siRNA (500 pmol) groups (n = 6 per group). Scr siRNA (SR30004, OriGene Technologies, Rockville, MD), FGFR1 siRNA (AM16708, Thermo Fisher Scientific Inc., Waltham, MA), and FGFR3 siRNA (AM16708, Thermo Fisher Scientific Inc., Waltham, MA) were administered i.c.v. 48 h before, while rFGF-2 was administered intranasally 30 min after SAH induction. After mortality, neurobehavioral scores, and SAH severity 24 h after modeling were evaluated, the expression of FGFR1 and FGFR3 and the downstream proteins in the left hemisphere samples were analyzed using WB (n = 6 per group).
Experiment 3.3
To investigate the effects of endogenous FGF-2 on post-SAH, 24 rats were randomly assigned to four groups: sham+normal mouse immunoglobulin G (IgG; 7.2 μg), sham+anti-FGF-2 neutralizing antibody (7.2 μg), SAH+normal mouse IgG, and SAH+anti-FGF-2 neutralizing antibody (n = 6 per group). Anti-FGF-2 neutralizing antibody (05-117, EMD Millipore Inc., Temecula, CA) and negative control normal mouse IgG (12-371, EMD Millipore Inc., Temecula, CA) were administered i.c.v. 1 h before SAH. Mortality, neurological scores, SAH severity, and WB in the left hemisphere sample were assessed 24 h after modeling.
SAH Severity and Exclusion Criteria
The severity of SAH was blindly assessed at each sacrifice as previously described [12]. Briefly, the basal cistern was divided into six segments, and each segment was allotted a grade from 0 to 3 depending on the amount of SAH. The rats received a total score ranging from 0 to 18 by adding the scores from all six segments. In the analysis, mild SAH (SAH grade ≤ 7) rats were excluded in the experiments except in experiment 2.2, because mild SAH did not induce neurological impairments in the previous study [12]. In experiment 2.2, no animals were excluded except mortal models.
Intranasal Administration
Intranasal administration 30 min after modeling was carried out as previously described [13]. Briefly, rats were placed in a supine position under 2% isoflurane anesthesia. A total volume of 27 μl PBS or rFGF-2 at three different dosages (3, 9, and 27 μg) dissolved in PBS was administered alternately into left and right nares, 4.5 μl in one nare every 5 min for a period of 30 min. The dosage of rFGF-2 was determined from a previous report that described 9 μg of rFGF-2 being administered intranasally to rats, and its concentration in the cerebrum, hippocampus, and cerebellum peaked at 30 min, ameliorating memory impairment [14]. Assuming that a different dosage of rFGF-2 should be needed to be effective for the brain, we administered 3 and 27 μg of rFGF-2 to have a set dosage window for this study.
Intracerebroventricular Infusion
An i.c.v. infusion was performed as previously described [15]. Briefly, rats were placed in a stereotaxic apparatus under 2–2.5% isoflurane anesthesia. The needle of a 10-μl Hamilton syringe (Hamilton Company Inc., Reno, NV) was inserted through a burr hole into the right lateral ventricle using the following coordinates relative to the bregma: 0.9 mm posterior, 1.5 mm lateral, and 3.3 mm below the horizontal plane of the bregma. Drug and siRNA were infused at a rate of 1 μl/min. The needle was removed at 5 min after i.c.v. infusion, and we quickly sealed the skull hole with bone wax, and then the incision site was sutured.
PD173074 was prepared at a concentration of 10 μg/μl in DMSO, and 8 μl of DMSO and PD173074 were infused at 1 h before SAH. Negative control Scr siRNA, FGFR1 siRNA, and FGFR3 siRNA were prepared at a concentration of 100 pmol/μl in RNase-free resuspension buffer. A total volume of 5 μl siRNA was injected 48 h before SAH induction. Negative control normal mouse IgG and anti-FGF-2 neutralizing antibody were prepared at a concentration of 1 μg/μl in buffer containing 0.1 μmol/μl Tris-glycine, 0.15 μmol/μl NaCl, 0.05% sodium azide, and 30% glycerol. A total volume of 7.2 μl normal mouse IgG and anti-FGF-2 neutralizing antibody were injected 1 h before modeling. The dosage of PD173074 was determined from a previous study [7]. In the study, 9.2–11.8 μg PD173074 was injected i.c.v. into mice (the volume was dependent on the body weight of each mouse in the said study) and reversed rFGF-2 treatment effect entirely. As the cerebrospinal fluid volume of rats is considered to be 4 to 9 times of that of mice [16, 17], we assumed that 80 μg PD173074 was enough to get an equivalent cerebrospinal fluid PD173074 concentration in rats. The dosage of siRNA was determined on the basis of our previous report [13]. The neutralizing antibody against FGF-2 was determined from a previous report [18]. In the study, the continuous i.c.v. infusion of anti-FGF-2 neutralizing antibody in a dose of 7.2 μg/day induced a significant decrease in hippocampal neurons in ischemic gerbils. Therefore, 7.2 μg anti-FGF-2 neutralizing antibody was tentatively selected for our study.
Neurological Scores
Neurological impairments were blindly evaluated using the modified Garcia’s neurological score and beam balance score system as previously described [19]. Briefly, the evaluation consisted of six categories (spontaneous activity; spontaneous movement of four limbs; forepaw outstretching; climbing; body proprioception; and response to whisker stimulation) that could be scored 0 to 3 or 1 to 3. Neurological score was determined by adding the six scores to get a final score between 2 (which is the worst) and 18 (which would be the highest score). The median score of 3 consecutive trials in a 5-min interval was calculated. A beam balance test investigated the animal’s ability to walk on a narrow wooden beam for 60 s, and we blindly marked a grade from 0 to 4 points as follows: 4 points, walking ≥ 20 cm; 3 points, walking < 20 cm; 2 points, walking but falling; 1 point, not walking while remaining on beam; and 0 points, falling without walking. The median score of 3 consecutive trials in a 5-min period was calculated.
Brain Water Content
Brain edema was determined using the wet/dry method as previously described [19]. Briefly, after sacrificing rats under deep anesthesia, the brain was quickly removed, separated into four segments (left and right cerebral hemispheres, cerebellum, and brain stem), and weighed immediately as wet weight. The brain specimens were dried in an oven at 105 °C for 72 h and weighed again as dry weight. The water content of each specimen was calculated according to the following formula: [(wet weight − dry weight) / wet weight] × 100%.
Rotarod Test
The rotarod (Columbus Instruments, Columbus, OH) was used to assess sensorimotor coordination and balance as previously described [20]. Briefly, the cylinder started at 5 and 10 revolutions per minute (rpm), respectively, accelerating by 2 rpm every 5 s. The duration that rats were able to stay on the accelerating rotating cylinder was recorded by a photo beam circuit.
Morris Water Maze Test
Water maze was used to evaluate spatial learning capacity and reference memory as previously described [21]. Briefly, the water maze test was performed on day 22 after SAH, which was used as a control to assess any sensorimotor and/or motivational deficits that could affect performance during the spatial water maze test. The spatial water maze test was performed on days 23–26 after SAH. On day 27, the animals were tested with a 60-s probe trial in which the platform was removed from the water. A video recording system traced the activities of the animals and the swim patterns were measured for quantification of distance, latency, and swimming speed by Video Tracking System SMART-2000 (San Diego Instruments Inc., San Diego, CA).
Immunofluorescence Staining
Under deep anesthesia, rats were sacrificed by transcardial perfusion with 100 ml PBS followed by 2 min of 10% neutral buffered formalin. Brains were fixed in 10% neutral buffered formalin for 24 h at 4 °C followed by 30% sucrose solution for another 72 h. After being frozen at − 80 °C, the brain was cut into 10-μm-thick coronal sections at 1.0 mm posterior to the bregma using a cryostat (LM3050S; Leica Microsystems, Bannockburn, III, Germany). Slides were washed with 0.01 M of PBS 3 times for 10 min then incubated in 0.3% Triton X-100 in 0.01 M of PBS for 30 min at room temperature. After being blocked with 10% donkey serum in 0.01 M of PBS for 1 h at room temperature, the sections were incubated at 4 °C overnight with primary antibody as follows: mouse anti-neuron-specific nuclear protein (NeuN; 1:400; ab104224), rabbit anti-NeuN (1:400; ab177487) (Abcam Inc., Cambridge, MA), anti-FGFR1 (1:200; 9740s, Cell Signaling Technology Inc., Danvers, MA), anti-FGFR3 (1:50; sc-390423, Santa Cruz Biotechnology Inc., Dallas, TX), and anti-cleaved caspase-3 antibody (1:400; #9661, Cell Signaling Technology Inc., Danvers, MA). Then, the sections were washed with 0.01 M of PBS and incubated with appropriate fluorescence-conjugated secondary antibodies (1:500; Jackson ImmunoResearch Inc., West Grove, PA) for 1 h at room temperature. The slides were observed and photographed under a fluorescence microscope (DMi8; Leica Microsystems Inc., Germany; × 20), and the average number of cleaved caspase-3-positive cells in the four continuous pictures of left temporal cortex as cells/mm2 was calculated by Image-Pro Plus 6.0 software (Media Cybernetics Inc., Rockville, MD).
Fluoro-Jade C Staining
Coronal brain sections were prepared in the same way as immunofluorescence staining. FJC staining was performed using Fluoro-Jade C Ready-to-Dilute Staining Kit (Biosensis Inc., Thebarton, SA, Australia) for identifying degenerating neurons according to the manufacturer’s instructions. Four continuous pictures of the left temporal cortex were photographed under a fluorescence microscope (× 20), and the average number of FJC-positive cells in the fields as cells/mm2 was calculated by Image-Pro Plus 6.0 software.
TUNEL Staining
Coronal brain sections were prepared in the same way as immunofluorescence staining. For quantification of neuronal apoptosis, double staining of neuron marker NeuN and TUNEL staining was performed using in situ cell death detection kit, TMR red according to the manufacturer’s instructions. Four continuous pictures of the left temporal cortex were photographed under a fluorescence microscope (× 20), and the average number of TUNEL-positive cells and NeuN-positive neurons in the fields as cells/mm2 was calculated by Image-Pro Plus 6.0 software. Data were presented as the ratio of TUNEL-positive neurons (%).
Nissl Staining
Coronal brain sections were prepared in the same way as immunofluorescence staining. Sections were immersed in 0.1% cresyl violet for 2 min, dehydrated in the Flex tissue specimen system (Richard-Allan Scientific Inc., San Diego, CA), cleared in xylenes, and coverslipped with Permount. Three continuous pictures of the left hippocampal cornu ammonis (CA) 1, CA3, and dentate gyrus (DG) were photographed under a light microscope (OLYMPUS BX51; Olympus Inc., Tokyo, Japan; × 20), and the average number of Nissl-positive cells was calculated by Image-Pro Plus 6.0 software. Data was expressed as the number of neurons per field.
Western Blot
The left cerebral hemisphere was separated and used. Equal amounts of protein samples (30 μg) were loaded on SDS-PAGE gels, electrophoresed, and transferred onto a polyvinylidene difluoride membrane. The membranes were blocked with 5% nonfat blocking grade milk for 1 h at room temperature followed by incubation overnight at 4 °C with the following antibodies: anti-FGF-2 (1:100; sc-74412), anti-FGFR3 (1:100; sc-390423), anti-β-actin (1:5000; sc-47778) (Santa Cruz Biotechnology Inc., Dallas, TX), anti-FGFR1 (1:1000; 9740s), anti-PI3k (1:1000; 4249s), anti-p-Akt (1:1000; 9271s), anti-Akt (1:1000; 9272s) (Cell Signaling Technology Inc., Danvers, MA), anti-phosphorylated-FGFR (p-FGFR; 1:1000; PA5-64626, Thermo Fisher Scientific Inc., Waltham, MA), anti-Bax (1:1000; ab182734), and anti-Bcl-2 antibody(1:2500; ab59348, Abcam Inc., Cambridge, UK). On the following day, the membranes were incubated with the appropriate secondary antibody (1:5000; sc-516102, Santa Cruz Biotechnology Inc., Dallas, TX) and (1:5000; 12-348, MilliporeSigma Inc., Temecula, CA) at room temperature for 2 h. Immunoreactive bands were detected with a chemiluminescence reagent kit (ECL Prime; Amersham Biosciences Inc., Arlington Heights, IL) and quantified by densitometry with ImageJ software (NIH, Bethesda, MD).
Statistical Analysis
Neurological score and SAH grade were expressed as median ± 25th–75th percentiles and were compared with Kruskal–Wallis tests followed by Steel–Dwass multiple comparisons. Brain water content, immunostaining, WB, rotarod test, and Morris water maze test results were evaluated for the normality of variable distribution and homogeneity of variances by Shapiro–Wilk test and Levene’s test, respectively, and were considered violated when this test yielded P < 0.05. The data were expressed as mean ± standard deviation and were compared with one-way analysis of variance (ANOVA) followed by Tukey–Kramer post hoc tests. Statistical analyses were performed using SPSS version 23.0 (IBM, Tokyo, Japan). A value of P < 0.05 was considered significant.
Availability of Data and Material
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Results
General Observation
Comparisons of physiological parameters revealed no significant differences among the groups (data not shown). The mortality was 0 of 62 (0%) in the sham-operated rats and it was 31 of 227 (14%) in the SAH-operated rats in total during the observation period in all experiments. A total of 42 rats were not used for the analyses because of mild (≤ 7) SAH grade (Additional File 1: Fig. S2a). There was no significant difference in SAH grade between all SAH groups at 24 h after SAH (Additional File 1: Fig. S2b, c). Brain water content, all immunostaining, WB, rotarod test, and Morris water maze test results were normally distributed and homogeneity of variance.
Time Course and Spatial Expression of FGF-2, FGFR1, and FGFR3 After SAH
WB results showed that expression of FGF-2 elevated 3 h after SAH induction and the elevation prolonged until 24 h when compared to the sham group (Fig. 1a). In contrast, there were significant elevations of the expression of FGFR1 in the period between 6 and 12 h and the expression of FGFR3 in the period between 6 and 24 h after SAH. Double immunofluorescence staining showed that FGFR1 and FGFR3 were expressed in neurons in the cerebral cortex in both sham and SAH groups 24 h after modeling (Fig. 1b).

Expression of endogenous fibroblast growth factor (FGF)-2, FGF receptor (FGFR) 1, and FGFR3. a Representative western blot images and densitometric quantification of time-dependent expression of FGF-2, FGFR1, and FGFR3 after subarachnoid hemorrhage (SAH). Expression levels of each protein are expressed as a ratio of β-actin levels for normalization and as mean ± standard deviation. *P < 0.05 and **P < 0.01 vs. the sham group (n = 6 per group). b Colocalization of FGFR1 and FGFR3 with neuron-specific nuclear protein (NeuN) in the left temporal cerebral cortex at 24 h after modeling. Nuclei are stained with DAPI. Scale bar = 20 μm (n = 4 per group)
FGF-2 Induction Improved Neurological Scores and Brain Edema via Attenuation of Neuronal Apoptosis
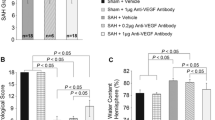
After 9 μg of rFGF-2 treatment, post-SAH aggravation of neurological scores was prevented during both modified Garcia’s and beam balance test significantly (Fig. 2a). In addition, brain water content in the left and right cerebral hemispheres 24 h post-SAH significantly improved in the 9-μg rFGF-2 treatment compared to the SAH+PBS group, while 3 and 27 μg of rFGF-2 administration improved brain water content only in the left cerebral hemisphere after SAH (Fig. 2b). Based on these findings, the following experiments were performed using 9 μg of rFGF-2 for treatment.

Effects of recombinant fibroblast growth factor-2 (rFGF-2) on short-term neurobehavioral function and brain edema. a Nine micrograms of rFGF-2 improved modified Garcia’s score and beam balance score at 24 h after subarachnoid hemorrhage (SAH). Data are expressed as median ± 25th–75th percentiles (n = 6–10 per group). †P < 0.01 vs. sham+PBS, ‡P < 0.05 and §P < 0.01 vs. SAH+phosphate-buffered saline (PBS). b rFGF-2 treatment improved brain water content at 24 h after SAH. Data are expressed as mean + standard deviation (n = 6 per group). *P < 0.05 and †P < 0.01 vs. sham+PBS, ‡P < 0.05 and §P < 0.01 vs. SAH+PBS
FJC staining for the severity of neuronal degeneration and cleaved caspase-3 for the expression of apoptotic marker were performed in the left cerebral cortex 24 h after SAH. SAH resulted in a significant increase in FJC-positive cells and cleaved caspase-3-positive cells 24 h post-SAH, which were significantly suppressed by rFGF-2 administration (Figs. 3 and 4).

Effects of recombinant fibroblast growth factor-2 (rFGF-2) on short-term neuronal degeneration after subarachnoid hemorrhage (SAH). a Representative images of Fluoro-Jade C (FJC)-positive cells in the left temporal cerebral cortex at 24 h after modeling. Scale bar = 50 μm. b Representative image demonstrating the location of staining (small black box). c Quantitative analysis of FJC-positive cells per mm2 in the left cerebral cortex at 24 h after modeling. Data are expressed as mean ± standard deviation (n = 4 per group). *P < 0.05, **P < 0.01. PBS, phosphate-buffered saline

Effects of recombinant fibroblast growth factor-2 (rFGF-2) on short-term apoptotic marker after subarachnoid hemorrhage (SAH). a Representative images demonstrating double immunofluorescent staining for cleaved caspase-3 and neuron-specific nuclear protein (NeuN) in the left temporal cerebral cortex at 24 h after modeling. Scale bar = 50 μm. b Representative image demonstrating the location of staining (small black box). c Quantitative analysis of number of cleaved caspase-3-positive cells per mm2 in the left cerebral cortex at 24 h after modeling. Data are expressed as mean ± standard deviation (n = 4 per group). *P < 0.05. PBS, phosphate-buffered saline
FGF-2 Prevented Long-term Neurological Deficits at 4 Weeks After SAH
Over the 21-day experimental window, SAH resulted in persistent neurologic deficits as seen in the rotarod test. Treatment with rFGF-2 significantly improved falling latency in the rotarod test on post-SAH 7 days at 5 rpm and 7 to 21 days at 10 rpm acceleration compared to the SAH+PBS group (Fig. 5a). In the water maze test, the escape latency and path length to find platform prolonged (Fig. 5b), and there was less time spent in probe quadrant (Fig. 5c, d) in the SAH+PBS group when compared to the sham group. SAH rats treated with rFGF-2 significantly improved memory recovery, reduced escape latency, reduced path length to platform, and spent more time in target quadrant as compared to SAH rats treated with PBS (Fig. 5b–d). There was no significant difference in swimming velocity between the groups on 27 days (Fig. 5e). Nissl staining of left hippocampal sections revealed the number of neurons that survived in the SAH+PBS group was significantly lower than that in the sham and SAH+rFGF-2 groups (Fig. 6a–c).

Effects of recombinant fibroblast growth factor-2 (rFGF-2) on long-term neurobehavioral function after subarachnoid hemorrhage (SAH). a Rotarod tests of 5 and 10 rpm. *P < 0.05 and **P < 0.01 vs. sham, †P < 0.05 vs. SAH+phosphate-buffered saline (PBS). b Escape latency and swimming distance of Morris water maze. *P < 0.05 and **P < 0.01 vs. sham, †P < 0.05 vs. SAH+PBS. c Representative heat maps of the probe trial. The white circles indicate the positions of the probe platform. d Quantification of the probe quadrant duration in the probe trial. *P < 0.05 vs. sham, †P < 0.05 vs. SAH+PBS. e Swimming velocities of different groups in probe trial. Data are expressed as mean ± standard deviation (n = 8 per group)

Effects of recombinant fibroblast growth factor-2 (rFGF-2) on long-term neuronal degeneration after subarachnoid hemorrhage (SAH). a Representative Nissl staining pictures in different regions of the hippocampus. Original magnification × 200. b Representative image demonstrating the location of interest area (small black boxes) in the left hippocampus. c Quantification of the Nissl-positive neurons per area in dentate gyrus (DG), cornu ammonis (CA) 1, and CA3 at 28 days after modeling. Data are expressed as mean ± standard deviation (n = 8 per group). *P < 0.05, **P < 0.01. PBS, phosphate-buffered saline
The FGFR/PI3k/Akt Pathway Made a Greater Contribution to rFGF-2-Mediated Anti-apoptotic Effect After SAH
The pan-FGFR inhibitor PD173074 and rFGF-2 administration did not affect neurological scores in sham-operated rats. However, PD173074 significantly reduced the neuroprotective effects of post-SAH rFGF-2 treatment compared to DMSO (Fig. 7a). WB results showed that rFGF-2 treatment significantly increased the expression of p-FGFR in SAH-operated rats as compared to the SAH+PBS+DMSO group. PD173074 significantly reversed the upregulation of the expression of p-FGFR by rFGF-2 treatment (Fig. 7b). Next, the expression of intracellular mediators of FGFR signaling PI3k, p-Akt, and Akt; anti-apoptotic marker Bcl-2; and apoptotic marker Bax were evaluated by WB. PD173074 and rFGF-2 administration did not affect the expression of PI3k, p-Akt, Bcl-2, and Bax in sham-operated rats. However, treatment with rFGF-2 significantly activated the expression of PI3k, p-Akt, and Bcl-2 and inhibited the expression of Bax compared to PBS at 24 h after SAH induction. PD173074 abolished the upregulation of the expression of PI3k, p-Akt, and Bcl-2 and the downregulation of the expression of Bax compared to DMSO in SAH rats treated with rFGF-2 (Fig. 7b). TUNEL and cleaved caspase-3 staining revealed that rFGF-2 reduced the number of TUNEL-positive neurons and cleaved caspase-3-positive cells after SAH induction compared to the SAH+PBS+DMSO group, and the anti-apoptotic effect of rFGF-2 was reversed by PD173074 administration (Figs. 8 and 9).

Effects of pan-fibroblast growth factor receptor (FGFR) inhibitor after subarachnoid hemorrhage (SAH). a Effects of inhibition of FGFR on neurobehavioral scores at 24 h after modeling (n = 6–10 per group). *P < 0.05, **P < 0.01. b Representative western blot images and densitometric quantification of expression of PI3k, phosphorylated Akt (p-Akt), Bcl-2, and Bax. Expression levels of each protein are expressed as mean ± standard deviation (n = 6 per group). *P < 0.05, **P < 0.01. DMSO, dimethyl sulfoxide; PBS, phosphate-buffered saline; PD173074, pan-FGFR inhibitor; rFGF-2, recombinant fibroblast growth factor-2

Effects of pan-fibroblast growth factor receptor (FGFR) inhibitor on neuronal apoptosis by TUNEL assay after subarachnoid hemorrhage (SAH). a Representative microphotographs of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive neurons in the left temporal cerebral cortex at 24 h after modeling. Scale bar = 50 μm. b Representative image demonstrating the location of staining (small black box). c Quantitative analysis of TUNEL-positive neurons per mm2 in the left cerebral cortex at 24 h after modeling. Data are expressed as mean ± standard deviation (n = 4 per group). **P < 0.01. DMSO, dimethyl sulfoxide; PBS, phosphate-buffered saline; PD173074, pan-FGFR inhibitor; rFGF-2, recombinant fibroblast growth factor-2

Effects of pan-fibroblast growth factor receptor (FGFR) inhibitor on apoptotic marker after subarachnoid hemorrhage (SAH). a Representative images demonstrating double immunofluorescent staining for cleaved caspase-3 and neuron-specific nuclear protein (NeuN) in the left temporal cerebral cortex at 24 h after modeling. Scale bar = 50 μm. b Representative image demonstrating the location of staining (small black box). c Quantitative analysis of number of cleaved caspase-3-positive cells per mm2 in the left cerebral cortex at 24 h after modeling. Data are expressed as mean ± standard deviation (n = 4 per group). *P < 0.05. DMSO, dimethyl sulfoxide; PBS, phosphate-buffered saline; PD173074, pan-FGFR inhibitor; rFGF-2, recombinant fibroblast growth factor-2
FGFR3-Regulated Signaling Pathway Made a Greater Contribution on Anti-apoptotic Effect Rather than FGFR1
The gene silencing efficacy of FGFR1 and FGFR3 siRNA was demonstrated by WB in SAH animals. FGFR1 and FGFR3 siRNA injection significantly reduced the expression of FGFR1 and FGFR3 in the left hemisphere sample compared to the SAH+Scr siRNA group, respectively (Fig. 10b). FGFR3 knockdown significantly reduced post-SAH neuroprotective effects of rFGF-2, while FGFR1 knockdown did not affect neurological function (Fig. 10a). In addition, FGFR3 knockdown suppressed the expression of PI3k, p-Akt, and Bcl-2 and enhanced the expression of Bax compared to the SAH+Scr siRNA group at 24 h after SAH induction, while FGFR1 knockdown did not affect these proteins.

Effects of fibroblast growth factor receptor (FGFR) 1 and FGFR3 knockdown after subarachnoid hemorrhage (SAH). a Effects of knockdown of FGFR1 and FGFR3 on neurobehavioral scores at 24 h after modeling (n = 6 per group). *P < 0.05. b Representative western blot images and densitometric quantification of expression of PI3k, phosphorylated Akt (p-Akt), Bcl-2, and Bax. Expression levels of each protein are expressed as mean ± standard deviation (n = 6 per group). *P < 0.05, **P < 0.01. rFGF-2, recombinant fibroblast growth factor-2; siRNA, small interfering ribonucleic acid; Scr siRNA, scrambled siRNA
Endogenous FGF-2 also Alleviated Post-SAH Neurological Deficits via PI3k/Akt signaling Pathway
A neutralizing antibody against FGF-2 administration did not affect neurological scores in sham-operated rats. However, a neutralizing antibody against FGF-2 treatment significantly reduced neurobehavioral function in SAH-operated rats (Fig. 11a). Expression of FGF-2, PI3k, p-Akt, Akt, Bcl-2, and Bax was evaluated by WB. SAH rats treated with normal mouse IgG did not significantly induce the expression of PI3k and p-Akt as compared to sham-operated rats treated with normal mouse IgG. In contrast, anti-FGF-2 neutralizing antibody induction inhibited the expression of FGF-2 as compared to normal mouse IgG in SAH rats. In addition, anti-FGF-2 neutralizing antibody significantly suppressed the expression of PI3k and p-Akt and enhanced the expression of Bax compared to normal mouse IgG in SAH-operated rats. In contrast, there was no significant difference in the expression of Bcl-2 between anti-FGF-2 neutralizing antibody and normal mouse IgG after SAH (Fig. 11b).

Effects of neutralizing endogenous fibroblast growth factor-2 (FGF-2) after subarachnoid hemorrhage (SAH). a Effects of anti-FGF-2 neutralizing antibody on neurobehavioral scores at 24 h after SAH (n = 6 per group). *P < 0.05, **P < 0.01. b Representative western blot images and densitometric quantification of expression of PI3k, phosphorylated Akt (p-Akt), Bcl-2, and Bax. Expression levels of each protein are expressed as mean ± standard deviation (n = 6 per group). *P < 0.05, **P < 0.01. IgG, immunoglobulin G; Nab, neutralizing antibody
Discussion
The novel findings in the present study are as follows: (1) The expression of FGF-2, FGFR1, and FGFR3 upregulated following endovascular perforation induction of SAH in rats; (2) rFGF-2 improved neuronal degeneration in the long term as well as short term through alleviation of apoptosis via upregulation of the expression of PI3k and p-Akt, which leads to the suppression of the expression of Bax after SAH; (3) neuroprotective effects of FGF-2 enhanced through FGFR3-dependent signaling pathway rather than FGFR1 after SAH; and (4) endogenous FGF-2 also alleviated the neurobehavioral impairments after SAH.
Previous studies demonstrated that FGFR1 through FGFR5 are expressed in astrocytes, and FGFR1 to FGFR4 are expressed in neurons and microglia as well [4]. These FGFR families induce PI3k/Alt signaling pathway mediated by src homology-2, growth factor receptor-bound protein 2 (GRB2), and son of sevenless (SOS) adaptor proteins [22]. Akt might be involved in the mechanism for EBI after SAH [3, 23, 24]. Activated Akt inhibits apoptosis-inducing proteins including Bax and Bad and promotes cyclic AMP response element-binding protein phosphorylation, which induces anti-apoptotic protein Bcl-2 [23]. Tang et al. [8] demonstrated that administration of rFGF-2 alleviated neuronal apoptosis after rat traumatic brain injury via activation of the PI3k/Akt signaling pathway. Wang et al. [5] demonstrated that FGFR activation by rFGF-2 exerts anti-apoptotic effects via enhancement of the expression of extracellular signal-regulated kinase (ERK) 1/2 as well as the expression of PI3k and p-Akt after cerebral ischemia–reperfusion injury in mice. In the present study, we demonstrated, for the first time, that the expression of FGFR1 and FGFR3 was enhanced in neurons after SAH, and FGF-2 therapy attenuated EBI involving neuronal apoptosis via the FGFR/PI3k/Akt signaling pathway after SAH in rats. In addition, less neuronal loss of CA1, CA3, and DG of the hippocampus was found even on 28 days after rFGF-2 administration in SAH rats, suggesting that FGF-2 treatment targeting EBI contributed to improvement of long-term functional outcomes as well. Alternatively, the newly born neurons of neurogenesis may contribute to the increased number of neurons in the hippocampus after SAH. Evidence for functional neuronal replacement through endogenous neurogenesis has been reported in a model of ischemic stroke that affects the hippocampus predominantly. Infusion of FGF-2 led to regeneration of CA1 neurons derived from the DG, which were thought to have ameliorated neurological deficits [25]. Of the FGFR subtypes, FGFR3 would make contribution to FGF-2-mediated neuronal survival after SAH. In the present study, we demonstrated that FGFR3 knockdown reversed the activation of the PI3k/Akt signaling pathway as well as the neuroprotective effects of rFGF-2 treatment after SAH, while FGFR1 knockdown did not change the expression of PI3k and Akt as well as neurobehavioral function. FGFR3 shares approximately 70% amino acid sequence identity with FGFR1 in its cytoplasmic domain [22]. However, the FGFR3 signal pathway has unique signaling pathway utilizing two GRB2-containing complexes (i.e., src homologous and collagen-GRB2-SOS and 80K-H-GRB2-SOS), which can link to Ras leading to induction of mitogen-activated protein kinase (MAPK) signaling pathway including ERK1/2 [22]. Noda et al. [4] demonstrated that the FGFR3–ERK signaling pathway led to microglia-mediated neuronal survival, while neutralizing antibodies for FGFR1, FGFR2, FGFR4, and FGFR5, and the selective FGFR1 blocker had no effect on neuronal survival when damaged by glutamate or oligomeric amyloid β1–42 in vitro.
It has been proposed that apoptotic cells or degenerating neurons release endogenous FGF-2, which activates FGFR of brain cells including neurons in the CNS [26]. However, the role of endogenous FGF-2 in CNS injury is not entirely understood. As far as we know, few studies have investigated if endogenous FGF-2 exerts neuroprotective effects after CNS injury. Rowntree and Kolb [27] demonstrated that the expression of endogenous FGF-2 increased after motor cortex injury, and neutralizing antibody against FGF-2 retards functional recovery. Fagan et al. [28] demonstrated that anti-FGF-2 neutralizing antibodies displayed a reduction in axonal sprouting in rat denervated hippocampus. Wen et al. [18] demonstrated that anti-FGF-2 neutralizing antibody induced a significant shortening of the response latency and a decrease in hippocampal neurons in ischemic gerbils. Our present study demonstrated that anti-FGF-2 neutralizing antibody aggravated neurobehavioral function, suppressed the expression of the PI3k and p-Akt, and enhanced the expression of Bax in post-SAH rats. The results suggest that endogenous FGF-2 attenuate neuronal impairments via the PI3k/Akt signaling pathway after SAH. However, the change of expression level of p-Akt following SAH is controversial. Our previous studies demonstrated that the expression of p-Akt was significantly decreased 24 h after SAH [20, 29]. In contrast, Endo et al. [24] showed that the expression of p-Akt significantly increased at 6 h and returned to baseline at 24 h after SAH. In the present study, the expression of PI3k and p-Akt was not significantly induced 24 h post-SAH as compared to sham. Also, in the present study, the expression of endogenous FGF-2 peaked at 3 h and declined after that, although it was significantly higher until 24 h after SAH compared to sham. The concentration of FGF-2 protein was around 38 to 111 ng/mg in the neonatal rat pup brain [30]. The amount of the enhanced endogenous FGF-2 may not be high enough to activate Akt at least at 24 h after SAH, and the anti-FGF-2 neutralizing antibody could eliminate the neuroprotective effects by initial elevation of endogenous FGF-2.
This study is somewhat limited. First, inclusion and exclusion were performed using SAH grading score. However, criteria on the basis of cerebral perfusion pressure (CPP) may be better in the study regarding EBI after SAH. Although CPP reductions in animals were not always associated with poor neurological outcomes after SAH, early brain injury linearly correlated with reduction in cerebral perfusion pressure during the hyperacute phase of SAH [31, 32]. In addition, intracranial pressure elevation and blood pressure variation were not monitored in the present experiments. Therefore, SAH without CPP elevation may not be excluded appropriately. Second, only immediate intranasal treatment with FGF-2 at 30 min after SAH was tested in this study. Most of the patients cannot receive FGF-2 treatment within 30 min after SAH. To assess the therapeutic time window of FGF-2 treatment, the effects of multiple treatments at different dosages or time courses should be examined in future preclinical evaluations. Third, of the FGFR subtypes, only FGFR1 and FGFR3 were compared using siRNA in this study. Although FGF-2 has high affinity with FGFR1 and FGFR3 in general condition, we cannot exclude the possibility that other subtypes exert more effectively on anti-apoptosis due to change of affinity with FGF-2 under the hyperacute phase of SAH. Fourth, this study demonstrated that rFGF-2 prevented neuronal apoptosis via upregulation of the expression of PI3k and p-Akt. However, we cannot exclude the possibility that FGFRs’ activation might induce alternative neuroprotective pathways against EBI such as neuronal apoptosis, inflammation, and blood–brain barrier disruption. Fifth, there is the possibility that anti-FGF-2 neutralizing antibody inactivates other neuroprotective receptors or ligands rather than FGF-2. Lastly, endogenous ligands that activated FGFR other than FGF-2 were not investigated in this study. Though there are potential limitations, the study presents FGF-2 as a novel treatment option targeting SAH by suppressing neuronal apoptosis. Further studies to reveal the mechanism underlying the neuroprotective effects of FGF-2 are needed to reinforce the findings.
Conclusions
This study demonstrated for the first time that intranasal administration of rFGF-2 prevented neuronal loss both short and long term after SAH through attenuation of neuronal apoptosis possibly via the enhancement of the FGFR3/PI3k/Akt signaling pathway.
Abbreviations
- BWC :
-
brain water content
- CA :
-
cornu ammonis
- CNS :
-
central nervous system
- DG :
-
dentate gyrus
- DMSO :
-
dimethyl sulfoxide
- EBI :
-
early brain injury
- ERK :
-
extracellular signal-regulated kinase
- FGF :
-
fibroblast growth factor
- FGFR :
-
fibroblast growth factor receptor
- FJC :
-
Fluoro-Jade C
- GRB2 :
-
growth factor receptor-bound protein 2
- i.c.v.:
-
intracerebroventricular
- IgG :
-
immunoglobulin G
- IHC :
-
immunohistochemistry
- Nab :
-
neutralizing antibody
- NeuN :
-
neuron-specific nuclear protein
- p-Akt :
-
phosphorylated Akt
- PBS :
-
phosphate-buffered saline
- p-FGFR :
-
phosphorylated fibroblast growth factor receptor
- rFGF-2 :
-
recombinant fibroblast growth factor-2
- rpm :
-
revolutions per minute
- SAH :
-
subarachnoid hemorrhage
- Scr siRNA :
-
scrambled small interfering ribonucleic acid
- siRNA :
-
small interfering ribonucleic acid
- SOS :
-
son of sevenless
- TUNEL :
-
terminal deoxynucleotidyl transferase dUTP nick end labeling
- WB :
-
western blot
References
Okada T, Suzuki H (2017) Toll-like receptor 4 as a possible therapeutic target for delayed brain injuries after aneurysmal subarachnoid hemorrhage. Neural Regen Res 12:193
Prunell GF, Svendgaard NA, Alkass K, Mathiesen T (2005) Delayed cell death related to acute cerebral blood flow changes following subarachnoid hemorrhage in the rat brain. J Neurosurg 102:1046–1054
Zhou XM, Zhou ML, Zhang XS, Zhuang Z, Li T, Shi JX, Zhang X (2014) Resveratrol prevents neuronal apoptosis in an early brain injury model. J Surg Res 189:159–165
Noda M, Takii K, Parajuli B, Kawanokuchi J, Sonobe Y, Takeuchi H et al (2014) FGF-2 released from degenerating neurons exerts microglial-induced neuroprotection via FGFR3-ERK signaling pathway. J Neuroinflamm 11:1–11
Wang Z, Zhang H, Xu X, Shi H, Yu X, Wang X, Yan Y, Fu X et al (2012) BFGF inhibits ER stress induced by ischemic oxidative injury via activation of the PI3K/Akt and ERK1/2 pathways. Toxicol Lett 212:137–146
Wang ZG, Cheng Y, Yu XC, Ye LB, Xia QH, Johnson NR, Wei X, Chen DQ et al (2016) bFGF protects against blood-brain barrier damage through junction protein regulation via PI3K-Akt-Rac1 pathway following traumatic brain injury. Mol Neurobiol 53:7298–7311
Huang B, Krafft PR, Ma Q, Rolland WB, Caner B, Lekic T, Manaenko A, le M et al (2012) Fibroblast growth factors preserve blood-brain barrier integrity through RhoA inhibition after intracerebral hemorrhage in mice. Neurobiol Dis 46:204–214
Tang C, Shan Y, Hu Y, Fang Z, Tong Y, Chen M, Wei X, Fu X et al (2017) FGF2 attenuates neural cell death via suppressing autophagy after rat mild traumatic brain injury. Stem Cells Int 2017:1–14. https://doi.org/10.1155/2017/2923182
Reilly JF, Kumari VG (1996) Alterations in fibroblast growth factor receptor expression following brain injury. Exp Neurol 140:139–150
Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier F et al (1996) Receptor specificity of the fibroblast growth factor family. J Biol Chem 271:15292–15297
Enkhjargal B, McBride DW, Manaenko A, Reis C, Sakai Y, Tang J et al (2017) Intranasal administration of vitamin D attenuates blood–brain barrier disruption through endogenous upregulation of osteopontin and activation of CD44/P-gp glycosylation signaling after subarachnoid hemorrhage in rats. J Cerebr Blood F and Met 37:2555–2566
Sugawara T, Ayer R, Jadhav V, Zhang JH (2008) A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model. J Neurosci Meth 167:327–334
Mo J, Enkhjargal B, Travis ZD, Zhou K, Wu P, Zhang G, Zhu Q, Zhang T et al (2019) AVE 0991 attenuates oxidative stress and neuronal apoptosis via Mas/PKA/CREB/UCP-2 pathway after subarachnoid hemorrhage in rats. Redox Biol 20:75–86
Feng C, Zhang C, Shao X, Liu Q, Qian Y, Feng L, Chen J, Zha Y et al (2012) Enhancement of nose-to-brain delivery of basic fibroblast growth factor for improving rat memory impairments induced by co-injection of β-amyloid and ibotenic acid into the bilateral hippocampus. Int J Pharm 423:226–234
Zhou K, Enkhjargal B, Xie Z, Sun C, Wu L, Malaguit J, Chen S, Tang J et al (2018) Dihydrolipoic acid inhibits lysosomal rupture and NLRP3 through lysosome-associated membrane Protein-1/Calcium/Calmodulin-Dependent Protein Kinase II/TAK1 pathways after subarachnoid hemorrhage in rat. Stroke 49:175–183
Zappaterra MW, LaMantia AS, Walsh CA, Lehtinen MK (2013) Isolation of cerebrospinal fluid from rodent embryos for use with dissected cerebral cortical explants. J Vis Exp 73:e50333.
Westerhout J, Ploeger B, Smeets J, Danhof M, de Lange ECM (2012) Physiologically based pharmacokinetic modeling to investigate regional brain distribution kinetics in rats. The AAPS J 14:543–553
Wen TC, Matsuda S, Yoshimura H, Aburaya J, Kushihata F, Sakanaka M (1995) Protective effect of basic fibroblast growth factor-heparin and neurotoxic effect of platelet factor 4 on ischemic neuronal loss and learning disability in gerbils. Neuroscience 65:513–521
Okada T, Kawakita F, Nishikawa H, Nakano F, Liu L, Suzuki H (2019) Selective toll-like receptor 4 antagonists prevent acute blood-brain barrier disruption after subarachnoid hemorrhage in mice. Mol Neurobiol 56:976–985
Xie Z, Enkhjargal B, Wu L, Zhou K, Sun C, Hu X, Gospodarev V, Tang J et al (2018) Exendin-4 attenuates neuronal death via GLP-1R/PI3K/Akt pathway in early brain injury after subarachnoid hemorrhage in rats. Neuropharmacology 128:142–151
Bromley-Brits K, Deng Y, Song W (2011) Morris water maze test for learning and memory deficits in Alzheimer’s disease model mice. J Vis Exp 53:e2920
Kanai M, Go M, Tsunekawa S, Podolsky DK (1997) Signal transduction pathway of human fibroblast growth factor receptor 3. J Biol Chem 272:6621–6628
Moisa H, Palade C, Nica D, Savu R, Ciurea A (2013) Interference of apoptosis in the pathophysiology of subarachnoid hemorrhage. Asian J Neurosurg 8:106–111
Endo H, Nito C, Kamada H, Yu F, Chan PH (2007) Reduction in oxidative stress by superoxide dismutase overexpression attenuates acute brain injury after subarachnoid hemorrhage via activation of Akt/glycogen synthase kinase-3beta survival signaling. J Cerebr Blood F Met 27:975–982
Nakatomi H, Kuriu T, Okabe S, Yamamoto S, Hatano O, Kawahara N, Tamura A, Kirino T et al (2002) Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural progenitors. Cell 110:429–441
Logan A, Frautschy SA, Gonzalez AM, Baird A (1992) A time course for the focal elevation of synthesis of basic fibroblast growth factor and one of its high-affinity receptors (flg) following a localized cortical brain injury. J Neurosci 12:3828–3837
Rowntree S, Kolb B (1997) Blockade of basic fibroblast growth factor retards recovery from motor cortex injury in rats. Eur J Neurosci 9:2432–2441
Fagan AM, Suhr ST, Lucidi-Phillipi CA, Peterson DA, Holtzman DM, Gage FH (1997) Endogenous FGF-2 is important for cholinergic sprouting in the denervated hippocampus. J Neurosci 17:2499–2511
Zhu Q, Enkhjargal B, Huang L, Zhang T, Sun C, Xie Z et al (2018) Aggf1 attenuates neuroinflammation and BBB disruption via PI3K/Akt/NF-ΚB pathway after subarachnoid hemorrhage in rats. J Neuroinflamm. 15:1–13
Kim J, Gale K, Kondratyev A (2010) Effects of repeated minimal electroshock seizures on NGF, BDNF and FGF-2 protein in the rat brain during postnatal development. Int J Dev Neurosci 28:227–232
Sehba FA, Hou J, Pluta RM, Zhang JH (2012) The importance of early brain injury after subarachnoid hemorrhage. Prog Neurobiol 97:14–37
Marbacher S, Neuschmelting V, Andereggen L, Widmer HR, von Gunten M, Takala J, Jakob SM, Fandino J (2014) Early brain injury linearly correlates with reduction in cerebral perfusion pressure during the hyperacute phase of subarachnoid hemorrhage. Intensive Care Med Exp 2:30. https://doi.org/10.1186/s40635-014-0030-1
Acknowledgments
The authors acknowledge the technical assistance provided by Desislava Doycheva, Jerry Flores, Marcin Gamdzyk, Helen Huang, Prativa Sherchan, Orhan Altay, Weilin Xu, Yuchun Zuo, Ganz Zuo, and Jun Peng.
Funding
This article is supported partially by grants from the National Institutes of Health (NS081740 and NS082184) to Dr. Zhang.
Author information
Authors and Affiliations
Contributions
TO, BE, SH, JT, and JHZ were involved in the study design. TO, BE, and JHZ were involved in data interpretation and writing of the manuscript; TO performed the majority of the laboratory work and contributed to the analysis of data; BE was responsible for the animal model; ZDT was involved in manuscript editing. UO was responsible for the rotarod and water maze test; ZDT was involved in manuscript editing.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
The Institutional Animal Care and Use Committee (IACUC) of Loma Linda University approved the study protocol, and all experiments were conducted in accordance with the NIH Guidelines for Use of Animals in Neuroscience Research. All authors have read and approved the submitted manuscript.
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(DOCX 948 kb)
Rights and permissions
About this article

Cite this article
Okada, T., Enkhjargal, B., Travis, Z.D. et al. FGF-2 Attenuates Neuronal Apoptosis via FGFR3/PI3k/Akt Signaling Pathway After Subarachnoid Hemorrhage. Mol Neurobiol 56, 8203–8219 (2019). https://doi.org/10.1007/s12035-019-01668-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-019-01668-9