
Abstract
Many traumatic brain injury (TBI) survivors sustain neurological disability and cognitive impairments due to the lack of defined therapies to reduce TBI-induced blood-brain barrier (BBB) breakdown. Exogenous basic fibroblast growth factor (bFGF) has been shown to have neuroprotective function in brain injury. The present study therefore investigates the beneficial effects of bFGF on the BBB after TBI and the underlying mechanisms. In this study, we demonstrate that bFGF reduces neurofunctional deficits and preserves BBB integrity in a mouse model of TBI. bFGF suppresses RhoA and upregulates tight junction proteins, thereby mitigating BBB breakdown. In vitro, bFGF exerts a protective effect on BBB by upregulating tight junction proteins claudin-5, occludin, zonula occludens-1, p120-catenin, and β-catenin under oxygen glucose deprivation/reoxygenation (OGD) in human brain microvascular endothelial cells (HBMECs). Both the in vivo and in vitro effects are related to the activation of the downstream signaling pathway, PI3K/Akt/Rac-1. Inhibition of the PI3K/Akt or Rac-1 by specific inhibitors LY294002 or si-Rac-1, respectively, partially reduces the protective effect of bFGF on BBB integrity. Overall, our results indicate that the protective role of bFGF on BBB involves the regulation of tight junction proteins and RhoA in the TBI model and OGD-induced HBMECs injury, and that activation of the PI3K/Akt /Rac-1 signaling pathway underlies these effects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Traumatic brain injury (TBI) is a leading cause of death and disability in the Western World [1]. More than 1.7 million new cases of TBI occur in the USA each year, causing 60 % of all trauma-related deaths [2]. TBI damage is highly heterogeneous and can also trigger other neurological complications including epilepsy, depression, and dementia. The initial injury often leads to the development of secondary sequelae including neurovascular dysfunction, inflammation, oxidative stress, and apoptosis [3–6]. Among all these pathological events, blood-brain barrier (BBB) breakdown is one key mechanism that leads to progression of brain injury and long-term neurological deficits. The BBB integrity is compromised soon after TBI due to mechanical breach or functional breakdown of endothelial cells and other essential BBB components. BBB disruption results in uncontrolled efflux of ions and proteins from the intravascular space to the interstitial brain compartments with water accumulation, vasogenic brain edema, elevated intracerebral pressure, and secondary ischemic injuries [5]. Therefore, targeting the molecular mechanisms that regulate BBB permeability may lead to more efficacious therapeutic strategies for TBI [7, 8]. In the previous study, bFGF treatment shows the efficacy to reduce cerebral edema and neurological deficits after ischemia/reperfusion injury in stroke models [9]. In our study, we first reported that the protective role of bFGF on TBI-induced BBB breakdown is related to the upregulation of tight junction proteins and inhibition of RhoA via Rac-1.
Tight junctions (TJs) are the hallmark of BBB integrity that essentially contributes to its structural inviolacy. The paracellular flux of hydrophilic molecules across the BBB is thereby limited which is essential for the functional integrity of the CNS [10]. TJs are composed of transmembrane proteins such as occludin, claudins, and junctional adhesion molecules. All of these proteins are anchored to endothelial cells by cytoplasmic protein complexes comprising zonula occludens-1 [ZO-1], zonula occludens-2 [ZO-2], and cingulin [11, 12]. The TJs limit the flux of hydrophilic molecules across the BBB while smaller lipophilic substances such as O2 and CO2 diffuse freely across plasma membranes along their concentration gradient [13]. The development of vasogenic brain edema after TBI is caused by BBB rupture and consists of protein-rich fluid [11]. The alteration of TJ assemblies may contribute to the loss of BBB integrity and BBB breakdown [14]. A wide array of growth factors, cytokines, and drugs influence TJs and their barrier function. For example, steroids [15] or unsaturated fatty acids [16] enhance TJ tightness by increasing the expression of occludin. Cytokines, VEGF [17], and tumor necrosis factor-ɑ [18] perturb TJ integrity by decreasing occludin and ZO-1 expression and causing cl5 and ZO-1 protein disruption. It is safe to suggest that any pathologic stimulus affecting the expression and localization of TJ proteins will profoundly affect the integrity of the BBB.
Basic fibroblast growth factor (bFGF or FGF-2) is a member of the fibroblast growth factor family which regulates a variety of biological functions including proliferation, morphogenesis, and suppression of apoptosis during development via a complex signal transduction system [19–21]. bFGF is highly expressed in the nervous system where it has multiple roles including the regulation of vascular integrity. Inhibition of fibroblast growth factors receptor (FGFR) signaling resulted in a decrease in cell-cell adhesions through disintegration of the p120-catenin/VE-cadherin complex [22]. Furthermore, previous studies demonstrated that bFGF inhibited RhoA by activating Ras-related C3 botulinum toxin substrate 1 (Rac1) through the phosphatidylinositol 3-kinase (PI3K)-Akt signaling pathway in human endothelial cells of the cornea [23], as well as in bone marrow stromal cells [24]. Activated Rac1 inhibits RhoA [25] which may preserve BBB integrity and reduce the development of vasogenic brain edema after TBI.
The goal of the present study was to explore the mechanisms by which exogenous bFGF treatment preserves BBB integrity, specifically by upregulating TJs proteins, attenuating neurofunctional deficits in TBI mice. We demonstrate that bFGF activation of the PI3K-Akt-Rac1 signaling pathway and consequent inhibition of RhoA results in preservation of tight junction proteins. Collectively, our results suggest that the bFGF may be an effective and feasible target for drug development of TBI both in vivo and in vitro.
Material and Methods
Reagents and Antibodies
Recombinant human basic fibroblast growth factor (bFGF) was purchased from Sigma (Sigma-Aldrich, St. Louis, MO), and PI3K inhibitor LY294002 (Sigma-Aldrich, St Louis, MO) was dissolved in 25 % dimethylsulfoxide solution (DMSO). FITC-dextran and Evans Blue were purchased from Sigma-Aldrich, St. Louis, MO. Anti-β-catenin, anti-p120-catenin, and anti-CD31 were purchased from Abcam, Cambridge, MA. GTP-Rac1, total-Rac1, GTP-RhoA, and total-RhoA were detected using Rac1/Cdc42 and Rho Activation Assay Kits (Millipore, Temecula, CA). Anti-Akt and anti-p-Akt (Ser473), anti-claudin-5, anti-occludin, and anti-zonula occludens-1 were from Proteintech. Human brain microvascular endothelial cells (HBMECs) and endothelial cell medium (ECM) were purchased from Sciencell (Carlsbad, CA, USA). The appropriate secondary antibodies were obtained from Santa Cruz Biotechnology.
Animals and Surgical Procedures
C57BL/6N male mice (20–25 g) were purchased from the Animal Center of the Chinese Academy of Sciences. The animal use and care protocol conformed to the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health and was approved by the Animal Care and Use Committee of Wenzhou Medical University. The animals were housed under standard conditions, including adequate temperature and humidity (60 %) control with a 12-h light/12-h dark cycle, and free access to water and food. All procedures used in this study were approved by the ethics committee for the use of experimental animals at Wenzhou Medical University. All animals remained in the animal care facility for a minimum of 7 days prior to the experiments. The TBI model was used as previously described [26]. The C57BL/6N mice were anesthetized with 4 % choral hydrate (10 ml/kg IP), and surgery was performed under aseptic conditions and mounted in a stereotaxic system (David Kopf Instruments, Tujunga, California). The following steps were all performed using aseptic techniques. A midline incision on the scalp exposed the skull, without requiring muscle retraction. Craniotomy was performed by hand-held trephine. For the trephine method, a 3-mm-diameter manual trephine (Roboz Surgical Instrument Co., Gaithersburg, MD) was carefully used to penetrate the skull for removal of the bone flap. Mice were subjected to TBI on the right part of the brain between the lambda and the bregma about 1 mm from the midline. The stereotaxic frame was inclined so as to make the plane of the cortex perpendicular to the impactor tip. The impact velocity was set at 4 m/s with the penetration depth at 1.5 mm and the impactor dwell time was 600 ms, as described previously [27]. The craniotomy which did not significantly affect physiological parameters (arterial pressure, heart rate, or body weight) was closed immediately after TBI. For the sham operation group, only the surgical procedure was performed on the animals without cortical impact. Inhibitor LY294002 (50 nmol/Kg) was injected into the left striatum at a rate of 2 μl/min. After completed injection, the needle was left in place for an additional 10 min to prevent backflow of collagenase along the needle tract, before being withdrawn at a rate of 1 mm/min. To explore the effect of bFGF in the TBI mouse, a dose of 0.5 μg/g bFGF was intranasally administrated 1 h before induction of the TBI model. Vehicle animals received same volume of PBS. After the surgical incisions were closed, the mice were allowed to recover for 24 h. Animals were housed under a 12-h light/dark cycle in a pathogen-free area with free access to water and food. All efforts were made to minimize the number of animals used and their suffering.
Evans Blue Extravasation Assay
Evans Blue [13] extravasation assays were conducted 24 h after surgery. Briefly, mice received an intraperitoneal injection of 0.25 ml EB dye (2 %) 22 h after TBI. Two hours after EB dye injection (24 h after TBI), the anesthetized animals were perfused with saline to wash away any remaining dye in the blood vessels prior to sample collection. The right hemisphere was weighed. The EB dye was extracted by formamide, and the supernatant was allowed to incubate for 3 days at 72 °C and was then re-centrifuged. The quantity of extravasated Evans Blue dye was detected by spectrophotometer at an excitation wavelength of 610 nm and an emission wavelength of 680 nm and quantified according to a standard curve.
Determination of BBB Vascular Permeability
TBI or sham procedure was performed (n = 6 per group). Twenty-two hours later, the mice were injected with 0.2 ml 70 kDa FITC-dextran (100 μg/ml). Two hours later, the animals were subjected to systemic intracardiac perfusion with 1 USP U/mL of heparin in saline to flush the intravascular FITC-dextran out of the vasculature. The perfused brains were then harvested. Thereafter, relative fluorescence passing through the tissues was determined using an EnSpire Manager (PerkinElmer Company, USA) multimode plate reader at an excitation wavelength of 485 nm and an emission wavelength of 535 nm.
Behavior Assessments
The sensorimotor Garcia Test [28] was conducted in a blinded fashion and was used to assess neurofunctional deficits in mice at 24 after surgery. The Garcia Test has been modified and consisted of 7 individual tests, examining spontaneous activity (1), axial sensation (2), vibrissae proprioception (3), limb symmetry (4), as well as the animal's ability of lateral turning (5), forelimb outstretching (6), and climbing (7). A score of 0 (worst performance) to 3 (best performance) was given for each sub-test, and a total Garcia score was calculated as the sum of all sub-tests (maximum score of 21).
Immunofluorescence Staining
Brain tissues were embedded in OCT and cut into 10-mm sections. The sections were then stained with specific antibodies for analysis. To determine claudin-5, ZO-1, CD31, and occluding activities, sections were incubated with 3 % H2O2 in methanol for 10 min, followed by blocking with 5 % bovine albumin in PBS for 30 min incubated 37 °C. Next, sections were incubated at 4 °C overnight with a primary antibody against claudin-5 (1:100), ZO-1 (1:100), occludin (1:100), or CD31 (1:500), followed by incubation with Alexa-Fluor594/647 donkey anti-mouse/rabbit, Alexa-Fluor488/594 donkey anti-rabbit/mouse, or Alexa-Fluor488/594 donkey anti-goat secondary antibody (1:500; Invitrogen Corporation, Carlsbad, CA, USA) for 1 h at 37 °C. Cellular nuclei were counterstained with Hoechst 33258. The saline injection group was considered to be the negative control. The results were imaged at ×400 magnifications using a Nikon ECLPSE 80i.
Cell Culture and In Vitro Oxygen Glucose Deprivation/Reoxygenation Model
HBMECs were incubated at 37 °C in a humidified atmosphere of 5 % CO2 and 95 % air and cultured in endothelial cell medium (ECM). Normal growth medium was then replaced with ECM without FBS, and the cells were incubated in an anaerobic chamber for 24 h. This ensured that the oxygen level remained below 0.5 %. After oxygen glucose deprivation (OGD), the cells were incubated under normal culture conditions for 12 h. Basic fibroblast growth factor (bFGF) (50 ng/ml) was added 1 h before of OGD and maintained during the reoxygenation process. To further evaluate the effect of PI3K/Akt activation on OGD. Cells were pre-treated for 1 h with specific inhibitor LY294002 (20 μM). After 12 h, cells were detached and collected for further study. All experiments were performed in triplicate.
Paracellular Permeability Assay
Flux of 70 kDa FITC-dextran across HBMECs was analyzed. Briefly, HBMECs was seeded at a density of 2 × 104 cells/well in 100 μl medium onto polycarbonate 24-well transwell chambers with a 0.4-mm mean pore size and a 0.3-cm2 surface area (Millicell Hanging Cell Culture Inserts, USA). Cells were incubated with FITC-dextran (1 mg/ml) in medium for 4 h. Thereafter, relative fluorescence passing through the chamber (in the lower chambers) was determined by using an EnSpire Manager (PerkinElmer Company, USA) multimode plate reader at an excitation wavelength of 485 nm and an emission wavelength of 520 nm.
siRNA Preparation and Transfections
Transient transfection of siRNA was carried out using Lipofectamine 2000 (Invitrogen). One day before transfection, cells were trypsinized and plated on a 6-well plate at 2 × 105 cells/well in 1 ml of DMEM 10 % serum without antibiotics. In all, 100 pmol of siRNA diluted in 250 μl of serum-free DMEM and 3.5 μl of Oligofectamine diluted in 250 μl of serum-free DMEM was pre-incubated for 5 min. The two mixtures were combined and incubated for 20 min at room temperature for complex formation. After the addition of 500 μl of serum-free DMEM, the entire mixture (1 ml) was added to each well. Cells were assayed 1 day after transfection.
Western Blot Analysis
Total proteins were purified using protein extraction reagents for the right brain cortical region and HBMEC. The equivalent of 60 μg of protein was separated by 12 % gel and then transferred onto a PVDF membrane. After blocking with 5 % fat-free milk, the membranes were incubated with the following antibodies: claudin-5 (1:500), ZO-1 (1:200), occluding (1:500), p-Akt (1:500), p120-catenin (1:1000), β-catenin (1:1000), CD31 (1:1000), GTP-Rac1 (1:1000), total-Rac1 (1:1000), GTP-RhoA (1:1000), and total-RhoA (1:1000) overnight. The membranes were washed with TBS and treated with horseradish peroxidase-conjugated secondary antibodies for 2 h at room temperature. The signals were visualized with the ChemiDicTM XRS+ Imaging System (Bio-Rad Laboratories, Hercules, CA, USA), and the band densities were quantified with Multi Gauge Software of Science Lab 2006 (FUJIFILM Corporation, Tokyo, Japan).
Statistical Analysis
The data were expressed as the mean ± SEM. Statistical significance was determined with Student’s t test when there were two experimental groups. For more than two groups, statistical evaluation of the data was performed using the one-way analysis of variance (ANOVA) test, followed by Dunnett’s post hoc test with values of P < 0.05 being considered significant.
Results
bFGF Treatment Reduces Neurofunctional Deficits After TBI in Mice
In order to evaluate the role of bFGF in TBI, neurofunctional deficits and BBB disruption were evaluated at 24 h after TBI. As evaluated by the Garcia Test, the Garcia neuroscore of mice subjected to TBI significantly decreased compared to sham-operated animals at 24 h after surgery. bFGF (15 μg) treatments significantly ameliorated neurofunctional deficits compared with the TBI control group (Fig. 1a). BBB breakdown results in cerebral edema and secondary neuronal injury after brain ischemia or trauma [29]. In our previous study, bFGF treatment demonstrated efficacy at reducing cerebral edema and neurological deficits after ischemia/reperfusion injury in stroke models; we were therefore motivated to investigate the effects of bFGF on BBB integrity after TBI. EB leakage, an indicator of BBB injury, was also prominent in brain specimens at 24 h after TBI (Fig. 1b). Compared to vehicle-treated animals, bFGF-treated animals had significantly reduced EB content following TBI, as demonstrated by fluorescence intensity quantification (Fig. 1c). Consistent with the EB content test, the level of FITC-dextran that leaked through the BBB was also statistically significantly increased after TBI, but reduced by treatment with bFGF (Fig. 1d). Taken together, our results suggest that bFGF treatment reduces neurological deficits by ameliorating BBB disruption following TBI.

Effects of exogenous bFGF (15 μg) or PBS on Garcia scores (a) at 24 h after TBI. Effects of bFGF on Evans Blue fluorescence (b, c) and FITC-dextran permeability (d) at 24 h after TBI. *P < 0.05 versus the sham group. # P < 0.05 versus the TBI group. Data are the mean values ± SEM, n = 6
The Protective Role of bFGF on TBI-Induced BBB Breakdown Is Mediated by the Activation of TJs, Adherens Junction Proteins, and PI3K/Akt Pathways in Mice
To determine whether bFGF protects the BBB from disruption by regulating TJ proteins, Western blot analysis of the ipsilateral brain cortex was conducted. We analyzed the effect of bFGF on the activity of TJ proteins claudin-5, occluding, and zonula occludens-1, and adherens junction (AJ) proteins p120-catenin and β-catenin, 24 h after TBI. As shown in Fig. 2, bFGF significantly increased the expression of these TJ and AJ proteins compared to the TBI control group. Consistent with the Western blot results, dual-label immunofluorescence shows that the co-localization of ZO-1, claudin-5, occludin, and the microvessel marker CD31 were markedly increased after bFGF treatment compared to vehicle-treated TBI animals (Fig. 3). These results suggest that bFGF treatment preserves BBB integrity after TBI, at least in part by increasing these TJ proteins. To further evaluate whether the PI3K/Akt pathway is involved in the neuroprotective effect of bFGF, the PI3K/Akt inhibitor LY294002 was injected into the left striatum. Neurofunctional deficits, EB fluorescence, and FITC-dextran fluorescence were analyzed at 24 h after TBI. LY294002 co-administration reversed the neuroprotection of bFGF as shown by the Garcia Test results. As expected, bFGF + LY294002 treated animals showed significant lower neuroscores than bFGF-treated animals and the bFGF + DMSO group (Fig. 4a). Furthermore, mice receiving bFGF + LY294002 showed significantly more EB dye extravasation than bFGF treated animals (Fig. 4b–d). The protein expression of TJ and AJ proteins claudin-5, occludin, zonula occludens-1, p120-catenin, and β-catenin were also tested by Western blot. As shown in Fig. 5d–f, LY294002 treatment inhibited the activation by bFGF of the levels of these junction proteins. The DMSO control group showed no significant difference compared to the bFGF group (p > 0.05). Taken together, all of these results suggested that bFGF treatment protects against BBB breakdown after TBI, at least in part by regulating the PI3K/Akt pathway.

Protein expression of claudin-5, occludin, zonula occludens-1, p120-catenin, and β-catenin for the sham, TBI, and bFGF treatment groups. GAPDH was used as the loading control and for band density normalization (a). The optical density analysis of claudin-5, occludin, zonula occludens-1, p120-catenin and β-catenin protein (b, c). *P < 0.05 versus the sham group. # P < 0.05 versus the TBI group. Data are the mean values ± SEM, n = 6

Dual-label immunofluorescence staining results of endothelial cell marker (a) CD31 (red) and different tight junction proteins (b) Claudin-5 (c) Occludin, and (d) ZO-1 in the mouse brain 1 day after TBI. The nuclei are labeled by Hoechst. Scale bar = 10 μm. The proteins with obvious bright signals are labeled. Magnification was ×40

Effects of the PI3K/Akt inhibitor LY294002 (50 nmol/kg) on bFGF induced attenuation of brain injury at 24 h after TBI. Evaluation of Garcia test (a) and Evans Blue fluorescence (b, c) and FITC-dextran permeability (d) in mice subjected to TBI. *P < 0.05 versus the sham group. # P < 0.05 versus the TBI group. &P < 0.05 versus the TBI + bFGF group. Data are the mean values ± SEM, n = 6

The protein expression of p-Akt, GTP-Rac1, and GTP-RhoA after TBI-induced BBB destruction treated with bFGF and LY294002. GAPDH was used as the loading control and for band density normalization (a). The optical density analysis of p-Akt, GTP-Rac1, and GTP-RhoA protein (b, c). The protein expression of claudin-5, occluding, ZO-1, p120-catenin, and β-catenin after TBI-induced BBB destruction treated with bFGF and LY294002. GAPDH was used as the loading control and for band density normalization (d). The optical density analysis of pclaudin-5, occluding, ZO-1, p120-catenin, and β-catenin protein (e, f). *P < 0.05 versus the sham group. # P < 0.05 versus the TBI group. &P < 0.05 versus the TBI + bFGF group. Data are the mean values ± SEM, n = 6
bFGF Treatment Inhibits RhoA Activity via the PI3K-Akt-Rac1 Signaling Pathway in Mice
RhoA and Rac1 are members of the Rho subfamily of small GTPases which play crucial roles in the regulation of cytoskeletal organization in many cell types. It has been reported that Rac1 maintains and stabilizes the barrier function of microvascular endothelial cells, whereas RhoA antagonistically impairs endothelial barrier properties. To evaluate the role of RhoA and Rac1 in the protective effect of bFGF on BBB integrity, Western blot analysis was performed using the ipsilateral hemispheres of animals 24 h after TBI and subsequent co-administration of bFGF and LY294002. The results of the quantification of all target proteins were compared to groups treated with vehicle, bFGF, and bFGF + DMSO. As shown in Fig. 5a, LY294002 treatment reversed the activation by bFGF of the protein level of p-Akt as well as the GTP-Rac-1/Total-Rac-1 ratio. The ratio of GTP-RhoA/Total-RhoA was significantly increased in the bFGF + LY group compared to the bFGF alone group. Taken together, these results demonstrate that the protective role of bFGF on BBB integrity is related to the inhibition of RhoA through activation of the PI3K/Akt/Rac-1 signaling pathway in TBI mice.
bFGF Ameliorates OGD-Induced BBB Injury and Inhibition of the PI3K/Akt Pathway Partially Reverses the Protective Effect of bFGF In Vitro
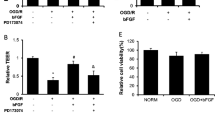
To further confirm the hypothesis that bFGF can ameliorate OGD-induced BBB injury in a cellular model, HBMECs were subjected to OGD conditions, followed by treatment with bFGF or bFGF combined with LY294002. As shown in Fig. 6a, paracellular permeability of HBMECs increased dramatically after OGD; however, bFGF administration significantly decreased the FITC-dextran permeability. On the other hand, the Akt inhibitor LY294002 partially reversed the protective effect of bFGF. In our previous study, BBB integrity was shown to be mainly dependent on the presence of TJs. Therefore, we hypothesized that OGD-induced alterations in TJs and AJs may be responsible for the increased BBB permeability. We measured the protein levels of CD31, claudin-5, occludin, zonula occludens-1, p120-catenin, and β-catenin after OGD and their expression was significantly decreased when compared with the control group. bFGF treatment significantly increased the expression of these proteins while Akt inhibitor LY294002 reversed their activation by bFGF (Fig. 6b–f). Consistent with the Western blot results, immunostaining of claudin-5, ZO-1, β-catenin, and p120-catenin proteins also showed that OGD treatment decreased expression of these TJ and AJ proteins while bFGF administration significantly increased their expression. As shown in Fig. 7, LY294002 incubation partially reversed the activation effect of bFGF on TJ and AJ proteins. All of these findings illustrate that bFGF ameliorates OGD-induced BBB destruction by regulating TJ and AJ proteins via the PI3K/Akt pathway in vitro.

HBMECs were pre-treated with 50 ng/ml bFGF with or without inhibitor LY294002 (20 μM) for 1 h, and then OGD conditions for 24 h and reperfusion for 12 h. The cells were analyzed for FITC-dextran transport (a). The cell lysates were analyzed by western blotting for the expression of CD31, claudin-5, occluding, ZO-1, p120-catenin, and β-catenin, and GAPDH was used as the loading control and for band density normalization (b, c). The optical density analysis of CD31, claudin-5, occluding, ZO-1, p120-catenin and β-catenin protein (c, e, f). *P < 0.05 versus the control group. # P < 0.05 versus the OGD group. &P < 0.05 versus the OGD + bFGF group. Data are the mean values ± SEM, n = 6

HBMECs were pre-treated with bFGF for 1 h, and then OGD conditions for 24 h, and reperfusion for 12 h. Immunofluorescence staining of confluent HBMECs monolayers for (a) claudin-5, (b) occluding, (c) p120-catenin, and (d) β-catenin. Nuclei were labeled by Hoechst. The proteins with obvious bright signals are labeled. Magnification was ×40
bFGF Treatment Preserves BBB Integrity by Inhibiting RhoA Activity via the PI3K-Akt-Rac1 Signaling Pathway
In order to evaluate whether the PI3K/Akt pathways is involved in the preservation of BBB in vitro, HBMECs under OGD conditions were treated with bFGF. Our data shows that bFGF improved the expression of p-Akt and GTP-Rac1 and decreased the expression of GTP-RhoA under OGD conditions. Co-treatment with Akt inhibitor LY294002 and bFGF significantly reversed the activation effect of bFGF on protein levels of p-Akt, GTP-Rac1, and GTP-RhoA (Fig. 8). These data suggest that the PI3K-Akt-Rac1 pathway is involved in the protective effect of bFGF and is dependent on the inhibition of RhoA.

The protein expression of p-Akt, GTP-Rac1 and GTP-RhoA in OGD-induced HBMECs treated with bFGF and LY294002 inhibitor. GAPDH was used as the loading control and for band density normalization (a). The optical density analysis of p-Akt, GTP-Rac1 and GTP-RhoA protein (b, c, d). *P < 0.05 versus the control group. # P < 0.05 versus the OGD group. &P < 0.05 versus the OGD + bFGF group. Data are the mean values ± SEM, n = 6
To further confirm the role of Rac1 in the protective effect of bFGF on TBI-induced BBB breakdown, RNA interference by transfection with Lipofectamine 2000 was used in HBMECs. The results showed that after treatment with Rac1 siRNA, the expression of TJs (claudin-5, occludin, zonula occludens-1), AJs (p120-catenin and β-catenin), and GTP-Rac1 were decreased significantly when compared with the control group while the expression of GTP-RhoA was significantly increased (Fig. 9a–d). As expected, silencingRac1 markedly reduced the stimulatory effect on these TJ and AJ protein levels by bFGF and fully abolished the inhibitory effect by bFGF on GTP-RhoA (Fig. 9a, e). All of these results further confirmed that the protective effect of bFGF on BBB integrity is mediated by inhibiting RhoA signaling through the PI3K-Akt-Rac1 pathway in vitro.

The protein expression of claudin-5, occluding, ZO-1, p120-catenin, β-catenin, GTP-Rac1 and GTP-RhoA in HBMECs was determined by Western blotting. GAPDH was used as the loading control and for band density normalization (a). The optical density analysis of all these proteins (b, c, d, e). *P < 0.05 versus the corresponds group. # P < 0.05 versus the control group. &P < 0.05 versus the OGD group. Data are the mean values ± SEM, n = 6
Discussion
Patients experience TBI as a result of traffic accidents, sports injuries, or other types of trauma, which can lead to lifelong disability and significant economic costs [30]. After TBI, the initial traumatic injury to brain tissue is followed by a long period of secondary damage including neurovascular dysfunction, inflammation, oxidative stress, and cell apoptosis [31]. The breakdown of BBB is one of the main contributors interfering brain recovery from secondary damage. The BBB normally restricts free transcellular water movement from the vascular compartment to the brain interstitium and thereby supports the restricted and closely controlled environment necessary for normal brain function [32]. Although improved emergency medicine has led to decreases in TBI mortality in recent years, many survivors suffer sustained physical disability and cognitive impairments due to the lack of defined therapies to reduce TBI-associated long-term brain damage and neurological dysfunction. In the present study, we showed that post-injury bFGF treatment robustly reduced brain damage and improved neurological functions evaluated in the mouse model of TBI. Moreover, the results presented here suggest that preserved BBB integrity is a key underlying mechanism of bFGF-afforded neuroprotection against TBI. Understanding this mechanism will be crucial for translating these results to human clinical trials, in which most subjects have chronic neurological impairments.
In line with our findings, Huang et al. demonstrated that bFGF treatment can effectively preserve BBB integrity through RhoA inhibition after intracerebral hemorrhage in mice [9]. We therefore investigated the effects of bFGF on BBB integrity after TBI in the current study. Our results support the notion that BBB breakdown plays a key role in the pathogenesis of TBI and may be a potential therapeutic target [3]. The present study reveals that bFGF treatment can improve BBB integrity and attenuate neurological deficits following TBI. Although the exact mechanism of bFGF-conferred BBB protection is unclear, our results suggest that it is likely associated with the upregulation of TJ proteins. There is compelling evidence that the upregulation of TJ proteins mediates BBB disruption after TBI [33, 34]. The present study revealed that TBI animals have increased EB leakage (Fig. 1b), neurofunctional deficits (Fig. 1a), and EB fluorescence intensities (Fig. 1c). However, bFGF treatment significantly attenuated neurofunctional deficits and reduced EB extravasation following TBI (Fig. 1a–c). Western blot results showed that bFGF also upregulates TJ proteins (claudin-5, occludin, and zonula occludens-1) and AJ proteins (p120-catenin and β-catenin) at 24 h after TBI (Fig. 2). Consistent with the Western blot results, dual-label immunofluorescence showed that co-localization between ZO-1, claudin-5, occludin, and the microvessel marker CD31 was markedly increased after bFGF treatment compared to vehicle-treated TBI animals (Fig 3). Notably, bFGF also upregulated these TJ and AJ proteins after OGD conditions in HBMECs (Figs. 6 and 7). To the best of our knowledge, this is the first study demonstrating that bFGF treatment preserves BBB integrity after TBI, at least in part by increasing these TJ proteins.
As a main downstream signal activated by bFGF, PI3K/Akt has essential neruoprotective effects [35]. The PI3K/Akt pathway is particularly important for mediating neuronal survival under a wide variety of circumstances and plays an important role in cellular angiogenesis, protein synthesis, metabolism, and proliferation [36, 37]. Activation of the PI3K/Akt pathway is also essential for growth factor-mediated cell survival. In this study, we focused on the role of the PI3K/Akt pathway in order to understand the signaling mechanisms involved in TBI-induced BBB breakdown. We demonstrated that the role of bFGF in TBI-induced BBB breakdown recovery is related to the activation of the PI3K/Akt pathway in vivo. We used the pathway inhibitor LY294002 combined with bFGF in the TBI mouse model, and the results showed significantly more dye extravasation than animals receiving bFGF treatment only; LY294002 also reversed the neuroprotective effect of bFGF (Fig. 4). To further confirm that the PI3K/Akt pathway is essential for the protective effect of bFGF on the BBB, we used HBMECs under OGD conditions (and additionally with the PI3K/Akt inhibitor LY294002) to show that the BBB breakdown induced by OGD was inhibited by bFGF treatment and further abolished by the inhibitor (Fig. 6a). In order to investigate how the PI3K/Akt signal pathway preserves BBB integrity, TJ proteins (claudin-5, occludin, and zonula occludens-1) and AJ proteins (p120-catenin and β-catenin) were detected after treatment with bFGF combined with LY294002. Western blot revealed that LY294002 treatment reversed the TJ and AJ protein upregulation effect by bFGF (Fig. 6d). Finally, our immunostaining results reinforced those shown by Western blot (Fig. 7). Taken together, all of these results suggest that bFGF ameliorates TBI or OGD-induced BBB destruction by regulating TJ and AJ proteins via the PI3K/Akt pathway.
Members of the Rho subfamily of small GTPases, including Rho, Rac, and Cdc42, play crucial roles in the regulation of cytoskeletal organization in many cell types. It has been previously reported that Rac1 maintains and stabilizes the barrier function of microvascular endothelial cells whereas RhoA antagonistically impairs endothelial barrier properties [38]. There is accumulating evidence that Rho regulates endothelial permeability, which depends on the integrity of intercellular junctions and actomyosin contractility [39]. One report showed that inhibition of Rho-kinase activity, following cerebral ischemia, may represent a viable therapeutic option to neutralize a variety of phenomena, including BBB dysfunction [40]. RhoA can disintegrate AJs through ROCK phosphorylation and has further been identified to increase actomyosin contractility which also results in breakdown of intercellular junctions. In our present study, we found that administration of bFGF increased the protein level of the GTP-Rac-1/Total-Rac-1 ratio. Additionally, the ratio of GTP-RhoA/Total-RhoA was significantly decreased after bFGF treatment (Fig. 5a). In order to figure out how bFGF regulated these Rho-kinase proteins, the inhibitor LY was co-administered with bFGF and our results showed that the protein regulation effects of bFGF were reversed by LY (Fig. 5a). The results of our in vitro study (Fig. 8) were also consistent with the results showed in vivo. To further validate the role of Rac-1 in the protective effect of bFGF, siRNA-Rac-1 was used in HBMECs. As demonstrated in Fig. 9, silencing Rac-1 partially inhibited the ability of bFGF to upregulate these TJ proteins and reversed the RhoA inhibition effect by bFGF. These results indicate that the protective function of bFGF on the BBB may be involved in the inhibition of RhoA protein and upregulation of TJ proteins via the Rac-1 pathway.
There are certainly limitations of bFGF as a therapy for TBI-induced BBB breakdown and still require further study and investigation. For example, a single dose of bFGF was administered immediately after injury; however, post-injury treatment with an optimized dose and extended treatment time would provide a better evaluation of its therapeutic value. In this study, we looked at the 1-day outcome but the long-term neurological outcomes, such as 2–4 weeks or longer, also need to be evaluated in the future. Moreover, HBMECs used in vitro are informative, yet future transwell assays of the effect of bFGF on astrocytes combined with HBMECs in co-culture would be more persuasive. Nevertheless, the neuroprotective effect of bFGF on TBI-induced BBB breakdown is confirmed and it is feasible to elucidate the pharmacodynamic properties and underlying mechanisms in future studies.
In conclusion, bFGF significantly reduced the extent of damage and preserved the BBB integrity after TBI. We first reported that the protective role of bFGF on BBB is related to the upregulation of TJ proteins and the inhibition of RhoA via Rac-1. Furthermore, activation of the downstream signaling pathway PI3K/Akt/Rac-1 is essential for the protective effect of bFGF on BBB integrity both in vivo and in vitro. Our study demonstrates that therapeutic strategies using bFGF may be suitable for recovery from TBI.
References
Maas AI, Stocchetti N, Bullock R (2008) Moderate and severe traumatic brain injury in adults. Lancet Neurol 7(8):728–741. doi:10.1016/S1474-4422(08)70164-9
Coronado VG, Xu L, Basavaraju SV, McGuire LC, Wald MM, Faul MD, Guzman BR, Hemphill JD (2011) Surveillance for traumatic brain injury-related deaths—United States, 1997-2007. MMWR Surveill Summ 60(5):1–32
Shlosberg D, Benifla M, Kaufer D, Friedman A (2010) Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol 6(7):393–403. doi:10.1038/nrneurol.2010.74
Nag S, Kapadia A, Stewart DJ (2011) Review: molecular pathogenesis of blood-brain barrier breakdown in acute brain injury. Neuropathol Appl Neurobiol 37(1):3–23. doi:10.1111/j.1365-2990.2010.01138.x
Werner C, Engelhard K (2007) Pathophysiology of traumatic brain injury. Br J Anaesth 99(1):4–9. doi:10.1093/bja/aem131
Potts MB, Koh SE, Whetstone WD, Walker BA, Yoneyama T, Claus CP, Manvelyan HM, Noble-Haeusslein LJ (2006) Traumatic injury to the immature brain: inflammation, oxidative injury, and iron-mediated damage as potential therapeutic targets. NeuroRx 3(2):143–153. doi:10.1016/j.nurx.2006.01.006
Lin Y, Pan YH, Wang ML, Huang XJ, Yin YH, Wang Y, Jia F, Xiong WH et al (2012) Blood-brain barrier permeability is positively correlated with cerebral microvascular perfusion in the early fluid percussion-injured brain of the rat. Lab Invest 92(11):1623–1634. doi:10.1038/labinvest.2012.118
Loane DJ, Faden AI (2010) Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci 31(12):596–604. doi:10.1016/j.tips.2010.09.005
Huang B, Krafft PR, Ma QY, Rolland WB, Caner B, Lekic T, Manaenko A, Le M et al (2012) Fibroblast growth factors preserve blood-brain barrier integrity through RhoA inhibition after intracerebral hemorrhage in mice. Neurobiol Dis 46(1):204–214. doi:10.1016/j.nbd.2012.01.008
Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ (2010) Structure and function of the blood-brain barrier. Neurobiol Dis 37(1):13–25. doi:10.1016/j.nbd.2009.07.030
Huber JD, Egleton RD, Davis TP (2001) Molecular physiology and pathophysiology of tight junctions in the blood-brain barrier. Trends Neurosci 24(12):719–725. doi:10.1016/S0166-2236(00)02004-X
Engelhardt B, Sorokin L (2009) The blood-brain and the blood-cerebrospinal fluid barriers: function and dysfunction. Semin Immunopathol 31(4):497–511. doi:10.1007/s00281-009-0177-0
Grieb P, Forster RE, Strome D, Goodwin CW, Pape PC (1985) O-2 exchange between blood and brain-tissues studies with O-18(2) indicator-dilution technique. J Appl Physiol 58(6):1929–1941
Luh C, Kuhlmann CR, Ackermann B, Timaru-Kast R, Luhmann HJ, Behl C, Werner C, Engelhard K et al (2010) Inhibition of myosin light chain kinase reduces brain edema formation after traumatic brain injury. J Neurochem 112(4):1015–1025. doi:10.1111/j.1471-4159.2009.06514.x
Antonetti DA, Wolpert EB, DeMaio L, Harhaj NS, Scaduto RC (2002) Hydrocortisone decreases retinal endothelial cell water and solute flux coincident with increased content and decreased phosphorylation of occludin. J Neurochem 80(4):667–677. doi:10.1046/j.0022-3042.2001.00740.x
Jiang WG, Bryce RP, Horrobin DF, Mansel RE (1998) Regulation of tight junction permeability and occludin expression by polyunsaturated fatty acids. Biochem Bioph Res Co 244(2):414–420. doi:10.1006/bbrc.1998.8288
Harhaj NS, Antonetti DA (2004) Regulation of tight junctions and loss of barrier function in pathophysiology. Int J Biochem Cell B 36(7):1206–1237. doi:10.1016/j.biocel.2003.08.007
Wachtel M, Bolliger MF, Ishihara H, Frei K, Bluethmann H, Gloor SM (2001) Down-regulation of occludin expression in astrocytes by tumour necrosis factor (TNF) is mediated via TNF type-1 receptor and nuclear factor-kappa B activation. J Neurochem 78(1):155–162. doi:10.1046/j.1471-4159.2001.00399.x
Wang ZG, Zhang HY, Xu XL, Shi HX, Yu XC, Wang XJ, Yan YB, Fu XB et al (2012) bFGF inhibits ER stress induced by ischemic oxidative injury via activation of the PI3K/Akt and ERK1/2 pathways. Toxicol Lett 212(2):137–146. doi:10.1016/j.toxlet.2012.05.006
Wang ZG, Wang Y, Huang Y, Lu Q, Zheng L, Hu D, Feng WK, Liu YL, et al. (2015) bFGF regulates autophagy and ubiquitinated protein accumulation induced by myocardial ischemia/reperfusion via the activation of the PI3K/Akt/mTOR pathway. Sci Rep-Uk 5. doi:Artn 9287. Doi 10.1038/Srep09287
Zhang HY, Wang ZG, Wu FZ, Kong XX, Yang J, Lin BB, Zhu SP, Lin L et al (2013) Regulation of autophagy and ubiquitinated protein accumulation by bFGF promotes functional recovery and neural protection in a rat model of spinal cord injury. Mol Neurobiol 48(3):452–464. doi:10.1007/s12035-013-8432-8
Murakami M, Nguyen LT, Zhang ZW, Moodie KL, Carmeliet P, Stan RV, Simons M (2008) The FGF system has a key role in regulating vascular integrity. J Clin Invest 118(10):3355–3366. doi:10.1172/Jci35298
Lee JG, Kay EP (2011) PI 3-Kinase/Rac1 and ERK1/2 regulate FGF-2-mediated cell proliferation through phosphorylation of p27 at Ser10 by KIS and at Thr187 by Cdc25A/Cdk2. Invest Ophth Vis Sci 52(1):417–426. doi:10.1167/Iovs.10-6140
Kamura S, Matsumoto Y, Fukushi J, Fujiwara T, Iida K, Okada Y, Iwamoto Y (2010) Basic fibroblast growth factor in the bone microenvironment enhances cell motility and invasion of Ewing's sarcoma family of tumours by activating the FGFR1-PI3K-Rac1 pathway. Brit J Cancer 103(3):370–381. doi:10.1038/sj.bjc.6605775
Wojciak-Stothard B, Ridley AJ (2003) Shear stress-induced endothelial cell polarization is mediated by Rho and Rac but not Cdc42 or PI 3-kinases. J Cell Biol 161(2):429–439. doi:10.1083/jcb.200210135
Zhang MY, Shan HY, Wang T, Liu WL, Wang YQ, Wang L, Zhang L, Chang P et al (2013) Dynamic change of hydrogen sulfide after traumatic brain injury and its effect in mice. Neurochem Res 38(4):714–725. doi:10.1007/s11064-013-0969-4
Luo CL, Chen XP, Yang R, Sun YX, Li QQ, Bao HJ, Cao QQ, Ni H et al (2010) Cathepsin B contributes to traumatic brain injury-induced cell death through a mitochondria-mediated apoptotic pathway. J Neurosci Res 88(13):2847–2858. doi:10.1002/Jnr.22453
Garcia JH, Wagner S, Liu KF, Hu XJ (1995) Neurological deficit and extent of neuronal necrosis attributable to middle cerebral-artery occlusion in rats—statistical validation. Stroke 26(4):627–634
Lo EH, Dalkara T, Moskowitz MA (2003) Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci 4(5):399–415. doi:10.1038/Nrn1106
Zlokovic BV (2008) The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57(2):178–201. doi:10.1016/j.neuron.2008.01.003
Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB (2014) Transcranial amelioration of inflammation and cell death after brain injury. Nature 505(7482):223–8. doi:10.1038/Nature12808
Chodobski A, Zink BJ, Szmydynger-Chodobska J (2011) Blood-brain barrier pathophysiology in traumatic brain injury. Transl Stroke Res 2(4):492–516. doi:10.1007/s12975-011-0125-x
Vajtr D, Benada O, Kukacka J, Prusa R, Houstava L, Toupalik P, Kizek R (2009) Correlation of ultrastructural changes of endothelial cells and astrocytes occurring during blood brain barrier damage after traumatic brain injury with biochemical markers of blood brain barrier leakage and inflammatory response. Physiol Res 58(2):263–268
Higashida T, Kreipke CW, Rafols JA, Peng CY, Schafer S, Schafer P, Ding JY, Dornbos D et al (2011) The role of hypoxia-inducible factor-la, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury Laboratory investigation. J Neurosurg 114(1):92–101. doi:10.3171/2010.6.Jns10207
Zhang HY, Zhang X, Wang ZG, Shi HX, Wu FZ, Lin BB, Xu XL, Wang XJ et al (2013) Exogenous basic fibroblast growth factor inhibits ER stress-induced apoptosis and improves recovery from spinal cord injury. CNS Neurosci Ther 19(1):20–29. doi:10.1111/cns.12013
Hossain MS, Ifuku M, Take S, Kawamura J, Miake K, Katafuchi T (2013) Plasmalogens rescue neuronal cell death through an activation of AKT and ERK survival signaling. Plos One 8(12):e83508. doi:10.1371/journal.pone.0083508
Zhao J, Cheng YY, Fan W, Yang CB, Ye SF, Cui W, Wei W, Lao LX et al (2015) Botanical drug Puerarin coordinates with nerve growth factor in the regulation of neuronal survival and neuritogenesis via activating ERK1/2 and PI3K/Akt signaling pathways in the neurite extension process. CNS Neurosci Ther 21(1):61–70. doi:10.1111/Cns.12334
Gerhard R, John H, Aktories K, Just I (2003) Thiol-modifying phenylarsine oxide inhibits guanine nucleotide binding of Rho but not of Rac GTPases. Mol Pharmacol 63(6):1349–1355. doi:10.1124/mol.63.6.1349
Zandy NL, Playford M, Pendergast AM (2007) Abl tyrosine kinases regulate cell-cell adhesion through Rho GTPases. Proc Natl Acad Sci U S A 104(45):17686–17691. doi:10.1073/pnas.0703077104
Gibson CL, Srivastava K, Sprigg N, Bath PM, Bayraktutan U (2014) Inhibition of Rho-kinase protects cerebral barrier from ischaemia-evoked injury through modulations of endothelial cell oxidative stress and tight junctions. J Neurochem 129(5):816–826. doi:10.1111/jnc.12681
Author information
Authors and Affiliations
Corresponding authors
Additional information
Zhou-Guang Wang, Yi Cheng and Xi-Chong Yu contributed equally to this work.
Rights and permissions
About this article

Cite this article
Wang, ZG., Cheng, Y., Yu, XC. et al. bFGF Protects Against Blood-Brain Barrier Damage Through Junction Protein Regulation via PI3K-Akt-Rac1 Pathway Following Traumatic Brain Injury. Mol Neurobiol 53, 7298–7311 (2016). https://doi.org/10.1007/s12035-015-9583-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9583-6