
Abstract
Glioblastoma is an extremely aggressive and deadly brain tumor known for its striking cellular heterogeneity and capability to communicate with microenvironment components, such as microglia. Microglia-glioblastoma interaction contributes to an increase in tumor invasiveness, and Wnt signaling pathway is one of the main cascades related to tumor progression through changes in cell migration and invasion. However, very little is known about the role of canonical Wnt signaling during microglia-glioblastoma crosstalk. Here, we show for the first time that Wnt3a is one of the factors that regulate interactions between microglia and glioblastoma cells. Wnt3a activates the Wnt/β-catenin signaling of both glioblastoma and microglial cells. Glioblastoma-conditioned medium not only induces nuclear translocation of microglial β-catenin but also increases microglia viability and proliferation as well as Wnt3a, cyclin-D1, and c-myc expression. Moreover, glioblastoma-derived Wnt3a increases microglial ARG-1 and STI1 expression, followed by an upregulation of IL-10 mRNA levels, and a decrease in IL1β gene expression. The presence of Wnt3a in microglia-glioblastoma co-cultures increases the formation of membrane nanotubes accompanied by changes in migration capability. In vivo, tumors formed from Wnt3a-stimulated glioblastoma cells presented greater microglial infiltration and more aggressive characteristics such as growth rate than untreated tumors. Thus, we propose that Wnt3a belongs to the arsenal of factors capable of stimulating the induction of M2-like phenotype on microglial cells, which contributes to the poor prognostic of glioblastoma, reinforcing that Wnt/β-catenin pathway can be a potential therapeutic target to attenuate glioblastoma progression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioblastomas (GBMs) are the most aggressive human brain tumors with low survival rates due to the resistance of these tumors to standard treatments [1, 2]. They exhibit extense heterogeneity that is believed to contribute to their resistance and recurrence [3,4,5,6]. Specifically, glioma stem-like cells (GSC) ensure the production of several distinct cell phenotypes conferring an intrinsic heterogeneity to GBMs. Currently, the role of tumor microenvironment during GBM development has been highlighted. The extrinsic heterogeneity, generated by a group of cells that interact with the tumor, has distinct origins (i.e., microglia, macrophages, and endothelial cells) and also plays a major role on GBM development and aggressiveness [7]. The analysis of glioma biopsies and mice xenotransplants showed that tumor mass is composed of 30–50% of infiltrated microglia/macrophages [8,9,10].
It has been shown that the tumor cells create an immunosuppressive microenvironment to evade the immune system [9, 11,12,13], and in part, it relies on the release of cytokines such as IL-6, IL-10, TGF-β, and other growth factors [11, 14,15,16]. These molecules induce the acquisition of an M2-like phenotype by the microglial cells which, in turn, do not recognize tumor cells due to their low levels of MHC expression. [17]. On the other hand, microglial cells participate actively during tumorigenesis promoting survival and tumor growth through the production of interleukins, cytokines, growth factors, and metalloproteinases, i.e., IL-10, TGF-ß, MMP-2, and MMP-9 [18]. Several studies showed that microglia exhibit considerable plasticity concerning the acquisition of distinct phenotypes, and just like macrophages, they can switch between M1/M2 profiles depending on the stimuli received or the pathological context [17, 19]. It is already known that microglia surrounding tumors are CD11b+ and CD45−, and they express Arginase-1 (ARG-1), which is correlated with tumor proliferation [20]. Similarly, stress-inducible protein 1 (STI1) is released by microglia and induces tumor proliferation and migration through MMP-9 production [21]. Moreover, an in vivo study showed that tumor progression was accompanied by increased expression of STI1 in GFP-CX3C chemokine receptor 1 (CX3CR1GFP) microglial cells [9]. However, the microglia-GBM crosstalk is still far from being completely understood.
The Wnt/β-catenin signaling pathway is activated in gliomas and may play a pivotal role in carcinogenesis and progression of these tumors. It has been demonstrated that GBMs present an aberrant activation of Wnt signaling pathway, which is related to increased levels of Wnt3a expression when compared to normal brain tissue and astrocytomas of lower grades, mainly grades I and III. Moreover, β-catenin and the transcription factor 4 (TCF4) levels are exclusively expressed by higher-grade tumors, such as anaplastic astrocytomas and GBMs, suggesting a role for Wnt/β-catenin in tumor aggressiveness and malignancy [22].
Furthermore, new evidences suggest that Wnt signaling pathway is involved in the modulation of microglia activity [23, 24]. Microglia expresses high levels of Wnt receptors such as FDZ4, FDZ5, FDZ7, FDZ8, and their co-receptor LRP5/6 when stimulated with Wnt3a [23]. In silico analysis of The Cancer Genome Atlas (TCGA), transcript profiling of human GBM samples showed that the high expression of Wnt5 is correlated with increased levels of CX3CR1 (CX3 chemokine receptor R1), CD163, and Iba-1 receptors, which are frequently expressed by microglial cells. This data suggests that the activation of non-canonical Wnt signaling in tumors is associated with the presence and infiltration of microglial cells and also with the inflammatory response, due to the activation of signaling pathways such as Toll-like receptor (TLR) [25]. However, the role of canonical Wnt signaling has in microglia-GBM interactions is not clarified, more specifically the Wnt3a. We then hypothesized that the Wnt3a expressed by GBM cells could trigger the recruitment of microglial cells with an M2-like phenotype. Here, we present the first evidence that Wnt3a plays a major role in microglia-GBM crosstalk contributing to the formation of tumor extrinsic heterogeneity.
Material and Methods
Reagents
All culture reagents as well as the secondary antibodies, conjugated to Alexa Fluor 488, 546, 633, 647, and streptavidin-Cy3, were obtained from Life Technologies (Carlsbad, CA, USA). The Alexa Fluor 488 phalloidin was acquired from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Protease and phosphatase inhibitors and the Bromodeoxyuridine kit (BrdU) were purchased from Roche (Indianapolis, IN, USA). β-catenin antibody, anti-α-tubulin and the 4-6-diamino-2-phenylindole (DAPI) were purchased from Sigma-Aldrich Corp. (St. Louis, MO, USA). Glial fibrillary acidic protein and vimentin antibodies were purchased from DAKO (Glostrup, Denmark). Arginase-1, cyclin-D1, and c-myc antibodies were all purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). GAPDH and STI1 antibodies were purchased from Abcam (Cambridge, MA, USA). Wnt3a recombinant protein and Wnt3a human/mouse antibodies were purchased from R&D (Merck, Darmstadt, Germany). Biotinylated Griffonia simplicifolia lectin I (GSL) Isolectin B4 (IB4) was obtained from Vector (Burlingame, CA, USA).
Ethics Statement and Mice
This study was approved by the Ethics Committee at the Center for Health Sciences in the Federal University of Rio de Janeiro (Protocol No. 001/16) and by the Brazilian Ministry of Health Ethics Committee (CONEP Protocol No. 2340). The protocol on “Guide for the Care and Use of Laboratory Animals” (published by the National Academy of Science, National Academy Press, Washington, DC) was strictly followed in all experiments. Efforts were made to minimize the number of animals used and their suffering. Swiss mice were obtained from the Institute of Biomedical Sciences and the Bio Rio Foundation, Federal University of Rio de Janeiro.
Glioblastoma and N9 Cultures
The GBM02 and GBM95 glioblastoma cell lines were established and characterized in our laboratory, as previously described [5, 26, 27]. The GBM cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM/F12) supplemented with 10% fetal bovine serum (FBS), at 37 °C and in a controlled atmosphere containing 5% CO2.
The N9 microglial cell line was kindly provided by Dr. Behnam Badie and was maintained in DMEM/F12 supplemented with 5% FBS, at 37 °C and in a controlled atmosphere containing 5% CO2.
All cell lines were submitted to mycoplasma test using MycoAlert™ Mycoplasma Detection Kit (Lonza Group Ltd. Basel Switzerland). The GBM cell lines were used until 20 passages and microglial lineage was used until 10 passages after unfrozen vial.
Microglial Cultures
Microglial cultures were obtained from the brain cortex of newborn Swiss mice as previously described [28]. The cerebral tissues were dissociated in DMEM/F12 medium supplemented with 3.5 mg/ml glucose, 0.1 mg/ml penicillin, and 0.14 mg/ml streptomycin after removal of the meninges.
The cells were cultured in DMEM/F12 supplemented with 3.5 mg/ml glucose, 0.1 mg/ml penicillin, 0.14 mg/ml streptomycin, and 10% fetal bovine serum (FBS). First, the 75-cm2 culture flasks (Corning Glass) were pretreated with poly-lysine (Sigma Aldrich, St. Louis, MO, USA), and then, the cells were cultured at 37 °C and in a controlled atmosphere of 5% CO2 for 12 days. After 12 days in culture, microglial cells were isolated from astrocytes by shaking for 45–60 min and re-plating in DMEM/F12 medium supplemented with 5% FBS on glass coverslips at a density of 1 × 105 cells/well for immunostainings or on 6-well plates at a density of 50 × 105 cells/well for Western blottings. For lipopolysaccharide (LPS) treatments, microglial cells were treated with 1 μg/ml of LPS (Sigma Aldrich, St Louis, MO, USA) for 24 h.
MTT Assays
Cell viability was assessed using the 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) reduction colorimetric assay. Briefly, cells were plated onto 96-well plates at 0.2 × 104 cells/well and incubated for 24 h with either the conditioned medium originated from control GBM (called GBM-CM Wnt3a−) or the conditioned medium originated from GBM pretreated with Wnt3a (called GBM-CM Wnt3a+). The final concentration of 5% FBS was used in all conditions. Then, MTT (5 mg/ml) was added to each well at a final concentration of 10% and left for 2 h. The blue formazan crystals produced were dissolved by adding 100 μl of DMSO. The absorbance of the cell culture medium was measured by a microplate reader at 570 nm. The viability was analyzed using GraphPad Prism (GraphPad Software, Inc., San Diego, CA, USA).
BrdU Incorporation Assay
Microglial cells were treated for 24 h with GBM-CM. Their proliferation capacities were determined by quantification of the BrdU incorporation into the DNA of replicating cells using the Cell Proliferation ELISA, following the manufacturer’s instructions (Cell Proliferation ELISA for BrdU, Roche).
For the BrdU incorporation experiments, cells previously treated for 24 h with both GBM-CMs were incubated with BrdU labeling solution (0.1 μl/ml) for 120 min at 37 °C in a humidified atmosphere (5% CO2). The cells were then incubated with FixDenat solution and with anti-BrdU POD (anti-BrdU-FLUOS) according to the manufacturer’s instructions (Roche, USA). Colorimetric analyses were performed using a VICTOR X3 multi-label plate reader, and the absorbances were determined at 450 nm (Perkin-Elmer, Waltham, MA, USA).
Immunocytochemistry
GBM and microglial cells were cultured on coverslips in a 24-well plate. After 24 and 48 h of incubation in the absence or presence of treatment, the cells were fixed with 4% PFA/PBS for 20 min, permeabilized with 0.1% Triton X-100/PBS, and blocked with 5% BSA/PBS for 1 h. For the analysis of the expression of Wnt/β-catenin components (Wnt3a and β-catenin), M2 phenotype markers (Arg1 and STI1), and cytoskeleton proteins (F-actin and IB4) by immunofluorescence, the cells were incubated overnight at 4 °C with anti-wnt3a (1:200), anti-β-catenin (1:1000), anti-arginase 1(1:200), anti-STI1 (1:200), and IB4 (1:500), and 2 h with Alexa Fluor 555 phalloidin staining solution (1:400). Thereafter, the cells were washed with PBS and incubated with secondary antibodies conjugated with Alexa Fluor 488, 546, or 633 (1:400) and streptavidin-Cy3 (1:200) for 2 h at room temperature. Cells were then washed with PBS, stained with DAPI, and mounted in Fluoromount-G®. Negative controls were prepared with non-immune rabbit or mouse IgG. Cells were imaged at 40× and 63× using a DMi8 advanced fluorescence microscope (Leica Microsystems, Germany) and analyzed with Leica LA SAF Lite. Images were processed using the software ImageJ 1.49v (Wayne Rasband, National Institutes of Health, USA).
Immunoblotting
GBM and microglial cells were cultured in 6-well plates and incubated for 24 h in the presence of specific treatments. The cells were then washed with PBS and detached using a cell scraper, after which lysis buffer was added (10 mM Tris base, 0.25 M saccharose, and 1 mM EDTA, in the presence of protease inhibitors). The lysates were sonicated and then centrifuged at 4 °C, 10.000g for 10 min. The supernatants were analyzed for protein content using a bicinchoninic acid assay (BCA) kit (Bradford from BioRad.).Western blotting was performed, as described by Towbin et al. and with minor modifications [29]. For the immunodetection of proteins, 30 μg of total cell proteins were separated by electrophoresis on 10% SDS polyacrylamide gels and transferred to polyvinylidene difluoride (PVDF) membranes. The PVDF membranes were then blocked with 5% non-fat milk in Tris-buffered saline with 0.1% Tween-20 (TBS-T) for 1 h, incubated with specific primary antibodies overnight at 4 °C, washed with TBS-T, and incubated with peroxidase-conjugated antibodies. The signals of Wnt3a (1:1000, R&D), β-catenin (1:2000, Sigma), Arg-I (1:1000, Thermoscientific), STI1 (1:1000, Abcam), cyclin-D1 (1:1000, Cell Signaling), c-myc (1:1000, Cell Signaling Technology), α-tubulin (1:2000, Sigma) and GAPDH (1:5000, Abcam) were detected using chemiluminescence (ECL) in ChemiDoc MP imaging system (BioRad, Benicia, CA, USA). The densitometry analyses were performed using ImageJ software, and the values obtained represent the ratio between the immunodetected protein and the loading control (GAPDH or α-tubulin).
qPCR
Total RNA was extracted from GBM, primary microglial, and N9 cells using Purelink RNA Mini kit (Thermo Fisher Scientific) following the manufacturer’s instructions. One microgram of the total RNA, oligodT (12–18) primer (Thermo Fisher Scientific), and High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) were used to perform the cDNA synthesis. For quantitative PCR (qPCR) using the Power Sybr Green Master Mix (Thermo Fisher Scientific), we utilized 20 ng of cDNA per well of the microglial and N9 samples. The primers used were designed as follows: the mouse IL-10 primers (forward: 5′-ACCTGGTAGAAGTGATGCCC-3′; reverse: 5′-GAAATCGATGACAGCGCCTC-3′), the IL-1β primers (forward: 5′-TACAAGGAGAACCAAGCAACGA-3′; reverse: 5′-TGCCGTCTTTCATTACACAGGA-3′), the TNF-α primers (forward: 5′-AAGTTCCCAAATGGCCTCCC-3′; reverse: 5′- TGGTGGTTTGCTACGACGTG-3′), and β-actin primers (forward:5′-TGGATCGGTGGCTCCATCCTGG-3′; reverse:5′-GCAGCTCAGTAACAGTCCGCCTAGA-3′) and were used as the endogenous genes. Thermal cycling was carried out using CFX96 Touch Real-Time PCR detector (BioRad) according to the manufacturer’s recommendations. The relative fold variation in mRNA expression was calculated using the Pfaff method (Pfaff, 2011). The data was obtained from three independent experiments and analyzed using ANOVA.
Time-Lapse Video Microscopy
Video microscopies of GBM and microglia co-cultures were performed using culture-insert 2 well in μ-dish 35 mm (IBIDI, Germany) which were treated with Wnt3a at 10 ng/ml. Each of the samples was placed in a culture chamber with controlled temperature and CO2 environment (37 °C and 5%, respectively), attached to Nikon Eclipse TE300. During 24 h, phase contrast images were captured every minute using a Hamamatsu C2400 CCD camera (Hamamatsu, Japan). The wound healing was quantified after 12 and 24 h using MRI wound healing tool from Image J.
Scanning Electron Microscopy
GBM and microglia co-cultures were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2) for 1 h, washed in the same buffer and then dehydrated in an ethanol series ranging from 30 to 100%. Finally, the samples were critical point dried, coated with a thin gold layer in a Balzers gold sputtering system, and observed in a FEI-Quanta scanning electron microscope.
GBM Xenografts
Xenografts were performed as previously described [8]. Female Swiss mice of 10–14 weeks of age weighing 30–35 g were used. Mice were anesthetized with diazepam (5 mg/kg i.m.), ketamine (100 mg/kg i.m.), and xylazine (25 mg/kg i.m.), and then a brain midline incision was made on the scalp. A small hole was drilled in the skull at stereotaxic coordinates in the striatum of immunocompetent mice: 1 mm posterior to the bregma and + 2 mm mediolateral from the midline. 5 × 105 GBM cells were delivered in 3 μl DMEM-F12 at a depth of 3 mm with a Hamilton (Hamilton, Reno, Nevada, USA) syringe over 30 min. Animals were followed, and analysis was done 2 weeks after tumor cell injection. Three animals per group, (i) control—GBM cells non-treated with wnt3 (wnt3a−); (ii) treated group—GBM cells pre-treated with Wn3a (Wnt3a+), were used for each experiment described below.
Mouse Tissue Processing
The mice brains were dissected 2 weeks after xenotransplantation followed by overnight fixation in 4% PFA at 4 °C of which storage was done in PBS before processing. The brains were dehydrated in graded ethanol series (30, 40, 50, and 60% for 30 min each; then emerged in 70, 80, and 90% for 1 h; then 100% twice for 1 h each), being left overnight in xylene at room temperature. Next, the tissues were embedded in paraffin for 3 h at 67 °C. The brain sections were cut (5 um thick) on a microtome. The sections were stained with hematoxylin and eosin and photographed using DMi1 by Leica Microsystems.
Human Tissue Processing
A total of four different human brain samples of astrocytoma grade IV (GBM) were acquired from patients admitted in the Neuropathology Service of the Instituto Estadual do Cérebro Paulo Niemeyer (Rio de Janeiro, Brazil). The study was approved by Instituto Estadual do Cérebro Paulo Niemeyer Ethics Committee according to Helsinki Declaration protocol, whereby all patients signed an informed consent for the use of their biological samples for research investigation. The GBMs were fixed in 4% paraformaldehyde for 12 to 24 h, embedded in paraffin, and cut into 5-μm sections. The hematoxylin-eosin images obtained by DMi1 from Leica are representatives of the three independent experiments.
Immunohistochemistry
The brain sections after deparaffinization were washed with PBS and incubated with 10% NGS diluted in PBS with 0.3% Triton X-100 for 90 min. They were then incubated with GFAP (1:400), vimentin (1:400), Wnt3a (1:500) antibodies, and with biotinylated IB4 (1:100) overnight at 4 °C then washed again with PBS and incubated with secondary antibodies conjugated with Alexa Fluor 488, 546, or 633 (1:400) and streptavidin-Cy3 (1:200) for 2 h. The sections were counterstained with DAPI and coverslipped with fluoromount. Negative controls were performed with non-immune rabbit IgG. Slices were imaged using DMi8 advanced fluorescence microscope (Leica Microsystems, Germany). The plug-in Fibril Tool for ImageJ (NIH, USA) was used to image processing.
Statistical Analysis
All values were expressed as mean ± SD. The groups were compared by means of one-way ANOVA test, Dunnet’s test, and Bonferroni probabilities (except for qPCR, as mentioned above), with a significance threshold of p ≤ 0.05. All statistical analyses were performed using GraphPad Prism 6 (GraphPad Software Inc., San Diego, CA, USA).
Results
GBM and Microglial Cells Activate Wnt/ß-Catenin Pathway upon Wnt3a Stimulation
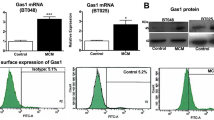
Our previous work showed that GBM cells present constitutive activation of Wnt/ß-catenin signaling [30]. Moreover, high levels of Wnt3a have already been described as activators of microglial cells in other contexts, such as neurodegenerative diseases [23, 24]. Wnt3a is known to stabilize β-catenin, the downstream protein involved in canonical Wnt signaling, and increases its translocation to the nucleus [31]. Here, we addressed whether the stimulation with recombinant Wnt3a protein could induce the activation of Wnt/ß-catenin signaling in GBM and microglial cells through the enhancement of the expression and rearrangement of the β-catenin subcellular localization and also upregulation of Wnt3a levels produced by GBM and microglial cells. Both cells were stimulated by Wnt3a at 10 ng/ml during 24 h, and immunofluorescence data showed that the β-catenin was translocated to the nucleus following Wnt3a administration (Fig. 1a–c). We also observed increased β-catenin and Wnt3a levels after stimulation with Wnt3a as verified by Western blot analysis when compared to controls (Fig. 1d–f). Moreover, we assessed the expression of Wnt target genes which have been associated with activation of Wnt/β-catenin pathway, such as c-myc and cyclin-D1 [32]. The expression of c-myc and cyclin-D1 was evaluated by Western blot analysis (Fig. 1g). The c-myc and cyclin-D1 expression was increased in N9 cells after Wnt3a stimulation. Our data suggests that Wnt3a is capable of inducing the activation of Wnt/β-catenin pathway in GBM and microglial cells, as compared to control, despite their constitutive activation.

Wnt3a induces activation of Wnt/β-catenin pathway in GBM and microglial cells. The components of Wnt/ β-catenin pathways in GBM cell line, microglia cell line (N9), and primary microglial cells were assessed by immunofluorescence and Western blot analysis. The expression of β-catenin increases after Wnt3a stimulation in GBM cells. The β-catenin translocation into the nucleus was observed by colocalizing signal of β-catenin (red) with DAPI (blue) in GBM cells (a), in N9 cells (b), and in primary microglia culture (microglia) (c) when compared with the control as verified by immunofluorescence. β-catenin and Wnt3a expression were detected by Western blot analysis in GBM cells (d), in N9 cells (e), and in microglia (f). The levels of Wnt target proteins c-myc and cyclin-D1 were detected in Wnt3a recombinant protein-treated N9 cells by Western blot (g). The β-catenin and Wnt3a were revealed using specific antibodies, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or a-tubulin was used as the loading control. The 30 μg of protein was loaded in each lane. Each value represents the mean ± SD of three independent experiments. *p < 0.05. Scale bar = 50 μm
Wnt3a GBM-Derived Induces Proliferation and Activation of Wnt/β-Catenin Signaling in Microglial Cells
We next verified whether GBM cells could release Wnt3a to the medium. GBM cells present a basal secretion of Wnt3a (Fig. 2a). Moreover, we verified that GBM cells pretreated with Wnt3a protein at 10 ng for 24 h (Wnt3a+) enhance this release into the medium (Fig. 2a). This result led us to prompt whether the conditioned medium produced by control GBM cells (GBM-CM Wnt3−) or GBM cells pretreated with Wnt3a (GBM-CM Wnt3+) could increase the viability (Fig. 2b) and proliferation (Fig. 2c) of microglial cells. We observed that both microglial cell types (N9 cell line and primary microglia cultures) increased the viability in the presence of GBM-CM Wnt3a+ when compared to GBM-CM Wnt3−. This result is in agreement with the increase in BrdU incorporation of microglial cells treated with GBM-CM Wnt3a+ as compared with the GBM-CM Wnt3a− (Fig. 2c), suggesting that GBM-CM Wnt3a+ increases microglial cell proliferation.

Wnt3a produced by GBM cells induces proliferation and activation of Wnt/β-catenin in microglial cells. GBM cells pretreated with 10 ng of recombinant Wnt3a during 24 h increased the expression and release of Wnt3a in the medium (GBM-CM Wnt3+) as compared with the normal conditioned medium (GBM-CM Wnt3−) (a). N9 cells and primary microglial cultures (microglia) were treated with both GBM-CM for 24 h, and the cell viability was determined by MTT assay (b) whereas the proliferation was assessed by BrdU incorporation (c). β-catenin and Wnt3a expression was observed under treatment with GBM-CM Wnt3− and GBM-CM Wnt3+ through immunofluorescence in N9 (d) and microglia (e) and also by Western blot (f, g). The expression of Wnt target proteins, cyclin-D1 and c-myc, was observed by Western blot analysis (h). Each value represents the mean ± SD of three independent experiments. *p < 0.05, **p < 0.01, and ***p < 0.001, compared to the control groups. Scale bar = 50 μm
We further evaluated if both GBM-CM Wnt3a− and GBM-CM Wnt3a+ could change the microglia β-catenin and Wnt3a levels by immunofluorescence (Fig. 2d, e) and Western blot analysis (Fig. 2f, g) in microglia cells. We found that GBM-CM Wnt3+-stimulated microglial cells express higher levels of β-catenin and Wnt3a when compared to the microglia treated with GBM-CM Wnt3−. The expression of Wnt target proteins, such as c-myc and cyclin-D1, was evaluated by Western blot analysis (Fig. 2h). The c-myc and cyclin-D1 expression was increased in N9 cells after GBM-CM Wnt3a+ stimulation.
These results show that the Wnt3a-treated GBM cells increase Wnt signaling by releasing high levels of Wnt3a into the medium. In turn, the GBM-CM Wnt3a+ strongly stimulates the Wnt signaling on microglial cells, compared to control.
Induction of M2-Like Phenotype in Microglia Treated with Wnt3a GBM-Derived
In some neurodegenerative diseases, such as Alzheimer’s disease, microglia present constitutively activated Wnt/β-catenin pathway and produce high levels of Wnt3a [23, 24]. It has been demonstrated that LiCl, an inhibitor of GSK3, induces Wnt/β-catenin activation [33]. Moreover, LiCl attenuates the M1 phenotype and induces the M2-like phenotype by increasing IL10 expression [34]. Since increased levels of Arg-I have been associated with M2-like phenotype [20, 35] and STI1 is produced by microglial cells to promote tumor proliferation, as previously described by our group [9, 21], we next determined whether GBM-derived Wnt3a could affect the expression of Arg-1 and STI1 in microglial cells. The N9 and primary microglial cells were treated with GBM-CM Wnt3+ or GBM-CM Wnt3− for 24 h, and the Arg-I and STI1 levels were assessed by immunofluorescence and Western blot analysis. Our data showed that GBM-CM Wnt3+-treated microglial cells increased Arg-1 and STI1 levels as compared to control and GBM-CM Wnt3a− (Fig. 3a, b). Furthermore, the expression of inflammatory cytokines involved in M1, such as IL-1β and TNF-α, and M2-like phenotype, such as IL-10, was measured by RT-qPCR in GBM-CM-treated groups and compared with LPS-treated N9 cells. The RT-qPCR results suggested that GBM-CM Wnt3a+ decreases the IL-1β expression (Fig. 3c). However, the levels of TNF-α remained unchanged of TNF-α remained unchanged after both treatments (Fig. 3d). On the other hand, the GBM-CM Wnt3a+ was effective in inducing the M2-like phenotype by increasing the mRNA levels of IL-10 (Fig. 3e). All these data suggest that Wnt3a expressed and secreted by GBM cells can modulate the phenotype of microglial cells, turning them into a M2-like phenotype.

Induction of M2-like phenotype in microglia treated with GBM-derived Wnt3a. After 24 h, immunofluorescence (a) and Western blot (b) were performed using specific antibodies against STI1 and Arg-1 to evaluate the effect of CM-GBM in microglia cells. GAPDH was used as the loading control. The mRNA levels of interleukins involved in M1 and M2-like phenotype were detected by qPCR in N9 cells after CM-GBMs stimulation and LPS (c–e). Each value represents the mean ± SD of three independent experiments. *p < 0.05, compared to the untreated cells. Scale bar = 50 μm
Wnt3a Increases Microglial and GBM Interaction
The Wnt/β-catenin pathway regulates the metastasis-associated phenotypes in cancer cells [36]. Besides, the aberrant migration is necessary for the invasiveness and metastasis of the tumor cells [37]. Moreover, to acquire migration abilities, tumor cells need cytoskeleton reorganization [37]. It has been demonstrated that Wnt3a induces the rearrangement of the actin filaments (F-actin), which play a pivotal role in cell motility [38, 39]. According to other studies, the Wnt3a is essential not only for the motility of tumor cells [40] but also for microglia motility [23], and its knockdown in GBM cells reduces tumor migration ability [40]. Thus, we next evaluated the changes in the cytoskeleton by observing F-actin and IB4 in co-cultures of GBM and microglial cells treated with Wnt3a. Our results showed an increased density of F-actin in Wnt3a-treated cells as compared to control cells (Fig. 4a–d). We also observed F-actin-based nanotubular projections, known as tunneling nanotubes (TNTs) or intercellular nanotubes, between GBM and microglial cells. Wnt3a increased the formation of TNTs between GBM and microglial cells (Fig. 4e–h). These structures have been described as a mechanism of cell-cell communication that promotes the exchange of vesicles and cytoplasmic materials, signals like microRNAs, chemokines, and oncogenes between cells [41,42,43]. The observation that Wnt3a promotes the reorganization of cytoskeleton led us to prompt whether Wnt3a could also modulate GBM and microglia migration capacities. Through 24 h of time-lapse microscopy, we observed that the migration of co-cultured cells was increased by the presence of Wnt3a (Fig. 4i–j). These findings support our hypothesis that Wnt3a plays a key role in microglia and GBM migration and interaction.

Wnt3a enhances microglial and GBM interaction. Co-cultures of microglial and GBM cells were performed to observe the interaction between both cells after 24 h of Wnt3a treatment. F-actin (red) and IB4 (green) were labeled and observed by immunofluorescence (a–d). The interaction between GBM and microglia under Wnt3a treatment was observed through electronic microscopy (e–h) and by time-lapse over 24 h (i, j). Wound healing assay was determined by ImageJ plugin. Each value represents the mean ± SD of three independent experiments. *p < 0.05. Scale bar = 50 μm
Wnt3a Colocalizes with IB4-Positive Cells both in GBM Human Samples and Xenografts
Our in vitro data described above suggest that Wnt3a induces activation and migration of microglial cells and play an important role during microglia-GBM crosstalk. Taking into account that the aberrant activation of Wnt/β-catenin pathway has been associated with malignancy of gliomas [22], we decided to investigate the expression of Wnt3a and correlate it with the presence of microglial cells in human GBM samples. In paraffin-embedded human GBM samples, we observed that microglial cells infiltrate the tumor mass (Fig. 5a) and express Wnt3a as well as the GBM cells (Fig. 5b). To confirm this observation, we xenografted Wnt3a-untreated GBM cells (Wnt3a−) and Wnt3a-treated GBM cells (Wnt3+) into the immunocompetent Swiss mice brain, as previously described by our group [8]. Histological analysis was performed 2 weeks after the graft (Fig. 5c) and revealed that Wnt3a+ GBM cells induced not only the formation of a much larger tumor mass (Fig. 5i, d, respectively) but also presented more aggressive features, such as increased hemorrhagic and necrotic areas with pseudopalisade figures (Fig. 5j) when compared to Wnt3a− GBM cells. Moreover, both tumor masses presented tumor-infiltrated microglia; however, this infiltration was more prominent in Wnt3+ (Fig. 5k) as compared to the Wnt3− (Fig. 5f). The human GBM cells were identified with a specific human vimentin antibody, and microglial/macrophage cells were stained with IB4 (Fig. 5g, l). We observed that IB4-positive cells express Wnt3a in the peritumoral zone on tumor control (Fig. 5h), whereas the Wnt3+ tumor has increased levels of Wnt3a across the whole tumor mass. Furthermore, we observed an enrichment in IB4+ cells colocalizing with Wnt3a in the tumor mass generated through the xenograft of Wnt3a+ GBM cells when compared to the Wnt3a− tumors (Fig. 5h). In addition, we also observed that the tumor-infiltrated microglia express Wnt3a in both the peritumoral zone and tumor core (Fig. 5m). These in vivo findings support our in vitro results and suggest that Wnt3a is not only essential for microglia-GBM interactions but also contributes to GBM progression.

Wnt3a induces tumor progression and contributes to GBM/microglia interaction in vivo. The immunohistochemistry analysis of human GBM paraffin-embedded samples revealed the GFAP expression (gray) and presence of IB4+ cells (red) (a). The Wnt3a expression (green) and human vimentin (hvim) staining (gray) and tumor-infiltrated microglia stained with IB4 (red) in human GBM samples (b). Schematic depiction of GBM untreated (Wnt3−) and Wnt3a-treated GBM cells (Wnt3a+) xenograft in striatum of Swiss brain mice (c). Histopathological features of the tumor after 2 weeks of Wnt3− GBM cells xenotransplantation by hematoxylin-eosin staining (d). Microscopic analysis showed anaplastic cells and small necrosis area (white asterisk) (e). Immunohistochemistry of Wnt3a− tumor mass (human vimentin, red) showed the presence of IB4-positive cells (green) mostly in peritumoral zone (f, g). Increased levels of Wnt3a (green) in peritumoral zone colocalized with IB4-positive cells (red) (h). Hematoxylin-eosin staining in Wnt3a+ tumor xenotransplanted in Swiss mouse brain after 2 weeks (i). The Wnt3a+ tumor exhibited larger tumor mass with anaplastic cells and pseudopalisade surrounding extensive necrosis foci (white asterisk) (j). Immunohistochemistry analysis showed a higher number and diffuse IB4-positive cells (green) in Wnt3a+ tumor (k, l) and also increased levels of Wnt3a (green) and colocalization with IB4 (red) (m). In all immunofluorescence images, the blue indicates nuclear staining (DAPI). Scale bar = 50 μm
Discussion
Glioblastoma-infiltrated microglia plays an important role in tumor progression through the release of factors that induce invasion, migration, and suppression of immunity [8, 9, 12, 13, 17,18,19]. There have been several attempts to find new factors that are involved in GBM and microglia interactions, and one of the signaling pathways involved in aggressiveness of GBM is the canonical Wnt/β-catenin pathway. It is known that this pathway is aberrantly activated in GBM, specifically when it comes to the levels of Wnt3a itself [22]. On the other hand, microglial cells express high levels of proteins involved in Wnt/β-catenin machinery pathway, including the receptors FDZ and their co-receptor LRP5/6 [23], which contribute to microglia activity stimulation when exposed to Wnt3a [23]. Since the GBMs express high levels of Wnt3a and microglia present receptors and components of its pathway, we hypothesized that Wnt3a produced by GBM cells could induce the recruitment of microglial cells and stimulate the switch to M2-like phenotype, promoting migration and proliferation.
First, we showed that Wnt/β-catenin pathway is activated in GBM and microglial cells, reflected by the high levels of β-catenin nuclear translocation in these cells in vitro. This result is in agreement with other works that have already noticed and characterized the activation of Wnt/β-catenin pathway in microglia and glioma cells [23, 30, 40]. Taking into account that Wnt3a induces the β-catenin stabilization and translocation into the nucleus [31], we induced an overactivation of Wnt/β-catenin pathway in GBM and microglial cells through stimulation with recombinant Wnt3a treatment. Our results confirmed that Wnt3a was able to induce β-catenin translocation to the nucleus, despite their basal constitutive activation. In response to this pathway overactivation, we observed that microglia, as well as GBM cells, increased the expression of Wnt3a, probably because β-catenin translocation induces the expression of WNT and other target genes, such as c-myc and cyclin-D1 [32]. In this sense, we also observed that c-myc and cyclin-D1 expression was increased in both cells after Wnt3a stimulation.
The conditioned medium of cells stimulated with recombinant Wnt3a (GBM-CM Wnt3a+) induces more production of Wnt3a that is further secreted into the medium [44]. This fact prompted us to investigate whether GBM cells could produce and secrete higher amounts of soluble Wnt3a upon recombinant Wnt3a stimulation. Our in vitro results showed that the Wnt3a-treated cells released higher levels of Wnt3a as compared to untreated cells. Since GBM-CM induces microglial cell activation and is enriched with Wnt3a that, in turn, is involved with tumor cells proliferation [40], we verified that the microglial viability and proliferation were increased in the presence of GBM-CM Wnt3a+ when compared to the GBM-CM Wnt3−. Moreover, c-myc and cyclin-D1 levels were also increased in microglial cells after GBM-CM Wnt3a+. These results are in accordance with our observations that microglial cells stimulated with GBM-CM Wnt3+ express higher levels of β-catenin and Wnt3a when compared with cells treated with GBM-CM Wnt3−. In fact, GBM progression and aggressiveness have been associated to the extrinsic heterogeneity of the GBM, which involves the interactions of tumor cells with other entities from the brain parenchyma, such as endothelial and microglial cells [7]. This aggressiveness is also associated with the aberrant Wnt activity which culminates on the exacerbation of WNT target genes expression, such as c-myc, cyclin-D1, and WNT itself [32], exacerbating this pathway through a loop of activation. Since Wnt3a induces microglia activation [23, 24] and the amount of tumor-infiltrated microglia/macrophages is strictly related to tumor growth [9, 10, 41], we addressed the question whether Wnt3a produced by GBM cells increased inflammatory response through induction of M2-like phenotype in microglial cells. Others works already demonstrated that GBM release factors, such as chemokines and interleukins, induce M2-like phenotype in microglial cells [17]. Thus, we hypothesized that Wnt3a could be one of these factors. To evaluate the induction of M2-like phenotype in microglial cells, we investigated the expression of Arg-I, STI1, and IL-10 due to their effect of promoting tumor cells proliferation [17,18,19, 21, 35, 41]. Our results showed that Wnt3a provided from GBM cells induced the expression of the Arg-I and STI1 levels in microglial cells. Moreover, CM-GBM Wnt3a+ increased the mRNA levels of IL-10 and consequently reduced IL-1β levels. These findings support our hypothesis that Wnt3a released by GBM cells modulates microglial phenotype through the induction of a M2-like phenotype.
To the best of our knowledge, we are the first to show that microglia-GBM co-cultures exhibit intercellular or tunneling nanotubes between cells and that Wnt3a stimulation increases the appearance of these structures. It has been previously demonstrated that the communication between cells can be made through tunneling nanotubes, which allows the transmission or exchange of vesicles, cytoplasmic materials, signals like miRNAs, chemokines, and oncogenes [41, 42, 45]. Furthermore, the invasive behavior of the tumor cells has been associated with the formation of these nanotubes [45]. Therefore, the formation of these nanotubes may be one of the causes responsible for the aggressiveness of tumor cells. In addition, the induction of immunosuppressive microenvironment can be through the transport of proteins/molecules via tunneling nanotubes, such as Wnt proteins.
Further, the Wnt3a is essential for the motility of GBM cells [40] but also for microglial activation [23]. Moreover, the knockdown of Wnt3a in GBM cells reduces the migration ability of glioma cells [40]. In addition, we assessed the migration ability of microglial and GBM cells co-cultured under stimulation of Wnt3a during 24 h. Our data showed that migration capacities of both GBM and microglia were increased by the presence of Wnt3a, as expected.
The oncogenic potential of Wnt3a was already described [40], and in accordance with this fact, our xenograft experiments showed that GBM cells pre-treated with Wnt3a generate a tumor mass much larger than the untreated. Even more, we observed a greater amount of infiltrated microglia colocalized with Wnt3a in the Wnt3+ tumor mass. This finding is in agreement with what we observed in the biopsies of patients with GBM, highlighting the same pattern and distribution of microglial subpopulations.
Altogether, our in vitro and in vivo results corroborate our hypothesis by showing that Wnt3a produced by GBM cells stimulate the activation of Wnt/β-catenin pathway in microglial cells, which in turn induces the M2-like phenotype on these cells and increases the microglia and tumor crosstalk (Fig. 6). Thus, our work highlights that the Wnt/β-catenin pathway plays a role in the heterogeneity of the GBMs, specifically concerning the extrinsic heterogeneity, and we verified for the first time that Wnt3a is essential to GBM and microglia interactions. These findings are important to better understand how GBM Wnt pathways can modulate the tumor parenchyma cells in M2-like profile, which in turn contribute to a more aggressive behavior and a worse prognostic.

Wnt3a is essential to GBM and microglia crosstalk. Wnt3a released by GBM cells enhances the expression and rearranges the β-catenin subcellular localization and also increase the M2-like phenotype in microglia cells. Consequently, there is a greater recruitment and infiltration of the microglial cells into tumor mass in tumors stimulated with wnt3a (Wnt3a+). Macroscopically, the Wnt3a+ tumors have a larger tumor mass compared to control tumors (Wnt3a−). Histological analysis demonstrated that Wnt3+ tumors are highly aggressive compared to the tumor mass Wnt3a−
References
Stupp R, Hegi ME, Gilbert MR, Chakravarti A (2007) Chemoradiotherapy in malignant glioma: standard of care and future directions. J Clin Oncol 25:4127–4136. https://doi.org/10.1200/JCO.2007.11.8554
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109. https://doi.org/10.1007/s00401-007-0243-4
Friedmann-Morvinski D (2014) Glioblastoma heterogeneity and cancer cell plasticity. Crit Rev Oncog 19:327–336
Albini A, Bruno A, Gallo C, Pajardi G, Noonan DM, Dallaglio K (2015) Cancer stem cells and the tumor microenvironment: interplay in tumor heterogeneity. Connect Tissue Res 56:414–425. https://doi.org/10.3109/03008207.2015.1066780
Balça-Silva J, Matias D, Do Carmo A, Dubois LG, Gonçalves AC, Girão H, Silva Canedo NH, Correia AH et al (2017) Glioblastoma entities express subtle differences in molecular composition and response to treatment. Oncol Rep 38:1341–1352. https://doi.org/10.3892/or.2017.5799
Abou-Antoun TJ, Hale JS, Lathia JD, Dombrowski SM (2017) Brain cancer stem cells in adults and children: cell biology and therapeutic implications. Neurotherapeutics 14:372–384. https://doi.org/10.1007/s13311-017-0524-0
Roos A, Ding Z, Loftus JC, Tran NL (2017) Molecular and microenvironmental determinants of glioma stem-like cell survival and invasion. Front Oncol 7:120. https://doi.org/10.3389/fonc.2017.00120
Garcia C, Dubois LG, Xavier AL, Geraldo LH, da Fonseca ACC, Correia AH, Meirelles F, Ventura G et al (2014) The orthotopic xenotransplant of human glioblastoma successfully recapitulates glioblastoma-microenvironment interactions in a non-immunosuppressed mouse model. BMC Cancer 14:923. https://doi.org/10.1186/1471-2407-14-923
Carvalho da Fonseca AC, Wang H, Fan H, Chen X, Zhang I, Zhang L, Lima FRS, Badie B (2014) Increased expression of stress inducible protein 1 in glioma-associated microglia/macrophages. J Neuroimmunol 274:71–77. https://doi.org/10.1016/j.jneuroim.2014.06.021
Olah M, Raj D, Brouwer N, de Haas AH, Eggen BJL, den Dunnen WFA, Biber KPH, Boddeke HWGM (2012) An optimized protocol for the acute isolation of human microglia from autopsy brain samples. Glia 60:96–111. https://doi.org/10.1002/glia.21251
Razavi S-M, Lee KE, Jin BE, Aujla PS, Gholamin S, Li G (2016) Immune evasion strategies of glioblastoma. Front Surg 3:11. https://doi.org/10.3389/fsurg.2016.00011
da Fonseca ACC, Amaral R, Garcia C et al (2016) Microglia in cancer: for good or for bad? Adv Exp Med Biol 949:245–261. https://doi.org/10.1007/978-3-319-40764-7_12
Audia A, Conroy S, Glass R, Bhat KPL (2017) The impact of the tumor microenvironment on the properties of glioma stem-like cells. Front Oncol 7:143. https://doi.org/10.3389/fonc.2017.00143
Van Meir E, Sawamura Y, Diserens AC et al (1990) Human glioblastoma cells release interleukin 6 in vivo and in vitro. Cancer Res 50:6683–6688
Woiciechowsky C, Asadullah K, Nestler D, Glockner F, Robinson PN, Volk HD, Vogel S, Lanksch WR (1997) Different release of cytokines into the cerebrospinal fluid following surgery for intra- and extra-axial brain tumours. Acta Neurochir 139:619–624
Constam DB, Philipp J, Malipiero UV et al (1992) Differential expression of transforming growth factor-beta 1, -beta 2, and -beta 3 by glioblastoma cells, astrocytes, and microglia. J Immunol 148:1404–1410
Hambardzumyan D, Gutmann DH, Kettenmann H (2016) The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci 19:20–27. https://doi.org/10.1038/nn.4185
Markovic DS, Glass R, Synowitz M, Rooijen N, Kettenmann H (2005) Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J Neuropathol Exp Neurol 64:754–762
Matias D, Predes D, Niemeyer Filho P, Lopes MC, Abreu JG, Lima FRS, Moura Neto V (2017) Microglia-glioblastoma interactions: new role for Wnt signaling. Biochim Biophys Acta 1868:333–340. https://doi.org/10.1016/j.bbcan.2017.05.007
Zhang I, Alizadeh D, Liang J, Zhang L, Gao H, Song Y, Ren H, Ouyang M et al (2016) Characterization of arginase expression in glioma-associated microglia and macrophages. PLoS One 11:e0165118. https://doi.org/10.1371/journal.pone.0165118
Fonseca ACC, da Romão L, Amaral RF et al (2012) Microglial stress inducible protein 1 promotes proliferation and migration in human glioblastoma cells. Neuroscience 200:130–141. https://doi.org/10.1016/j.neuroscience.2011.10.025
Denysenko T, Annovazzi L, Cassoni P et al (2016) WNT/β-catenin signaling pathway and downstream modulators in low- and high-grade glioma. Cancer Genomics Proteomics 13:31–45
Halleskog C, Mulder J, Dahlström J, Mackie K, Hortobágyi T, Tanila H, Kumar Puli L, Färber K et al (2011) WNT signaling in activated microglia is proinflammatory. Glia 59:119–131. https://doi.org/10.1002/glia.21081
Zheng H, Jia L, Liu C-C, Rong Z, Zhong L, Yang L, Chen XF, Fryer JD et al (2017) TREM2 promotes microglial survival by activating Wnt/β-catenin pathway. J Neurosci 37:1772–1784. https://doi.org/10.1523/JNEUROSCI.2459-16.2017
Dijksterhuis JP, Arthofer E, Marinescu VD, Nelander S, Uhlén M, Pontén F, Mulder J, Schulte G (2015) High levels of WNT-5A in human glioma correlate with increased presence of tumor-associated microglia/monocytes. Exp Cell Res 339:280–288. https://doi.org/10.1016/j.yexcr.2015.10.022
Faria J, Romão L, Martins S, Alves T, Mendes FA, de Faria GP, Hollanda R, Takiya C et al (2006) Interactive properties of human glioblastoma cells with brain neurons in culture and neuronal modulation of glial laminin organization. Differentiation 74:562–572. https://doi.org/10.1111/j.1432-0436.2006.00090.x
Kahn SA, Biasoli D, Garcia C et al (2012) Equinatoxin II potentiates temozolomide- and etoposide-induced glioblastoma cell death. Curr Top Med Chem 12:2082–2093
Lima FR, Gervais A, Colin C et al (2001) Regulation of microglial development: a novel role for thyroid hormone. J Neurosci 21:2028–2038
Towbin H, Staehelin T, Gordon J (1992) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. 1979 Biotechnology 24:145–149
Amado NG, Fonseca BF, Cerqueira DM, Neto VM, Abreu JG (2011) Flavonoids: potential Wnt/beta-catenin signaling modulators in cancer. Life Sci 89:545–554. https://doi.org/10.1016/j.lfs.2011.05.003
Oloumi A, Syam S, Dedhar S (2006) Modulation of Wnt3a-mediated nuclear beta-catenin accumulation and activation by integrin-linked kinase in mammalian cells. Oncogene 25:7747–7757. https://doi.org/10.1038/sj.onc.1209752
Liao DJ, Thakur A, Wu J, Biliran H, Sarkar FH (2007) Perspectives on c-Myc, cyclin D1, and their interaction in cancer formation, progression, and response to chemotherapy. Crit Rev Oncog 13:93–158
Clément-Lacroix P, Ai M, Morvan F et al (2005) Lrp5-independent activation of Wnt signaling by lithium chloride increases bone formation and bone mass in mice. Proc Natl Acad Sci U S A 102:17406–17411. https://doi.org/10.1073/pnas.0505259102
Ajmone-Cat MA, D’Urso MC, di Blasio G, Brignone MS, de Simone R, Minghetti L (2016) Glycogen synthase kinase 3 is part of the molecular machinery regulating the adaptive response to LPS stimulation in microglial cells. Brain Behav Immun 55:225–235. https://doi.org/10.1016/j.bbi.2015.11.012
Crain JM, Nikodemova M, Watters JJ (2013) Microglia express distinct M1 and M2 phenotypic markers in the postnatal and adult central nervous system in male and female mice. J Neurosci Res 91:1143–1151. https://doi.org/10.1002/jnr.23242
Dey N, Barwick BG, Moreno CS, Ordanic-Kodani M, Chen Z, Oprea-Ilies G, Tang W, Catzavelos C et al (2013) Wnt signaling in triple negative breast cancer is associated with metastasis. BMC Cancer 13:537. https://doi.org/10.1186/1471-2407-13-537
Yamaguchi H, Condeelis J (2007) Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta 1773:642–652. https://doi.org/10.1016/j.bbamcr.2006.07.001
Shibamoto S, Higano K, Takada R, Ito F, Takeichi M, Takada S (1998) Cytoskeletal reorganization by soluble Wnt-3a protein signalling. Genes Cells 3:659–670
Holmes KC (2009) Structural biology: actin in a twist. Nature 457:389–390. https://doi.org/10.1038/457389a
Kaur N, Chettiar S, Rathod S, Rath P, Muzumdar D, Shaikh ML, Shiras A (2013) Wnt3a mediated activation of Wnt/β-catenin signaling promotes tumor progression in glioblastoma. Mol Cell Neurosci 54:44–57. https://doi.org/10.1016/j.mcn.2013.01.001
Zhang Z-M, Yang Z, Zhang Z (2015) Distribution and characterization of tumor-associated macrophages/microglia in rat C6 glioma. Oncol Lett 10:2442–2446. https://doi.org/10.3892/ol.2015.3533
O’Connor T, Borsig L, Heikenwalder M (2015) CCL2-CCR2 signaling in disease pathogenesis. Endocr Metab Immune Disord Drug Targets 15:105–118
Pontes B, Viana NB, Campanati L, Farina M, Neto VM, Nussenzveig HM (2008) Structure and elastic properties of tunneling nanotubes. Eur Biophys J 37:121–129. https://doi.org/10.1007/s00249-007-0184-9
Hooper C, Sainz-Fuertes R, Lynham S, Hye A, Killick R, Warley A, Bolondi C, Pocock J et al (2012) Wnt3a induces exosome secretion from primary cultured rat microglia. BMC Neurosci 13:144. https://doi.org/10.1186/1471-2202-13-144
Nawaz M, Fatima F (2017) Extracellular vesicles, tunneling nanotubes, and cellular interplay: synergies and missing links. Front Mol Biosci 4:50. https://doi.org/10.3389/fmolb.2017.00050
Acknowledgements
This study was supported by the Brazilian agencies Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Pró-Saúde Associação Beneficente de Assistência Social e Hospitalar, and Ary Frauzino Foundation for Cancer Research.
We would like to acknowledge Dra. Juliana Coelho Aguiar for giving us some primers, Dra. Graziella Ventura for helping us with the confocal microscopy acquisitions, and Geralda Cardoso for the lab technical support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
We confirm that this manuscript has been approved by all authors and that there are no known conflicts of interest associated with this publication.
Electronic Supplementary Material
ESM 1
(AVI 83542 kb)
Rights and permissions
About this article

Cite this article
Matias, D., Dubois, L.G., Pontes, B. et al. GBM-Derived Wnt3a Induces M2-Like Phenotype in Microglial Cells Through Wnt/β-Catenin Signaling. Mol Neurobiol 56, 1517–1530 (2019). https://doi.org/10.1007/s12035-018-1150-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-018-1150-5