
Abstract
Activated autophagy/mitophagy has been intensively observed in ischemic brain, but its roles remain controversial. Peroxynitrite (ONOO−), as a representative of reactive nitrogen species, is considered as a critical neurotoxic factor in mediating cerebral ischemia-reperfusion (I/R) injury, but its roles in autophagy/mitophagy activation remain unclear. Herein, we hypothesized that ONOO− could induce PINK1/Parkin-mediated mitophagy activation via triggering dynamin-related protein 1 (Drp1) recruitment to damaged mitochondria, contributing to cerebral I/R injury. Firstly, we found PINK1/Parkin-mediated mitophagy activation was predominant among general autophagy, leading to rat brain injury at the reperfusion phase after cerebral ischemia. Subsequently, increased nitrotyrosine was found in the plasma of ischemic stroke patients and ischemia-reperfused rat brains, indicating the generation of ONOO− in ischemic stroke. Moreover, in vivo animal experiments illustrated that ONOO− was dramatically increased, accompanied with mitochondrial recruitment of Drp1, PINK1/Parkin-mediated mitophagy activation, and progressive infarct size in rat ischemic brains at the reperfusion phase. FeTMPyP, a peroxynitrite decomposition catalyst, remarkably reversed mitochondrial recruitment of Drp1, mitophagy activation, and brain injury. Intriguingly, further study revealed that ONOO− induced tyrosine nitration of Drp1 peptide, which might contribute to mitochondrial recruitment of Drp1 for mitophagy activation. In vitro cell experiments yielded consistent results with in vivo animal experiments. Taken together, all above findings support the hypothesis that ONOO−-induced mitophagy activation aggravates cerebral I/R injury via recruiting Drp1 to damaged mitochondria.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autophagy is characterized as the process of “self-eating” to digest aggregated proteins or damaged organelles via degradation within autolysosomes [1, 2]. Activated autophagy has been intensively observed in ischemic brains with the evidences from both basic studies [3, 4] and clinical investigations [5, 6]. Although basal level autophagy is undoubtedly indispensable for cell survival, the roles of activated autophagy in cerebral ischemia-reperfusion (I/R) injury remain controversial [7, 8].
Neurons are highly dependent on mitochondrial function [9, 10]. Damaged mitochondria with deceased membrane potential in the ischemic brain are sophisticatedly removed by autophagy, termed mitophagy [11, 12]. Dynamin-related protein 1 (Drp1) is a key factor for mitochondrial fission/fragmentation [13, 14]. Subsequently, PINK1/Parkin signaling pathway plays critical roles in the mitophagic process under cerebral ischemia injury [15,16,17]. Similar to autophagy, basal-level mitophagy is essential for the removal of dysfunctional mitochondria to maintain mitochondrial homeostasis [9, 18]. Nevertheless, the roles of activated mitophagy in ischemic brain need to be further elucidated. For examples, mitophagy was reported to promote the clearance of injured mitochondria and then attenuate the inflammation in traumatic and ischemic brains [19, 20]. However, strategies for suppressing mitochondrial fragmentation/mitophagy showed the neuroprotective effects against cerebral I/R injury [21, 22]. When and how autophagy/mitophagy activation mediates neuroprotective or detrimental effects are still puzzles. We systematically reviewed current divergent literatures and suspected that reperfusion might be a critical turning point determining autophagy/mitophagy activation from the protective process to destructive procedure in ischemic brain [23]. Given that recanalization is the most effective therapeutic strategy for ischemic stroke [24], understanding the mechanisms how reperfusion-mediated autophagy/mitophagy contributes to ischemic brain injury would have therapeutic values.
Cerebral I/R insults induce free radical productions [25, 26]. Nitric oxide (NO) and superoxide anion (O2·−) are generated and subsequently form peroxynitrite (ONOO−) at diffusion rates during the reperfusion phase. ONOO− and its derivatives play critical roles in mediating oxidative and nitrosative stress, as well as neuronal cell death during cerebral I/R injury [27]. ONOO− could mediate inflammation, lipid membrane peroxidation, and mitochondrial dysfunction, exacerbating the blood-brain barrier (BBB) disruption and brain dysfunction [28, 29]. As a parent free radical of ONOO−, NO was reported to induce mitochondrial fission via S-nitrosylation of Drp1, resulting in excessive mitophagy and neuronal cell death [30, 31]. Actually, mitochondrial proteins are susceptible to ONOO−-triggered nitrative stress [32, 33]. From this perspective, we raised the hypothesis that ONOO− could induce mitophagy activation via recruiting Drp1 to damaged mitochondria under cerebral I/R insults. With the in vivo and in vitro experiments, we demonstrated that ONOO− could trigger mitochondrial recruitment of Drp1 and initiate PINK1/Parkin-mediated mitophagy activation, aggravating brain damage during cerebral I/R injury.
Methods
Focal Cerebral Ischemia-Reperfusion Model
Adult male Sprague Dawley (SD) rats weighing 250–280 g were supplied by the Laboratory Animal Unit, The University of Hong Kong. Animal experimental protocols were approved and supervised by the Committee on the Use of Live Animals in Teaching and Research, The University of Hong Kong. SD rats were maintained at controlled temperature (22 ± 2 °C) and kept available to food and water. Rats underwent middle cerebral artery occlusion (MCAO) followed by reperfusion to mimic cerebral I/R injury. All rats were randomly divided into MCAO-operated and sham-operated groups. Briefly, rats were anesthetized with 4% isoflurane (Abbott, IL, USA) during the induction of anesthesia and maintained with 1.5% isoflurane. During the operation, the rats were placed on the heating pad for maintaining body temperature at 37 °C. Before incision, the skin around the neck was sterilized with 75% ethanol after shaving the fur. Under the microscope, the left common carotid artery (CCA), external carotid artery (ECA), and internal carotid artery (ICA) were exposed and isolated through making a 2-cm middle incision of neck. Notably, nerves, especially the vagus nerve, should be carefully dissected from the CCA and ICA. The ECA was tied with suture and cut to serve as a stump. The blood flow of the CCA and ICA was temporarily blocked with the knot or clamp. Then, a monofilament with a silicon coating on the tip and a diameter of 0.36 mm (L3600; Jialing Co. Ltd., China) was inserted into the ICA and advanced forward to occlude the origin of the MCA, inducing the cessation of blood flow. After 2 h of occlusion, the arteries were exposed again under anesthesia and then the monofilament was withdrawn to achieve reperfusion. Sham-operated rats were subjected to similar surgical operation except occluding the MCA with the monofilament. All rats recovered from the anesthesia in a recovery cage. With regard to postoperative care, all rats were individually housed. There was approximately 2% mortality rate among MCAO-operated rats. Yet, all rats in sham-operated group survived. Those rats were sacrificed after 6, 14, or 22 h of reperfusion.
Drug Treatment
To examine the roles of autophagy/mitophagy in MCAO model with 2 h of ischemia plus 22 h of reperfusion, we designed parallel groups treated with 3-methyladenine (3-MA, M9281; Sigma, MO, USA) and Mdivi-1 (M0199; Sigma). Briefly, SD rats were anesthetized with a mixture of 100 mg per kg body weight (mg/kg) ketamine hydrochloride (Alfasan, Woerden, Holland) and 10 mg/kg xylazine hydrochloride (Alfasan) and then fixed in a rat brain stereotaxic instrument (Stoelting Co., IL, USA). Five microliters of 3-MA (600 nmol), Mdivi-1 (3 mg/kg), and the same volume vehicle were administered at the onset of reperfusion after 2 h of ischemia. In order to minimize the side effects and improve the bioavailability, the drugs and vehicle were directly injected into the left ipsilateral cerebral ventricle by using a 10-μL microsyringe (Hamilton Co., Reno, NV, USA) at the rate of 1 μL/min. The stereotaxic coordinates for the lateral ventricle (Paxinos, G. and Watson, C.) were 1 mm posterior to the bregma (anterior-posterior), 1.2 mm lateral to the midline (medial-lateral), and 3.5 mm below the dura (dorsal-ventral). By the end of injection, the micro-injector remained for 5 min.
To elucidate the roles of ONOO− during cerebral I/R injury, FeTMPyP (75854; Cayman, MI, USA) was used as a positive control. FeTMPyP at 1 and 3 mg/kg was administered intravenously through the rat femoral vein once at the onset of reperfusion. Then, the monofilament was withdrawn to perform reperfusion. All animals received individual care after operation.
Cerebral Infarct Size Measurement
Cardiac perfusion with phosphate-buffered saline (PBS) was performed after anesthetizing rats with a mixture of 200 mg/kg ketamine hydrochloride and 20 mg/kg xylazine hydrochloride through intraperitoneal injection. Brain samples were collected and cut into 2-mm coronal slices with a rat brain matrix (Braintree, MA, USA). Obtained slices were 2–8 mm away from frontal pole. Then, the slices were immersed into 2% 2,3,5-triphenyltetrazolium chloride (TTC, T8877; Sigma) solution at 37 °C in the dark for 20 min. Staining images were captured by a camera, and the infarct size was measured and analyzed by ImageJ. To minimize the effect of brain edema, the infarct size percentage was calculated with following formula: the infarct size percentage = (the size of the right hemisphere − the red size of the left hemisphere) / the right hemisphere size × 100%.
Mitochondrial Isolation
Followed by cardiac perfusion with PBS, brain tissues were collected and stored in − 80 °C for further use. Mitochondrial isolation of rat brain was performed using a commercially available kit (89801; Thermo, IL, USA). We majorly chose a unique reagent-based method to enable simultaneous multi-sample processing. In detail, rat brain tissue was disrupted via four Dounce strokes to obtain a homogeneous suspension. Then, mitochondria were separated from cytosolic fraction using extraction reagents and differential centrifugation. After mitochondrial isolation, mitochondrial protein was then extracted with 2% CHAPS (C9426; Sigma) in Tris-buffered saline (TBS) (300 μL CHAPS buffer per 200 mg brain tissue). Further, mitochondrial and cytosolic proteins were stored in − 80 °C for western blot analysis. The integrity and purity of isolated mitochondria was tested with western blot using the mitochondrial marker VADC1/porin (ab15895; Abcam, Cambridge, UK).
Cell Line
Human SH-SY5Y cell line was purchased from ATCC (catalog CRL-2266™). SH-SY5Y cells were cultured with 10% fetal bovine serum, 1% l-glutamine, as well as 1% penicillin and streptomycin in Dulbecco’s modified Eagle’s medium (DMEM). Each medium was purchased from Life Technologies (Gibco; Carlsbad, CA, USA).
Cell Treatment
To explore the relationship between mitophagy and ONOO− production in cells, we applied ONOO− donor 3-morpholinosydnonimine (SIN-1, 82220; Cayman) and synthesized sodium ONOO− (81565; Cayman) to induce the nitrative stress in SH-SY5Y cells in vitro. In detail, SH-SY5Y cells were treated with 1, 2, and 4 mM SIN-1 and 20, 40, and 80 μM sodium ONOO−, respectively, for 2 h. In addition, 50 μM FeTMPyP was used as positive control in the cells challenged by 4 mM SIN-1 or 80 μM sodium ONOO−. Then, western blot analysis and immunofluorescence test were conducted.
Immunofluorescence Staining
SD rats were successively subjected to cardiac perfusion with 200 mL PBS and 400 mL of 4% paraformaldehyde (PFA) in PBS. Brain tissues were collected and fixed again with 4% PFA in PBS overnight. After dehydrated with 15 and 30% sucrose in PBS, the brains were embedded in optimal cutting temperature (OCT) compound (14020108926; Leica, Wetzlar, Germany) for frozen slices. Before staining, epitopes of the samples were retrieved with 10 mM sodium citrate buffer (pH = 6.0) by using the microwave oven for 10 min. After incubation with 5% goat serum (G9023; Sigma) and permeabilization with 3‰ Triton X-100 (22686; USB, OH, USA) in PBS for 2 h at room temperature (RT), brain sections were then labeled with primary antibodies including LC3, ATP subunit beta (ATPB), Drp1, and 3-nitrotyrosine (Supplementary Table 1) at 4 °C overnight, followed by the incubation with fluorescent dye-conjugated secondary antibodies (1:800; Life Technologies, CA, USA) for 2 h at RT. Nuclear was stained with 4′,6-diamidino-2-phenylindole (DAPI) (Thermo) for 10 min at RT. Finally, the slices were mounted with mounting medium (S302380; Dako, Agilent, CA, USA) for fluorescence detection.
For the cell staining, SH-SY5Y cells were firstly cultured on the microscope slides (0111500; GmbH & Co. KG, Germany), followed by corresponding treatment. After 20 min of fixation and dehydration with 4% PFA plus 4% sucrose in PBS at RT, cell slides were blocked and permeabilized with 5% goat serum and 0.1‰ Triton X-100 in PBS for 1.5 h. After labeling with primary and secondary antibodies, nuclear was stained with DAPI for another 10 min at RT.
Both stained brain sections and SH-SY5Y cells were visualized using a Zeiss 780 reverse laser-scanning confocal microscope (Germany).
TUNEL Assay
Apoptotic cell death was detected by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay. After preparing 4-μm paraffin-embedded brain sections, TUNEL reagents (Roche Diagnostics, IN, USA) were used to label apoptotic cells following deparaffinization and hydration. Briefly, the tissue slices were immersed into 0.1 M citrate buffer (pH = 6.0) for the microwave irradiation. After washing with PBS three times, the tissue samples were incubated with TUNEL reaction mixture (50 μL/tissue) for 1 h at 37 °C in the dark. Besides, DAPI was also used to mark the nucleus. The fluorescent images were obtained by the Carl Zeiss Fluorescence Imaging System.
MTT Assay
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was widely used to detect the cell viability. After cell treatment, MTT was added into the medium for 4 h at 37 °C. Of note, culture medium was replaced with fresh medium before adding MTT. After 4 h of incubation, culture medium was removed and 200 μL dimethyl sulfoxide (DMSO) was added into the wells. Then, the absorbance at 490 nm was measured with a multi-plate reader (model 680; Bio-Rad).
Immunoprecipitation
To detect Drp1 nitration modification, total Drp1 extracted from the rat brains and SH-SY5Y cells was firstly pulled down by Dynabeads using the Dynabeads® Protein G Immunoprecipitation Kit (10007D; Novex, Life Technologies). Briefly, 50 μL Dynabeads for each sample was incubated with Drp1 primary antibody (Supplementary Table 1) in 200 μL antibody (Ab) binding and washing buffer for 30 min at RT. After removing the supernatant and washing Dynabeads, a 500-μL antigen (Ag)-containing protein sample was incubated with the Dynabeads-Ab complex for another 2 h, keeping rotation. Then, the supernatant was collected using a magnet as a flow-through sample. Followed by washing three times, the Dynabeads-Ab-Ag complex was gently suspended again in 20 μL elution buffer for 2 min. Separating with the magnet, the supernatant was considered as the target Drp1 sample for further western blot analysis.
Western Blot Analysis
Mitochondrial and cytosolic proteins from the rat brains were prepared after mitochondrial isolation. Cellular proteins were extracted with RIPA (R0278; Sigma) lysis buffer plus 1% protease inhibitor and phosphatase inhibitor (Sigma). All procedures were referred to the standard western blot protocol. After quantitative analysis, an equal amount of proteins was loaded and separated with sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) by 8 to 12% gels and then transferred to 0.45-μm-pore size polyvinylidene difluoride membranes (IPVH00010; EMD Millipore, Germany). After blocking with 5% bovine serum albumin, membranes were incubated with primary antibodies (Supplementary Table 1) overnight at 4 °C followed by washing with TBS-Tween 20 buffer and incubating in secondary antibody (1:2000) for 2 h at 4 °C. Chemiluminescence detection was performed with enhanced chemiluminescence (ECL) reagents (RPN2235; GE Healthcare, IL, USA) using the Bio-Rad ChemiDoc™ XRS+ System (USA).
Drp1 Peptide Preparation
The target protein Drp1 pulled downed by immunoprecipitation was further analyzed with mass spectrometry (MS) analysis. Firstly, Drp1 protein was separated with 8% SDS-PAGE gel. Then, SDS-PAGE gel was stained with Coomassie brilliant blue R-250 solution (1610436; Bio-Rad, UK) for 2 h with gentle shaking at RT. After rinsing three times with deionized water, the stained gel was destained with the mixture of 25% ethanol, 10% acetic acid, and 65% deionized water for four times. The target gel band was cut as close to the edge as possible. The gel plugs were rehydrated with 12.5 ng/μL trypsin (Promega) in 10 mM ammonium bicarbonate for 16 h at 37 °C. After incubation, the peptides were successively extracted with 5% formic acid (FA)/50% acetonitrile (ACN) and 100% ACN. After desalting, the peptides were dissolved by applying 1 μL of 10 mg/mL α-cyano-4-hydroxycinnamic acid matrix (Fluka) in 0.1% FA/50% ACN and stored at 4 °C until MS analysis.
MALDI-TOF/TOF Tandem Mass Spectrometry Analysis
Drp1 peptide modification was analyzed using the 4800 MALDI-TOF/TOF Analyzer (AB Sciex). All mass spectra were acquired in positive ion reflector mode using the 4000 Series Explorer, version 3.5.28193, software (AB Sciex). Before each detection, MS was calibrated with the peptide calibration standard, 4700 Cal-Mix (AB Sciex). Characteristic spectra were attained by averaging 500 acquisitions in reflector mode. The scanning range of general peptides was 900–4000 m/z. The peak detection criteria used were a minimum S/N of 5. The peptide mass fingerprint search was then performed with the GPS Explorer algorithm (version 3.6; AB Sciex) against the Swiss-Prot database using the in-house MASCOT search engine, version 2.2. Further, MASCOT search analysis setting for protein nitration modification was listed as follows: a peptide with one single tyrosine nitrated was expected to have a mass increase of 45 Da [34].
Clinical Study Design and Analysis
This clinical observational study was designed to observe nitrotyrosine (NT) level in the plasma after ischemic stroke and evaluate the correlation between NT level and neurological deficits, providing the evidence for screening potential therapeutic target. This clinical study was approved by the Ethics Committee of Hong Kong (UW14-319). We recruited ischemic stroke patients from Huizhou First People’s Hospital, Guangdong Province, China, during June 2015 to April 2016. Informed consent was obtained from the patients and participants before enrolment. One hundred one ischemic stroke patients meeting the criteria were included. Twenty-six participants in the control group with no stroke history were also recruited. Ischemic stroke is defined according to the World Health Organization criteria [35]. The National Institutes of Health Stroke Scale (NIHSS) [36] was used to evaluate the stroke severity by two experienced neurologists on admission. The NIHSS score ranges from 0 to 42, in which a higher score reflects more severe damage. Blood was collected within 24 h after stroke symptom onset. Blood sample was collected in ethylenediaminetetraacetic acid (EDTA) tubes and centrifuged at 1000g for 15 min, and then plasma was stored at − 80 °C for enzyme-linked immunosorbent assay (ELISA) analysis. Plasma NT level was determined with a commercially available ELISA kit from USCN (Wuhan, China).
Statistics
Data were expressed as Mean ± SEM. Differences in measured variables between two groups were assessed by the unpaired two-sided t-test. One-way ANOVA followed by Dunnett’s multiple-comparison test was used for multiple comparisons. The numbers of rats used are described in the corresponding figure legends. For clinical study, no patient was excluded for analysis. Data were shown as the median with interquartile range. Unpaired t-test with Welch’s correction was used for the comparison between the control and ischemic stroke groups. One-way ANOVA followed by Bonferroni’s multiple-comparison test was applied for multiple comparisons. All data were analyzed using GraphPad Prism (version 6.0; GraphPad Software, Inc., CA, USA). Two-sided P < 0.05 was considered as statistical significant.
Results
Inhibition of Autophagy Activation Attenuates Cerebral I/R Injury
We first sought to detect autophagy level during cerebral I/R injury. SD rats were subjected to 2 h of cerebral ischemia plus 6, 14, or 22 h of reperfusion by occluding and releasing the middle cerebral artery. TTC staining revealed that the infarct size of rat brain was increased with the time length of reperfusion as expected (Fig. 1a, b). Cell death was also aggravated, as evidenced by the decreased ratio of Bcl-2/Bax and the enhanced cytochrome c releasing in ischemia-reperfused brains (Fig. 1c). Autophagy activation was simultaneously indicated by the degradation of p62, an indicator for the autophagic late phase, in the ischemic brains (Fig. 1c). Compared with the sham-operated group, the 2 h of ischemia plus 22 h of reperfusion (I2/R22) group had a change in the distribution of LC3, showing the cytosolic LC3 aggregation to form puncta in the ischemic brains (Fig. 1d).

Activated autophagy aggravates the infarct size and cell death of ischemia-reperfused rat brains. a 2,3,5-Triphenyltetrazolium chloride (TTC) staining of brain tissues after 2 h of ischemia plus 6 h (I2/R6) (n = 5 rats), 14 h (I2/R14) (n = 5 rats), and 22 h (I2/R22) (n = 7 rats) of reperfusion and sham operation (n = 5 rats). b The infarct size percentage was analyzed by ImageJ and quantitatively calculated with following formula: the infarct size percentage = (the size of the right hemisphere − the red size of the left hemisphere) / the right hemisphere size × 100%. Data are expressed as mean ± SEM. **P < 0.01; ***P < 0.001, relative to the sham group (one-way analysis of variance (ANOVA) followed by Dunnett’s multiple-comparison test). c Western blot images of the expression levels of indicated proteins involved in apoptosis (Bcl-2, Bax, and cytochrome c (Cyt c)) and autophagy (p62). d Representative confocal images of LC3 staining (green) (n = 3 rats/group). Scale bar (left), 20 μm; scale bar (right), 10 μm. e–g Autophagy inhibitor 3-methyladenine (3-MA) (600 nmol) was administered into SD rats at the onset of reperfusion via intracerebroventricular injection. TTC staining of brain tissues (e) and quantitative analysis of the infarct size measured by ImageJ (f) (n = 5 rats/group). Data are expressed as mean ± SEM. ***P < 0.001, versus the vehicle-I2/R22 group (one-way ANOVA followed by Dunnett’s multiple-comparison test). Western blot analysis on the expression levels of Bcl-2, Bax, and p62 proteins extracted from the rat brains in the sham, vehicle-I2/R22, and 3-MA-I2/R22 groups (g)
We next performed the experiments to investigate the effects of autophagy inhibitor 3-MA on the infarct size in the brains subjected to I2/R22. Treatment of 3-MA at the onset of reperfusion obviously decreased the infarct size (Fig. 1e, f), restored the ratio of Bcl-2/Bax, and upregulated the expression of p62 (Fig. 1g). These results suggest that activated autophagy contributes to brain injury at the stage of reperfusion after cerebral ischemia.
PINK1/Parkin-Mediated Mitophagy is Activated via Recruiting Drp1 to Damaged Mitochondria, Contributing to Cerebral I/R Injury
Mitochondrial recruitment of Drp1 is a hallmark of mitophagy fission/fragmentation, and PINK1/Parkin pathway subsequently mediates the mitophagic process to degrade damaged mitochondria in most cases [37, 38]. Firstly, we found the ratio of LC3-II to LC3-I (LC3-II/LC3-I) was relatively mild in total protein fraction from the infarcted hemisphere. Conversely, the ratio of LC3-II/LC3-I was markedly increased in mitochondrial fraction isolated from the I2/R22 group (Fig. 2a, b). We then monitored the dynamic level of mitophagy in the brains during cerebral I/R injury. The expression of indicated proteins from mitochondrial and cytosolic fractions was detected with western blot. The ratio of LC3-II/LC3-I in mitochondrial fraction was enhanced with the time length of reperfusion following cerebral ischemia (Fig. 2c). Meanwhile, Parkin translocation from the cytoplasm to the mitochondria was accompanied with increased Drp1 and decreased cytochrome c expression in the mitochondria (Fig. 2c). Immunofluorescence revealed that LC3 was evenly distributed in the cytoplasm of the sham group, yet it translocated to the mitochondria marked by ATPB to form intensive immunofluorescent puncta in the I2/R22 group (Fig. 2d). Similarly, Drp1 was also recruited from the cytosol to the mitochondria in the I2/R22 group (Fig. 2e). In addition, co-immunostaining images of neuron marker TuJ1 and Parkin revealed that Parkin translocation to mitochondria occurred in neurons during cerebral I/R injury (Supplementary Fig. 1). These results suggest that mitophagy is time-dependently activated with mitochondrial recruitment of Drp1 and Parkin during cerebral I/R injury.

PINK1/Parkin-mediated mitophagy, as a selective autophagy, dominates in ischemia-reperfused brains. a Western blot detection for the ratio of LC3-II/LC3-I in total and mitochondrial fractions from the rat brains suffering I2/R22. b Quantitative analysis of LC3-II/LC3-I ratio in the I2/R22 group. Data are expressed as mean ± SEM. *P < 0.05; ***P < 0.001 (t-test). c Mitochondrial and cytosolic fractions were isolated from brain tissues after 2 h of ischemia followed by different time length reperfusion. Involved protein expression levels were detected with western blot. d Co-staining images of mitochondrial marker ATPB (red), LC3 (green), and nuclear (blue). Subcellular locations of ATPB and LC3 in the sham and I2/R22 groups were visualized with a confocal microscope. Scale bar, 50 μm. Enlarged immunofluorescent images of representative cells are shown (right) (n = 3 rats/group). e Co-stained with ATPB (red), Drp1 (green), and nuclear (blue) to detect mitophagy level in the brains after I2/R22. Scale bar, 50 μm. Typical cell images were enlarged (right) (n = 3 rats/group)
Then, Mdivi-1, a selective inhibitor of Drp1, was used to test the roles of Drp1 in mitophagy activation during cerebral I/R injury. Mdivi-1 treatment significantly decreased the infarct size (Fig. 3a, b), upregulated Bcl-2, and downregulated Bax expression in ischemia-reperfused brains (Fig. 3c). For mitophagy, Mdivi-1 treatment inhibited the translocation of Drp1 and Parkin from the cytosol to the mitochondria, decreased mitochondrial PINK1 expression, and restored the expression of p62, consistent with the reduced ratio of mitochondrial LC3-II/LC3-I (Fig. 3c, d). Collectively, above findings indicate that PINK1/Parkin-mediated mitophagy is activated via recruiting Drp1 to damaged mitochondria, aggravating brain damages during cerebral I/R injury.

Mdivi-1 significantly attenuates cerebral I/R injury via inhibiting Drp1 recruitment and mitophagy activation. SD rats were intracerebroventricularly administered with the Drp1 inhibitor Mdivi-1 (3 mg/kg) at the onset of reperfusion following 2 h of cerebral ischemia. Brain samples were collected 22 h after reperfusion. TTC staining (a) and statistical analysis (b) for the infarct size in the sham, vehicle-I2/R22, and Mdivi-1-I2/R22 groups (n = 5 rats/group). Data are expressed as mean ± SEM. ***P < 0.001, versus the vehicle-I2/R22 group (one-way ANOVA followed by Dunnett’s multiple-comparison test). c, d Representative western blot images of indicated cytosolic and mitochondrial proteins from the rat brains related to cell death and mitophagy. GAPDH and VDAC1 were used as cytosolic and mitochondrial loading control, respectively. Western blot analysis was repeated at least three times
ONOO− Triggers Mitochondrial Recruitment of Drp1 and Induces Mitophagy Activation During Cerebral I/R Injury
In order to explore the clinical correlation of ONOO− production and neurological deficits in ischemic stroke, we detected NT, a footprint of ONOO−, in the plasma of acute ischemic stroke patients. Characteristics of ischemic stroke patients and control individuals were compared in Supplementary Table 2. Intriguingly, ELISA analysis revealed that ischemic stroke patients within 24 h after ischemic stroke onset had a higher NT level in the plasma than the control group (P < 0.0001) (Fig. 4a). The predictive ability of NT to distinguish ischemic stroke patients from control individuals was evaluated with receiver operating characteristic (ROC) curve analysis, indicated by an area under the ROC curve (AUC) at 0.71 (95% CI 0.61 to 0.80) (P < 0.01) (Fig. 4b). Furthermore, we evaluated the NIHSS for neurological deficits. Spearman correlation analysis showed that plasma NT level was positively correlated with an NIHSS score within 12 h after symptom onset (P = 0.04) (Fig. 4c). In addition, plasma NT level in stroke patients had an upward trend over time (Fig. 4d). In line with the results from ischemic stroke patients, animal experiments revealed a time-dependent increase in the expression of cytosolic iNOS, NADPH oxidase subunit p47phox, as well as mitochondrial 3-nitrotyrosine (3-NT) in the rat brains subjected to 2 h of ischemia plus different durations of reperfusion (Fig. 4e, f). These results from clinical investigations and animal experiments indicate the involvement of ONOO− in the process of cerebral I/R injury.

Nitrotyrosine/3-nitrotyrosine (NT/3-NT), as a footprint of peroxynitrite (ONOO−), is increased in the plasma of ischemic stroke patients and ischemia-reperfused rat brains. a ELISA analysis for NT level in the plasma of control individuals and ischemic stroke patients within 24 h after ischemic stroke onset (control, n = 26; ischemic stroke, n = 101). Data are shown as median with interquartile range. ***P < 0.0001, relative to the control (unpaired t-test with Welch’s correction). b Receiver operating characteristic curve analysis to test the predictive ability of NT with an area under ROC curve of 0.71 (95% CI 0.61 to 0.80). **P < 0.01. c The Spearman correlation analysis to assess the association between NT level and neurological NIHSS score in ischemic stroke patients within 12 h after symptom onset (n = 49). *P = 0.04 < 0.05. d Plasma NT level of ischemic stroke patients at different time points after symptom onset (control, n = 26; 3–6 h, n = 14; 6–12 h, n = 35; 12–24 h, n = 47). *P = 0.022 < 0.05, versus control (one-way ANOVA followed by Bonferroni’s multiple-comparison test). Western blot images (e) and quantitative analysis (f) for iNOS and p47phox proteins in cytosolic fraction and 3-NT in mitochondrial fraction of rat brains in the sham, I2/R6, I2/R14, and I2/R22 groups. Data are expressed as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001, versus the sham group (one-way ANOVA followed by Dunnett’s multiple-comparison test)
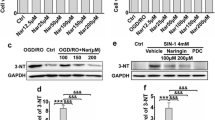
We then examined whether ONOO− was sufficient to induce mitophagy activation via triggering Drp1 mitochondrial recruitment of the rat brains in the I2/R22 group. FeTMPyP, a representative peroxynitrite decomposition catalyst (PDC), was intravenously administered into the rats at the onset of reperfusion after 2 h of cerebral ischemia. PDC treatment dose-dependently reduced the infarct size and inhibited apoptotic cell death in ischemia-reperfused brains (Fig. 5a–d). Meanwhile, PDC administration dose-dependently reduced 3-NT level and mitochondrial LC3-II/LC3-I ratio (Fig. 5e). Further, western blot analysis revealed that PDC inhibited the translocation of Drp1 and Parkin from cytosol to mitochondria during cerebral I/R injury (Fig. 5f, g). Immunofluorescence analysis also showed that PDC reversed the I/R-induced LC3 puncta accumulation around the mitochondria (Fig. 6a). Similarly, PDC intervention blocked the I/R-induced Drp1 redistribution from being ubiquitous in the cytosol to punctate around the mitochondria (Supplementary Fig. 2). Moreover, the Drp1 redistribution was simultaneously presented with increased 3-NT level in ischemia-reperfused brains, as illustrated by immunofluorescence (Fig. 6b). These results suggest that ONOO− could initiate mitophagy activation by recruiting Drp1 to damaged mitochondria, leading to cerebral I/R injury.

FeTMPyP (peroxynitrite decomposition catalyst, PDC) inhibits mitochondrial recruitment of Drp1 and mitophagy activation, attenuating cerebral I/R injury. Rats were treated with PDC (1 and 3 mg/kg) at the onset of reperfusion after 2 h of cerebral ischemia via femoral injection. Brain tissues were prepared 22 h after reperfusion for further detection. TTC staining images (a) and quantification (b) of the infarct size in the groups of sham (n = 5 rats), I2/R22 (n = 7 rats), and I2/R22 plus PDC (PDC 1 mg/kg, n = 5 rats; PDC 3 mg/kg, n = 6 rats). Data are expressed as mean ± SEM. *P < 0.05; ***P < 0.001, versus the vehicle-I2/R22 group (one-way ANOVA followed by Dunnett’s multiple-comparison test). c Representative TUNEL staining (green) for apoptotic cell death in the sham, I2/R22, and I2/R22 plus PDC (3 mg/kg) (n = 4 rats/group). Scale bar, 100 μm. d Quantitative analysis of TUNEL-positive cells. Data are expressed as mean ± SEM. ***P < 0.001, versus the vehicle-I2/R22 group (one-way ANOVA followed by Dunnett’s multiple-comparison test). e Representative western blot images of mitochondrial LC3 and 3-NT, as well as cytosolic 3-NT levels in the sham, vehicle-I2/R22, and PDC-I2/R22 groups. f Western blot analysis for cytosolic Drp1 and Parkin expression levels. g Western blot detection for mitochondrial Drp1, Parkin, and PINK1 expression levels

PDC reverses the redistribution of LC3 and Drp1 during cerebral I/R injury. Brain slices in the sham, vehicle-I2/R22, and PDC 3 mg/kg-I2/R22 groups were prepared 22 h after reperfusion for immunofluorescence. a Co-staining images of LC3 (green), ATPB (red), and nuclear (blue) in the brains of the sham, I2/R22, and I2/R22 plus PDC (3 mg/kg) groups. Scale bar, 50 μm. Immunofluorescent images of representative cells were enlarged (right) (n = 3 rats/group). b Representative co-staining images of Drp1 (green), 3-NT (red), and nuclear (blue) in the brains of corresponding groups. Scale bar, 50 μm. Typical cell images were enlarged (right) (n = 3 rats/group)
ONOO− Triggers Tyrosine Nitration of Drp1
Mitochondria are not only the pools for redox signaling and reactive species but also the targets of free radicals including ONOO− [39, 40]. We then tested how ONOO− triggered mitochondrial recruitment of Drp1 during mitophagy. Western blot analysis following Drp1 immunoprecipitation revealed a rise in the ratio of the nitrated Drp1 to total Drp1 in the I2/R22 brains (Fig. 7a, b), indicating the potential effects of ONOO− on the Drp1 nitration and translocation. Interestingly, comparing the brain samples between the sham and I2/R22 groups, MS analysis revealed that the tyrosine in peptide sequence YPSLANR could be a nitration modification target point of Drp1 protein (Table 1; Supplementary Fig. 3). Consistently, the PDC treatment group had a significant decrease in the nitrated Drp1, compared with the I2/R22 group (Fig. 7c, d). Thus, these data provide a clue that ONOO−-mediated Drp1 tyrosine nitration might initiate Drp1 recruitment in the ischemic brains.

ONOO− triggers Drp1 nitration modification in cerebral ischemia-reperfused rat brains, which is reversed by PDC treatment. Drp1 extracted from the rat brains was pulled down and relatively purified by the immunoprecipitation method. a, b Western blot images (a) and quantification (b) of the ratio of nitrated Drp1 to total Drp1 in the sham and I2/R22 groups with 3-NT antibody. Data are expressed as mean ± SEM. **P = 0.0062 < 0.01 (t-test). Western blot detection (c) and quantitative analysis (d) of Drp1 nitration level in the brains of the sham, vehicle-I2/R22, and PDC 3-I2/R22 groups. Values are expressed as mean ± SEM. **P < 0.01, versus the vehicle-I2/R22 group (one-way ANOVA followed by Dunnett’s multiple-comparison test). Above experiments were repeated three times
ONOO− Donors Initiate Mitochondrial Recruitment of Drp1 and Mitophagy Activation In Vitro
We then performed in vitro experiments to confirm the roles of ONOO− in mediating mitophagy by using ONOO− donor SIN-1 and synthesized sodium ONOO−. Immunofluorescent staining showed that SIN-1 treatment induced the punctate distribution of LC3 in SH-SY5Y cells (Fig. 8a). Concomitantly, SIN-1 treatment induced mitophagy activation, illustrated by the increased fluorescent co-localization of LC3 puncta and mitochondrial marker ATPB (Fig. 8a), as well as Drp1 accumulation around the mitochondria (Fig. 8b). SIN-1 treatment dose-dependently upregulated the level of 3-NT and the ratio of LC3-II/LC3-I and downregulated the level of Bcl-2 and p62 (Fig. 8c). Consistently, PDC treatment reversed these changes in the cells. Alternatively, 3-MA intervention also upregulated the expression of Bcl-2 in the SIN-1-treated cells (Supplementary Fig. 4).

3-Morpholinosydnonimine (SIN-1, ONOO− donor) induces mitochondrial recruitment of Drp1 and mitophagy activation, leading to SH-SY5Y cell death in vitro. a SH-SY5Y cells were treated with 4 mM SIN-1 and vehicle for 2 h. Fifty micromolars of PDC was used as a positive control drug. Representative confocal staining images of LC3 (green), ATPB (red), and nuclear (blue). Scale bar, 50 μm. b SH-SY5Y cells were incubated in SIN-1 at 1, 2, and 4 mM for 2 h. For the positive control group, cells were co-treated with 4 mM SIN-1 and 50 μM PDC. Representative staining images of Drp1 (green), ATPB (red), and nuclear (blue) in each group. Scale bar, 50 μm. c Western blot images of cellular proteins involved in cell death (Bcl-2), autophagy (LC3-II/LC3-I, p62), and ONOO− (3-NT) in the groups of control, SIN-1 (1, 2, and 4 mM), and SIN-1 (4 mM) plus PDC (50 μM). Above in vitro experiments were repeated at least three times
In addition, the experiments with synthesized sodium ONOO− yielded the similar results to SIN-1 donor experiments (Fig. 9). Synthesized sodium ONOO− dose-dependently reduced cell viability and increased cell death, which was attenuated by PDC treatment (Fig. 9a, b). Immunofluorescent analysis showed LC3 was recruited to the mitochondria in SH-SY5Y cells exposed to ONOO−, which was reversed by PDC treatment (Fig. 9c). Notably, PDC treatment reversed ONOO−-induced Drp1 nitration and mitochondrial recruitment in SH-SY5Y cells (Fig. 10). Those in vitro experiments further confirm the roles of ONOO− in Drp1-mediated mitophagic cell death.

Synthesized sodium ONOO− triggers nitrative stress and mitophagy activation, leading to SH-SY5Y cell death. SH-SY5Y cells were incubated in synthesized sodium ONOO− (20, 40, and 80 μM) for 2 h, similar to SIN-1. PDC (50 μM) was treated as a positive scavenger drug. a MTT assay for cell viability after 2 h of incubation with synthesized sodium ONOO−. Data are expressed as mean ± SEM. ***P < 0.001, relative to the control group (one-way ANOVA followed by Dunnett’s multiple-comparison test); ###P < 0.001, relative to ONOO− 80 μM group (t-test). b Western blot analysis for relevant protein expression levels including p62, LC3-II/LC3-I, Bcl-2, and active caspase-3. c Cell slices in each group were prepared after 2 h of treatment for immunofluorescent staining. Representative co-staining images of mitochondrial marker ATPB (red), autophagosome marker LC3 (green), and nuclear (blue). Scale bar, 50 μm. The experiments were repeated at least three times

Synthesized sodium ONOO− triggers Drp1 nitration and mitochondrial recruitment of Drp1, which is inhibited by PDC treatment. SH-SY5Y cells were incubated in synthesized sodium ONOO− (80 μM) and ONOO− (80 μM) plus PDC (50 μM) for 2 h. Then, cell slices and cellular proteins were collected for immunofluorescence and western blot analysis, respectively. a Co-staining images of mitochondrial marker VDAC1 (green), Drp1 (red), and nuclear (blue) in each group. Scale bar, 50 μm. Western blot images (b) and quantification (c) of the level of nitrated Drp1 in sham, ONOO−, and ONOO− plus PDC groups with 3-NT antibody following Drp1 immunoprecipitation. Data are expressed as mean ± SEM. **P < 0.01, versus the vehicle-ONOO− group (one-way ANOVA followed by Dunnett’s multiple-comparison test). Above all experiments were repeated at least three times
Discussion
To our knowledge, this is the first report that ONOO− could recruit Drp1 to damaged mitochondria and subsequently induce PINK1/Parkin-mediated mitophagy activation, contributing to cerebral I/R injury. Potentially, ONOO−-induced mitophagy could be a promising therapeutic target for cerebral I/R injury.
We previously reviewed current progress about the roles of autophagy/mitophagy during cerebral I/R injury and speculated that reperfusion after ischemia could be a critical turning point of autophagy/mitophagy from neuroprotection to neurotoxicity in ischemic brain injury [23]. To verify the speculation, we designed the experiments with different reperfusion time points after cerebral ischemia in the present study. We found that autophagy/mitophagy was increased in the ischemic brains with the extension of reperfusion time after cerebral ischemia. After I2/R22, the ischemic rat brains had a significant increase in the infarct size and autophagic level. Administration of the autophagy inhibitor 3-MA at the onset of reperfusion remarkably reduced the infarct size. Those data directly support the idea about the detrimental roles of activated autophagy at the reperfusion phase. However, it should be mentioned that the infarct size of rat brains in the I2/R22 group anesthetized with isoflurane (Figs. 1a and 5a) was larger than that of rats anesthetized with ketamine and xylazine hydrochloride (Figs. 1e and 3a). Increased infarct size might be attributed to the enhanced cerebral blood flow induced by isoflurane, compared with ketamine and xylazine hydrochloride [41, 42].
Mitochondria, as the power house of the cell, play a key role in many essential cellular functions, including energy metabolism, adenosine triphosphate production, and calcium ion homeostasis [43, 44]. However, under cerebral I/R insults, mitochondria are also the primary intracellular sources of free radicals, such as NO and O2·−. The simultaneous presentation of NO and O2·− rapidly induces ONOO− production. ONOO− possesses stronger lipid membrane permeability than O2·− and reflects both oxidative and nitrosative stress [45]. ONOO− plays a critical role in mediating neuronal cell death and the BBB disruption during cerebral I/R injury [46,47,48]. Due to its strong biomembrane permeability, it could be reflected by NT level, an ONOO− footprint marker, in the peripheral blood [49]. Consistently, we found that the NT level in the plasma was increased in ischemic stroke patients and had an upward trend over time (Fig. 4a, b, d). More importantly, plasma NT level was positively correlated with the neurological deficit score (Fig. 4c). Of note, age and hypertension (Supplementary Table 2), two major risk factors of stroke, have also been reported to have a positive correlation with NT/3-NT [50, 51]. Thus, more rigorous clinical data need to be collected in further study, excluding the potential influence of other factors like age and hypotension.
Mitochondria are often the major targets of ONOO− [32, 33]. The roles of ONOO− in mitochondria turnover and their impacts on brain injury remain unknown. Herein, we found that mitophagy activation was dominated in the I2/R22 rat brains (Fig. 2a, b). Akin to the effects of autophagy, inhibition of mitophagy remarkably reduced the infarct size of rat brains at the stage of reperfusion after cerebral ischemia (Fig. 3). Subsequently, we logically addressed whether ONOO− served as a potential factor contributing to mitophagy activation. Intriguingly, 3-NT level was increased in ischemia-reperfused rat brains, accompanied with mitochondrial recruitment of Drp1, PINK1/Parkin-mediated mitophagy activation, and enhanced apoptosis. Moreover, PDC dose-dependently reversed the increase in 3-NT level, suppressed Drp1 recruitment and mitophagy activation, and reduced apoptotic cell death after I2/R22 injury. These results conveyed an important message that ONOO− could play a crucial role in PINK1/Parkin-mediated mitophagy via triggering mitochondria recruitment of Drp1, leading to cerebral I/R injury.
The Drp1’s mitochondrial recruitment plays an important role in mitochondrial fission and fragmentation during mitophagy. The Drp1 recruitment is regulated by the activity of Drp1 via modification, such as phosphorylation [17] and nitrosylation [30]. Actually, mitochondrial proteins are comparatively vulnerable to nitrative stress [32, 33]. Thus, we investigated the effects of ONOO− on mitochondrial recruitment of Drp1 during cerebral I/R injury. The results showed that ONOO− recruited Drp1 to the damaged mitochondria and then activated mitophagy in the ischemic brain. More excitingly, we found the tyrosine nitration of Drp1 peptide YPSLANR might contribute to Drp1 recruitment in vivo (Table 1; Supplementary Fig. 3), and PDC significantly attenuated Drp1 nitration and recruitment (Fig. 7c, d; Supplementary Fig. 2). Besides, we confirmed these findings on SH-SY5Y cells after treatment of synthesized sodium ONOO− in vitro (Fig. 10). To our knowledge, this is the first evidence that ONOO− could induce Drp1 nitration and mitochondrial recruitment in the ischemia-reperfused brains. However, the precise impacts of Drp1 nitration on Drp1 mitochondrial recruitment and mitophagy activation remain obscure. Thus, more experiments need to be conducted to exclude other potential modification influences and elucidate exact effects of tyrosine nitration on Drp1 mitochondrial recruitment.
It has been widely accepted that ONOO− is a critical factor to induce DNA damage and cell apoptosis, contributing to the pathological process in ischemic stroke [52, 53]. However, it is needed to be further addressed about how ONOO− induces apoptotic cell death. In the present study, we found that ONOO− mediated excessive autophagy/mitophagy activation under cerebral I/R insults. Notably, direct and indirect evidences indicate that autophagy/mitophagy has a close interaction with apoptosis via a mechanistic overlap [54, 55]. The autophagy protein like Atg12 has been reported to initiate mitochondrial apoptosis via binding and inactivating prosurvival Bcl-2 family members in response to the stress [56]. Additionally, autophagosomal membrane was revealed to serve as a platform for the recruitment of caspase-8 to initiate the caspase-8/caspase-3 cascade [57]. Consistently, our data showed that apoptosis and autophagy/mitophagy were simultaneously activated during cerebral I/R injury, and the autophagy inhibitor 3-MA remarkably upregulated Bcl-2/Bax ratio in ischemia-reperfused brains (Fig. 1g). Moreover, the in vitro experiment also showed that 3-MA intervening led to an increase of Bcl-2 expression in SH-SY5Y cells exposed to ONOO− donor SIN-1 treatment (Supplementary Fig. 4). Of course, these findings just provide a clue to explain the relationship between autophagy/mitophagy and apoptosis during cerebral I/R injury. Obviously, further study is needed to elucidate the in-depth underlying mechanisms.
Of note, it is worthy to discuss the reliability of several experimental methods, including mitochondrial isolation referring to reagent-based protocol and 3-MA administration. It is critical to avoid the potential contamination with autophagosomes. Otherwise, the ratio of mitochondrial LC3-II/LC3-I may be due to the co-enrichment with autophagosomes in the mitochondrial fraction. To avoid the potential contamination of autophagosomes maximally, we chose the centrifuging at 3000×g, instead of 12,000×g. Centrifuging at 3000×g often results in less yield, but less contamination. Besides the reagent-based method, another mitochondrial isolation protocol with Dounce homogenization was also applied for quality control. The data showed that the ratio of LC3-II/LC3-I in mitochondrial fraction was enhanced in ischemia-reperfused rat brains after separating with above two protocols (Supplementary Fig. 5). Furthermore, immunofluorescent images also indicated the induction of mitophagy activation by co-staining of the mitochondria marker ATPB and the autophagosome marker LC3.
To date, efforts have been made for searching a single silver bullet for autophagy regulation, like finding a fitting shoe for Cinderella. But, no one is perfect yet [58]. 3-MA, as the first generation of autophagy inhibitor, has been applied for autophagy study [59]. However, it was reported that the concentration of 3-MA can influence its effects on autophagy. For instance, 3-MA at 2.5 mM did not show inhibitory effects on the basal level of autophagy but reversely triggered autophagic flux [60]. To some extent, well-designed experiments could overcome some limitations of 3-MA. To ensure its working solution and minimize the side effects of the peripheral system, we directly injected 3-MA (600 nmol, 5 μL) into the cerebral lateral ventricle. Western blot analysis also showed the inhibitory effects of 3-MA on autophagy (Fig. 1g). Of course, with the development of tools, more specific agents would pave a way for autophagy study in the future.
Taken together, we advanced the underlying mechanisms of activated mitophagy during cerebral I/R injury. As illustrated in the diagram (Fig. 11), ONOO− could induce PINK1/Parkin-mediated mitophagy activation via recruiting Drp1 to damaged mitochondria, leading to cerebral I/R injury. Thus, ONOO−-induced mitophagy could be a novel molecular target for seeking therapeutic drugs to protect the brain against I/R injury, which might have a significant implication for a combined therapeutic strategy with thrombolytic treatment.

ONOO− induces PINK1/Parkin-mediated mitophagy activation via recruiting Drp1 to damaged mitochondria, contributing to cerebral I/R injury. At the onset of reperfusion after cerebral ischemia, a large amount of ONOO− generates and induces nitrative stress. Then, ONOO− triggers Drp1 nitration modification and recruits Drp1 to damaged mitochondria, which are divided into functionally uneven two parts: healthy daughter mitochondria (green) and dysfunctional ones (red). Subsequently, PINK1/Parkin pathway is activated to initiate mitophagic flux to remove the daughter mitochondria with decreased membrane potential (red) by autophagosomes. Once mitophagy is over-activated by a consistent stimulus of ONOO−, it will aggravate cerebral I/R injury. Obviously, ONOO−-induced mitophagy could be a novel molecular target for seeking therapeutic drugs to protect the brain against I/R injury
References
Ohsumi Y (2014) Historical landmarks of autophagy research. Cell Res 24(1):9–23. https://doi.org/10.1038/cr.2013.169
Boya P, Reggiori F, Codogno P (2013) Emerging regulation and functions of autophagy. Nat Cell Biol 15(7):713–720. https://doi.org/10.1038/ncb2788
Tian F, Deguchi K, Yamashita T, Ohta Y, Morimoto N, Shang J, Zhang X, Liu N et al (2010) In vivo imaging of autophagy in a mouse stroke model. Autophagy 6(8):1107–1114. https://doi.org/10.4161/auto.6.8.13427
Lu Q, Harris VA, Kumar S, Mansour HM, Black SM (2015) Autophagy in neonatal hypoxia ischemic brain is associated with oxidative stress. Redox Biol 6:516–523. https://doi.org/10.1016/j.redox.2015.06.016
Li H, Qiu S, Li X, Li M, Peng Y (2015) Autophagy biomarkers in CSF correlates with infarct size, clinical severity and neurological outcome in AIS patients. J Transl Med 13(1):359. https://doi.org/10.1186/s12967-015-0726-3
Frugier T, Taylor JM, McLean C, Bye N, Beart PM, Devenish RJ, Crack PJ (2016) Evidence for the recruitment of autophagic vesicles in human brain after stroke. Neurochem Int 96:62–68. https://doi.org/10.1016/j.neuint.2016.02.016
Chen W, Sun Y, Liu K, Sun X (2014) Autophagy: a double-edged sword for neuronal survival after cerebral ischemia. Neural Regen Res 9(12):1210–1216. https://doi.org/10.4103/1673-5374.135329
Wei K, Wang P, Miao CY (2012) A double-edged sword with therapeutic potential: an updated role of autophagy in ischemic cerebral injury. CNS Neurosci Ther 18(11):879–886. https://doi.org/10.1111/cns.12005
Yuan Y, Zhang X, Zheng Y, Chen Z (2015) Regulation of mitophagy in ischemic brain injury. Neurosci Bull 31(4):395–406. https://doi.org/10.1007/s12264-015-1544-6
Shi RY, Zhu SH, Li V, Gibson SB, XS X, Kong JM (2014) BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci Ther 20(12):1045–1055. https://doi.org/10.1111/cns.12325
Dolman NJ, Chambers KM, Mandavilli B, Batchelor RH, Janes MS (2013) Tools and techniques to measure mitophagy using fluorescence microscopy. Autophagy 9(11):1653–1662. https://doi.org/10.4161/auto.24001
Liu K, Sun Y, Gu Z, Shi N, Zhang T, Sun X (2013) Mitophagy in ischaemia/reperfusion induced cerebral injury. Neurochem Res 38(7):1295–1300. https://doi.org/10.1007/s11064-013-1033-0
Michalska B, Duszynski J, Szymanski J (2016) Mechanism of mitochondrial fission—structure and function of Drp1 protein. Postepy Biochem 62(2):127–137
Cho B, Choi SY, Cho HM, Kim HJ, Sun W (2013) Physiological and pathological significance of dynamin-related protein 1 (Drp1)-dependent mitochondrial fission in the nervous system. Exp Neurobiol 22(3):149–157. https://doi.org/10.5607/en.2013.22.3.149
Zuo W, Yang PF, Chen J, Zhang Z, Chen NH (2016) Drp-1, a potential therapeutic target for brain ischaemic stroke. Br J Pharmacol 173(10):1665–1677. https://doi.org/10.1111/bph.13468
Zuo W, Zhang S, Xia CY, Guo XF, He WB, Chen NH (2014) Mitochondria autophagy is induced after hypoxic/ischemic stress in a Drp1 dependent manner: the role of inhibition of Drp1 in ischemic brain damage. Neuropharmacology 86:103–115. https://doi.org/10.1016/j.neuropharm.2014.07.002
Buhlman L, Damiano M, Bertolin G, Ferrando-Miguel R, Lombes A, Brice A, Corti O (2014) Functional interplay between Parkin and Drp1 in mitochondrial fission and clearance. Biochim Biophys Acta 1843(9):2012–2026. https://doi.org/10.1016/j.bbamcr.2014.05.012
Tang YC, Tian HX, Yi T, Chen HB (2016) The critical roles of mitophagy in cerebral ischemia. Protein Cell 7(10):699–713. https://doi.org/10.1007/s13238-016-0307-0
Lin C, Chao H, Li Z, Xu X, Liu Y, Hou L, Liu N, Ji J (2016) Melatonin attenuates traumatic brain injury-induced inflammation: a possible role for mitophagy. J Pineal Res 61(2):177–186. https://doi.org/10.1111/jpi.12337
Li Q, Zhang T, Wang J, Zhang Z, Zhai Y, Yang GY, Sun X (2014) Rapamycin attenuates mitochondrial dysfunction via activation of mitophagy in experimental ischemic stroke. Biochem Biophys Res Commun 444(2):182–188. https://doi.org/10.1016/j.bbrc.2014.01.032
Zhang XM, Zhang L, Wang G, Niu W, He Z, Ding L, Jia J (2015) Suppression of mitochondrial fission in experimental cerebral ischemia: the potential neuroprotective target of p38 MAPK inhibition. Neurochem Int 90:1–8. https://doi.org/10.1016/j.neuint.2015.06.010
Baek SH, Noh AR, Kim KA, Akram M, Shin YJ, Kim ES, SW Y, Majid A et al (2014) Modulation of mitochondrial function and autophagy mediates carnosine neuroprotection against ischemic brain damage. Stroke 45(8):2438–2443. https://doi.org/10.1161/STROKEAHA.114.005183
Feng J, Chen X, Shen J (2017) Reactive nitrogen species as therapeutic targets for autophagy: implication for ischemic stroke. Expert Opin Ther Targets 21(3):305–317. https://doi.org/10.1080/14728222.2017.1281250
Ansari S, Rahman M, Waters MF, Hoh BL, Mocco J (2011) Recanalization therapy for acute ischemic stroke, part 1: surgical embolectomy and chemical thrombolysis. Neurosurg Rev 34(1):1–9. https://doi.org/10.1007/s10143-010-0293-2
Gomis M, Davalos A (2014) Recanalization and reperfusion therapies of acute ischemic stroke: what have we learned, what are the major research questions, and where are we headed? Front Neurol 5:226. https://doi.org/10.3389/fneur.2014.00226
Shirley R, Ord EN, Work LM (2014) Oxidative stress and the use of antioxidants in stroke. Antioxidants (Basel) 3(3):472–501. https://doi.org/10.3390/antiox3030472
Chen XM, Chen HS, MJ X, Shen JG (2013) Targeting reactive nitrogen species: a promising therapeutic strategy for cerebral ischemia-reperfusion injury. Acta Pharmacol Sin 34(1):67–77. https://doi.org/10.1038/aps.2012.82
Kuhn DM, Sakowski SA, Sadidi M, Geddes TJ (2004) Nitrotyrosine as a marker for peroxynitrite-induced neurotoxicity: the beginning or the end of the end of dopamine neurons? J Neurochem 89(3):529–536. https://doi.org/10.1111/j.1471-4159.2004.02346.x
Ding R, Chen Y, Yang S, Deng X, Fu Z, Feng L, Cai Y, Du M et al (2014) Blood-brain barrier disruption induced by hemoglobin in vivo: Involvement of up-regulation of nitric oxide synthase and peroxynitrite formation. Brain Res 1571:25–38. https://doi.org/10.1016/j.brainres.2014.04.042
Nakamura T, Cieplak P, Cho DH, Godzik A, Lipton SA (2010) S-nitrosylation of Drp1 links excessive mitochondrial fission to neuronal injury in neurodegeneration. Mitochondrion 10(5):573–578. https://doi.org/10.1016/j.mito.2010.04.007
Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA (2009) S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 324(5923):102–105. https://doi.org/10.1126/science.1171091
Liu B, Tewari AK, Zhang L, Green-Church KB, Zweier JL, Chen YR, He G (2009) Proteomic analysis of protein tyrosine nitration after ischemia reperfusion injury: mitochondria as the major target. Biochim Biophys Acta 1794(3):476–485. https://doi.org/10.1016/j.bbapap.2008.12.008
Vattemi G, Mechref Y, Marini M, Tonin P, Minuz P, Grigoli L, Guglielmi V, Klouckova I et al (2011) Increased protein nitration in mitochondrial diseases: evidence for vessel wall involvement. Mol Cell Proteomics 10(4):M110 002964. https://doi.org/10.1074/mcp.M110.002964
Lozano-Juste J, Colom-Moreno R, Leon J (2011) In vivo protein tyrosine nitration in Arabidopsis thaliana. J Exp Bot 62(10):3501–3517. https://doi.org/10.1093/jxb/err042
Hatano S (1976) Experience from a multicentre stroke register: a preliminary report. Bull World Health Organ 54(5):541–553
Brott T, Adams HP Jr, Olinger CP, Marler JR, Barsan WG, Biller J, Spilker J, Holleran R et al (1989) Measurements of acute cerebral infarction: a clinical examination scale. Stroke 20(7):864–870. https://doi.org/10.1161/01.STR.20.7.864
Sebastian D, Palacin M, Zorzano A (2017) Mitochondrial dynamics: coupling mitochondrial fitness with healthy aging. Trends Mol Med 23(3):201–215. https://doi.org/10.1016/j.molmed.2017.01.003
Ashrafi G, Schwarz TL (2013) The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 20(1):31–42. https://doi.org/10.1038/cdd.2012.81
Grivennikova VG, Kareyeva AV, Vinogradov AD (2010) What are the sources of hydrogen peroxide production by heart mitochondria? Biochim Biophys Acta 1797(6–7):939–944. https://doi.org/10.1016/j.bbabio.2010.02.013
Kang J, Pervaiz S (2012) Mitochondria: redox metabolism and dysfunction. Biochem Res Int 2012:1–14. https://doi.org/10.1155/2012/896751
Horn T, Klein J (2013) Neuroprotective effects of lactate in brain ischemia: dependence on anesthetic drugs. Neurochem Int 62(3):251–257. https://doi.org/10.1016/j.neuint.2012.12.017
Bleilevens C, Roehl AB, Goetzenich A, Zoremba N, Kipp M, Dang J, Tolba R, Rossaint R et al (2013) Effect of anesthesia and cerebral blood flow on neuronal injury in a rat middle cerebral artery occlusion (MCAO) model. Exp Brain Res 224(2):155–164. https://doi.org/10.1007/s00221-012-3296-0
Zavodnik IB (2016) Mitochondria, calcium homeostasis and calcium signaling. Biomed Khim 62(3):311–317. https://doi.org/10.18097/PBMC20166203311
Klingenberg M (2008) The ADP and ATP transport in mitochondria and its carrier. Biochim Biophys Acta 1778(10):1978–2021. https://doi.org/10.1016/j.bbamem.2008.04.011
Ferrer-Sueta G, Radi R (2009) Chemical biology of peroxynitrite: kinetics, diffusion, and radicals. ACS Chem Biol 4(3):161–177. https://doi.org/10.1021/cb800279q
Gong J, Sun F, Li Y, Zhou X, Duan Z, Duan F, Zhao L, Chen H et al (2015) Momordica charantia polysaccharides could protect against cerebral ischemia/reperfusion injury through inhibiting oxidative stress mediated c-Jun N-terminal kinase 3 signaling pathway. Neuropharmacology 91:123–134. https://doi.org/10.1016/j.neuropharm.2014.11.020
Chen HS, Chen XM, Feng JH, Liu KJ, Qi SH, Shen JG (2015) Peroxynitrite decomposition catalyst reduces delayed thrombolysis-induced hemorrhagic transformation in ischemia-reperfused rat brains. CNS Neurosci Ther 21(7):585–590. https://doi.org/10.1111/cns.12406
Xu M, Chen X, Gu Y, Peng T, Yang D, Chang RC, So KF, Liu K et al (2013) Baicalin can scavenge peroxynitrite and ameliorate endogenous peroxynitrite-mediated neurotoxicity in cerebral ischemia-reperfusion injury. J Ethnopharmacol 150(1):116–124. https://doi.org/10.1016/j.jep.2013.08.020
Bas DF, Topcuoglu MA, Gursoy-Ozdemir Y, Saatci I, Bodur E, Dalkara T (2012) Plasma 3-nitrotyrosine estimates the reperfusion-induced cerebrovascular stress, whereas matrix metalloproteinases mainly reflect plasma activity: a study in patients treated with thrombolysis or endovascular recanalization. J Neurochem 123 Suppl 2:138–147. https://doi.org/10.1111/j.1471-4159.2012.07952.x
Shin CM, Chung YH, Kim MJ, Lee EY, Kim EG, Cha CI (2002) Age-related changes in the distribution of nitrotyrosine in the cerebral cortex and hippocampus of rats. Brain Res 931(2):194–199. https://doi.org/10.1016/S0006-8993(01)03391-1
Murdaugh LS, Wang Z, Del Priore LV, Dillon J, Gaillard ER (2010) Age-related accumulation of 3-nitrotyrosine and nitro-A2E in human Bruch’s membrane. Exp Eye Res 90(5):564–571. https://doi.org/10.1016/j.exer.2010.01.014
Ahmad R, Rasheed Z, Ahsan H (2009) Biochemical and cellular toxicology of peroxynitrite: implications in cell death and autoimmune phenomenon. Immunopharmacol Immunotoxicol 31(3):388–396. https://doi.org/10.1080/08923970802709197
Ramdial K, Franco MC, Estevez AG (2017) Cellular mechanisms of peroxynitrite-induced neuronal death. Brain Res Bull 133:4–11. https://doi.org/10.1016/j.brainresbull.2017.05.008
Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N (2013) Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta 1833(12):3448–3459. https://doi.org/10.1016/j.bbamcr.2013.06.001
Booth LA, Tavallai S, Hamed HA, Cruickshanks N, Dent P (2014) The role of cell signalling in the crosstalk between autophagy and apoptosis. Cell Signal 26(3):549–555. https://doi.org/10.1016/j.cellsig.2013.11.028
Rubinstein AD, Eisenstein M, Ber Y, Bialik S, Kimchi A (2011) The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol Cell 44(5):698–709. https://doi.org/10.1016/j.molcel.2011.10.014
Young MM, Takahashi Y, Khan O, Park S, Hori T, Yun J, Sharma AK, Amin S et al (2012) Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem 287(15):12455–12468. https://doi.org/10.1074/jbc.M111.309104
Nagelkerke A, Bussink J, Geurts-Moespot A, Sweep FC, Span PN (2015) Therapeutic targeting of autophagy in cancer. Part II: pharmacological modulation of treatment-induced autophagy. Semin Cancer Biol 31:99–105. https://doi.org/10.1016/j.semcancer.2014.06.001
Seglen PO, Gordon PB (1982) 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A 79(6):1889–1892. https://doi.org/10.1073/pnas.79.6.1889
Zheng XY, Li LJ, Li W, Jiang PF, Shen HQ, Chen YH, Chen X (2015) Low concentrations of chloroquine and 3-methyladenine suppress the viability of retinoblastoma cells synergistically with vincristine independent of autophagy inhibition. Graefes Arch Clin Exp Ophthalmol 253(12):2309–2315. https://doi.org/10.1007/s00417-015-3157-1
Acknowledgements
We thank the Faculty Core Facility, Li Ka Shing Faculty of Medicine, The University of Hong Kong, for providing the Carl Zeiss LSM 780 used in capturing confocal fluorescent images. We also appreciate the Centre for Genomic Sciences, Li Ka Shing Faculty of Medicine, The University of Hong Kong, for supplying the MALDI-TOF/TOF tandem mass spectrometry analysis service. Alternatively, we greatly appreciate Lingen Kong, Chunsheng Cai, and Qianwu Zhou in the Department of Neurology and the Clinical Laboratory of Huizhou First People’s Hospital, Huizhou.
Funding
This work was supported by the National Natural Science Foundation of China (No. 31570855) and the Research Grants Council, University Grants Committee (No. 776512M) and RGC Area of Excellence Sheme (AoE/P-705/16).
Author information
Authors and Affiliations
Contributions
J.-H.F. designed and performed the experiments, analyzed the data, and wrote the manuscript. X.-M.C. and B.-H.G. carried out the clinical trial, detected the ELISA analysis, and analyzed the data. C.-M.L. and J.-H.Q. supervised the clinical trial. J.-G.S conceived of and supervised this research, designed the experiments, and co-wrote the manuscript.
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Electronic supplementary material
ESM 1
(DOC 6162 kb)
Rights and permissions
About this article

Cite this article
Feng, J., Chen, X., Guan, B. et al. Inhibition of Peroxynitrite-Induced Mitophagy Activation Attenuates Cerebral Ischemia-Reperfusion Injury. Mol Neurobiol 55, 6369–6386 (2018). https://doi.org/10.1007/s12035-017-0859-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0859-x