
Abstract
The exposure to extremely low-frequency magnetic fields (ELF-MFs) has been associated to increased risk of neurodegenerative diseases, although the underlying molecular mechanisms are still undefined. Since epigenetic modulation has been recently encountered among the key events leading to neuronal degeneration, we here aimed at assessing if the control of gene expression mediated by miRNAs, namely miRs-34, has any roles in driving neuronal cell response to 50-Hz (1 mT) magnetic field in vitro. We demonstrate that ELF-MFs drive an early reduction of the expression level of miR-34b and miR-34c in SH-SY5Y human neuroblastoma cells, as well as in mouse primary cortical neurons, by affecting the transcription of the common pri-miR-34. This modulation is not p53 dependent, but attributable to the hyper-methylation of the CpG island mapping within the miR-34b/c promoter. Incubation with N-acetyl-l-cysteine or glutathione ethyl-ester fails to restore miR-34b/c expression, suggesting that miRs-34 are not responsive to ELF-MF-induced oxidative stress. By contrast, we show that miRs-34 control reactive oxygen species production and affect mitochondrial oxidative stress triggered by ELF-MFs, likely by modulating mitochondria-related miR-34 targets identified by in silico analysis. We finally demonstrate that ELF-MFs alter the expression of the α-synuclein, which is specifically stimulated upon ELF-MFs exposure via both direct miR-34 targeting and oxidative stress. Altogether, our data highlight the potential of the ELF-MFs to tune redox homeostasis and epigenetic control of gene expression in vitro and shed light on the possible mechanism(s) producing detrimental effects and predisposing neurons to degeneration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Over the last decades, the Western societies have been experiencing an increasing exposure to different sources of non-ionizing radiations arising from both environmental and domestic devices, as a consequence of urbanization, sustained economic progress, and widespread diffusion of technological applications. This represents a subject of growing concern to public health as whether the electromagnetic fields (EMFs) may elicit pathogenic effects is still matter of debate [1]. Based on an increased risk for childhood leukemia and glioma associated with residential extremely low-frequency (ELF) magnetic field (MF) and wireless phone use, both ELF and radiofrequency (RF) EMFs have been classified as “possibly carcinogenic to humans” by the International Agency for Research on Cancer (IARC) [2, 3]. Occupational exposure to ELF-MFs has been also associated with an increased risk of neurodegenerative diseases, mainly Alzheimer’s disease (AD) and amyotrophic lateral sclerosis (ALS), whereas a univocal association with Parkinson’s disease (PD) patients is still lacking [4,5,6,7,8]. A major drawback in assessing the possible impact of ELF-MFs on human pathologies relies on difficulty to establish a causal association between unfavorable health effects and radiations or whether it might be somehow influenced by additional/unrelated factors [9]. Furthermore, ELF-MFs are non-ionizing radiations, making the specific molecular targets and biological effectors hard to be identified.
In this context, ELF-MFs have been generating most attention as environmental factors playing relevant roles in neurodegenerative aetiopathogenesis, since their interaction with in vitro and in vivo neuronal systems directly implies the occurrence of oxidative stress. Indeed, oxidative stress is a major responsible for dysfunction and, eventually, death of brain cells, in which the high oxygen consumption and the rich-in-lipid content make them particularly vulnerable to oxidative damage [7, 10,11,12]. It has been demonstrated that ELF-MFs stabilize reactive oxygen and nitrogen species (ROS/RNS), and deplete neurons of their antioxidant defenses [13, 14]. In this context, we previously demonstrated that 50-Hz MF (1 mT) (i) directly induces redox imbalance and leads to severe protein carbonylation in proliferating and dopaminergic (DAergic) SH-SY5Y cells [15] and (ii) further sensitizes neurons to MPP+(1-methyl-4-phenylpyridinium) [15], a neurotoxin used to mimic PD symptoms, thus providing a clear-cut evidence for a detrimental role of ELF-MFs in an in vitro PD experimental model.
Notwithstanding redox imbalance through mitochondrial dysfunction exerts a key role in neurodegenerative diseases, the specific molecular mechanisms underlying PD have not yet been exhaustively elucidated. Both genetic and epigenetic components have been identified as co-players in the development of Parkinsonism through a complex network of expression control in response to environmental stimuli [16]. Both components synergistically concur to the accumulation of peculiar intra-cytoplasmic neuronal inclusions of α-synuclein (park1/SNCA) and progressive loss of DAergic neurons in the substantia nigra pars compacta). Along with the well-characterized mutations of the Park genes, the dysfunction of the epigenetic machinery—namely DNA methylation, histone modification and control of gene expression by small non-coding RNAs (sncRNAs)—has been also associated with PD aetiopathogenesis [17,18,19,20,21,22].
MicroRNAs (miRNAs/miRs) are sncRNAs molecules that post-transcriptionally regulate gene expression by binding to the 3’UTR (Un-Translated Region) of specific target mRNAs and trigger transcript degradation and/or translation inhibition [23]. A number of miRs have been so far associated with the DA phenotype of PD, mainly the miR-133b and the miR-7, targeting the Pitx3 transcriptional activator and the Snca, respectively [24,25,26]. Evidences for a direct epigenetic regulation of the typical familial PD-linked genes are indeed emerging, the Snca being the most studied target even in sporadic PD cases [27,28,29]. Moreover, a global miRNA profiling study disclosed a specific downregulation of both miR-34b and miR-34c in PD brain samples compared to healthy tissues, which was associated with a severe mitochondria dysfunction, oxidative stress and reduction of total cellular ATP content, early detectable in the disease course of untreated patients [30]. Interestingly, miRs-34 have been also linked with aging in Drosophila experimental models, with their loss leading to brain degeneration and decline in flies survival [31]. Furthermore, miR-34 family has recently been characterized for targeting the Snca 3’UTR in an in vitro neuroblastoma experimental model [32].
In both human and mouse genome, miR-34b and miR-34c locus is a gene cluster that undergoes transcription into a primary transcript (pri-miR) further processed to release mature 34b and 34c species. They mediate cell cycle arrest, apoptosis, and metabolic regulation and act as potential tumor suppressors [33,34,35,36]. MicroRNA-34b/c promoter displays different p53 response elements, as well as a conserved CpG island region often associated with aberrant hyper-methylation in cancer tumors. In addition, it has been reported to co-regulate the B cell translocation gene 4 (Btg4) through a bidirectional promoter activity [37,38,39,40,41,42,43].
We here aim at evaluating whether the ELF-MF (50 Hz, 1 mT) exposure affects the epigenetic machinery of neuronal cells in vitro, specifically in terms of miR-34b and 34c expression. We demonstrate that both proliferating and DAergic SH-SY5Y, as well as mouse primary cortical neurons (PCNs), are strongly depleted of both miRs upon ELF exposure, due to the inhibition of the pri-miR-34 expression at transcriptional level. miR-34b/c impairment is independent of p53 regulation and oxidative stress, whereas it is driven by promoter CpG island hyper-methylation, and directly contributes to the MF-induced oxidative damage. Exposure to the 50-Hz MF further prompts neuronal cells toward a degenerative phenotype via miR-34b/c direct stimulation of the Snca level. As overall, these findings strengthen the pro-oxidant and pro-degenerative properties of the 50-Hz MF in neuronal cells through the direct epigenetic regulation of the microRNA-34b/c; although so far achieved in in vitro experimental models, our data might help support the hypothesis of a possible pathogenic role of the ELF-MF in brain, specifically related to the PD phenotype.
Materials and Methods
Chemicals
Culture media, serum and supplements, trypsin-EDTA, phosphate buffer saline (PBS), and Hank’s balanced salt solution (HBSS) were obtained from Euroclone (Milan, Italy). B27 supplement, l-buthionine sulfoximine (BSO), 5-aza-2-deoxycytidine (DAC), 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI), diethylpyrocarbonate (DEPC), dimethyl sulfoxide (DMSO), doxorubicin, ethidium bromide, ethylene-diamine-tetra-acetic acid (EDTA), glutathione ethyl ester (GSHest), H2O2, N-acetyl-l-cysteine (NAC), Nutlin-3a, poly-d-lysine, phorbol 12-myristate 13-acetate (PMA), all-trans retinoic acid (RA), and trypan blue solution (0.4%) were purchased from SIGMA Aldrich (Milan, Italy). The fluorescent probes dihydroethidium (hydroethidine, DHE), 2′,7′-dichlorofluorescin diacetate (H2-DCFDA), and MitoTracker Green were obtained from Molecular Probes (Thermo Fisher Scientific, Waltham, MA, USA). Synthetic miRNA sequences (Mimic) and LNA knock-down miR (anti-miR) molecules were purchased from Dharmacon (Lafayette, CO, USA) and Exiqon (Denmark), respectively; the Interferin was obtained from Polyplus (Illkirch, France).
Exposure System
The ELF-MF exposure system consists of two couples of square coils (two coils for each sub-system, arranged coaxially in Helmholtz configuration), as previously detailed [15]. Briefly, the coils are connected to a Variac (40NC) for voltage feeding and current circulation within the cable turns. The two systems are used for inducing magnetic field (B-field) and sham exposure of the biological samples at the same time, thus allowing blind experimental conditions. The coil double wire configuration is used for sham exposure implementation, which allows to obtain a null B-field by using currents flowing in opposite directions. The B-field produced by these systems, at the operating frequency of 50 Hz, was set at a root mean square (RMS) amplitude of 1 mT for a supplied current of 3.4 A. The sham exposures were performed at a residual B-field amplitude of about 0.3 μT (RMS), this representing the background field emitted by the incubator electronics. B-field measurements were performed at the center of the exposure volume of each couple of coils (20 × 20 × 10 cm3) with an isotropic probe (ELT400; Narda, Pfullingen, Germany), in both sham and real exposure configurations. To assess the B-field homogeneity within the exposure setup and the induced electric field (E field) values within the culture samples contained in Petri dishes, numerical simulations were carried out using a finite element method [15]. Globally, our experimental and numerical data guaranteed a high homogeneity (95%) for B-field in the exposure volume, and the proper positioning of the biological samples for rigorously controlled and repeatable exposure conditions [15]. In order to guarantee the temperature stability (37 °C) inside the incubators, a refrigerating system, consisting of water from two separate thermostatic baths and circulating in plastic tubes surrounding the coils, was set-up to prevent heating due to ohmic losses, thus maintaining the temperature of both H-field and sham-exposed samples at 37.0 ± 0.2 °C. Temperature was monitored in the exposure volume of each incubator by two T-thermocouple probes (SENSORTEK Inc., Clifton, NJ, USA), placed in a dummy Petri dish and in air.
Human Cell Cultures and Mouse Primary Cortical Neurons
Human SH-SY5Y neuroblastoma cells were purchased from the European Collection of Cell Culture, cultured in complete Dulbecco’s modified Eagle’s medium/Ham’s F12 (DMEM/F12 (50:50 mix, Euroclone), supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 100 μg/ml penicillin-streptomycin, and kept in culture up to 15 passages. The cells were maintained at 37 °C in a 5% CO2 atmosphere in air and routinely trypsinized and plated at 4 × 104/cm2 on flasks. Cell viability was assessed by Trypan blue dye exclusion. At 60–70% confluence, cells were sub-cultured and differentiated into a dopaminergic phenotype with 5 μM RA for the first 3 days, followed by 100 nM PMA for further 3 days in reduced serum condition (5%) [15].
Mouse primary cortical neurons (PCNs) were obtained from cerebral cortices of E15 C57BL/6 mice embryos, as previously reported [44]. Animals were used in accordance with both international guidelines and Italian law and always applying the 3Rs principle; expert and trained staff routinely monitored the health status of all animals enrolled in the study. Minced cortices were digested with trypsin-EDTA 0.25% at 37 °C for 7 min. Cells were stained with 0.08% Trypan blue solution and only viable cells were counted and plated at the density of 1 × 105/cm2 onto poly-d-lysine coated multi-well plates in 25 mM glucose-containing MEM medium supplemented with 10% fetal bovine serum, 2 mM glutamine, and 0.1 mg/mL gentamicin. After 1 h, the medium was replaced with Neurobasal medium containing antioxidant-free B27 supplement, 2 mM glutamine, and 0.1 mg/mL gentamicin. Cell cultures were kept at 37 °C in a humidified atmosphere containing 5% CO2. Every 3 days, one third of the medium was replaced.
ELF Exposure, Cell Treatments, and Transfections
For ELF-MF exposure (50 Hz, 1 mT), human cells were seeded in 60-mm Petri dishes and placed in either ELF-MF or sham incubator (under blind condition) 24 h after plating. Mouse PCNs were seeded in 24-multiwell and exposed to the MF or Sham incubator (under blind condition) 5 days after plating. All cells underwent a continuous exposure to either ELF-MF (50-Hz, 1 mT) or sham over a time window of 4–72 h [15].
Nutlin-3a and DAC were dissolved in DMSO, freshly diluted in PBS before each experiment, and added at final concentration of 50 μM (Nutlin-3a) and 10 μM (DAC) to culture medium 6 h before MF exposure. BSO, NAC and GSHest were dissolved in PBS to 100 mM stock and filtered, then diluted with serum-free DMEM/F12 medium to final concentrations and maintained throughout the experiment. Since GSH solution is highly acidic, pH was adjusted with 7.5% NaHCO3. Cells were given GSHest (1 mM) and NAC (1 mM) 6 h before ELF-MF switch-on. In Sham-exposed cells, BSO (5 mM) was administered for 24 h; H2O2 was added at 5 and 25 μM for 2 h [15].
Transient transfection experiments with Mimic and anti-miR molecules were carried as previously reported [45]; lyophilized molecules were dissolved in DEPC-treated water, stocked and freshly diluted before experiments, according to manufacturer’s instructions. Twenty-four hours after plating, cells were transfected by Interferin with either anti-miR agents (5–20 nM) or Mimic molecules (25 nM), then exposed to the Sham/ELF-MF system.
RNA Extraction, Reverse Transcription, and Gene Expression Analysis
Total RNA was extracted from samples by Trizol® (Invitrogen, Thermo Fisher Scientific) protocol. The amount and purity of the extracted RNA was evaluated by fiber optic spectrophotometer (Nanodrop ND-1000, NanoDrop Technologies, Wilmington, DE, USA) calculating the 230/260 and 260/280 absorbance ratios. Two hundred nanograms of total RNA were retro-transcribed with random primers into total cDNA by TaqMan® Reverse Transcription Reagent (Applied Biosystems, Thermo Fisher Scientific), according to manufactures’ indications [45]. Analysis of the pri-miR-34b/c, p53, Btg4, and Snca expression was carried out with 1 μL of cDNA using SYBR Green master mix (Applied Biosystems) and analyzed on an Eco™ Real-Time PCR System (Illumina, San Diego, CA, USA). All reactions were run in quadruplicate and the relative abundance of the specific mRNA levels was calculated by normalizing to glyceraldehyde-3-phosphate dehydrogenase (GAPDH), β-actin and tubulin expression using the 2-ΔΔCt method [46]. The GAPDH has been included as reference gene in our analysis, as our experiments have been all performed within 72 h of continuous ELF-MF exposure; the time window used in our experimental set-up is much shorter that the 10-day threshold suggested by Falone and colleagues in terms of GAPDH activity stimulation by ELF-MF [47]. The complete list of primer sequence is reported as Online Resource 1.
microRNA Expression Analysis and Target Prediction
Analysis of mature miRNA expression was carried out on total RNA extracted with the miRcury RNA isolation kit (Exiqon). Ten nanograms of total RNA were retro-transcribed by the miRcury LNA universal RT microRNA kit (Exiqon); cDNA was diluted 1:80 and amplified by the miRcury LNA Sybr green master mix and miR-specific LNA PCR primer sets (Exiqon), according to manufacturer’s instructions. All reactions were run in quadruplicate and the relative abundance of each specific miR (133b, 34b, and 34c) was normalized to small nucleolar RNAs (RNUU6, RNU1A1) by applying the 2−ΔΔCt method [46].
The in silico prediction of putative miRNA targets was carried out by integrating the results obtained from the following genome browsers: Sanger miRbase (microrna.sanger.ac.uk), Target Scan (www.targetscan.org), and miRanda (www.microrna.org/microrna/home.do) [45].
Western Blotting
Cells were lysated in a buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% NP-40, and 12 mM Na-deoxycholate (SIGMA Aldrich) supplemented with protease and phosphatase inhibitors cocktail (SIGMA Aldrich). Lysates were centrifuged at 13,000 rpm for 10 min at 4 °C to discard cellular debris. Protein concentration was determined by the Lowry protein assay (Bio-Rad Laboratories, Hercules, CA, USA). Proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes (GE Healthcare Life Science, Europe) that were probed with the following antibodies: anti-P53 and anti-GAPDH (Santa Cruz Biotechnology, CA, USA), anti-SNCA (Cell Signaling Technologies, Danvers, MA, USA). After immuno-staining with appropriate secondary horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse antibodies (SIGMA Aldrich), bands were revealed by a Fluorchem Imaging system (Alpha Innotech, San Leandro, CA, USA), using the Amersham ECL detection system (GE Healthcare Life Science) and quantified by densitometry.
Flow Cytometric Analysis
A FACScan Flow cytometer (Becton Dickinson, Bedford, MA, USA) equipped with a 488-nm argon laser was used for the flow-cytometric analysis. Forward (FSC-H) and side scatters (SSC-H) were used to establish size and granularity parameters, respectively, and to exclude cellular debris from the analysis, in order to delimitate the integer/healthy cells gate (defined R1). Only cells contained in the R1 region were included in the fluorescence analysis. The excitation wavelength was set at 488 nm. The observation wavelength of 530 nm was chosen for green fluorescence and 585 nm for red fluorescence and the intensities of emitted fluorescence were collected on FL1 and FL2/3 channels, respectively. In each measurement, a minimum of 10,000 cells were analyzed. Data were acquired and analyzed using the Cell Quest software (Becton Dickinson). For fluorescence data analysis, the mean fluorescence intensity (MFI) was calculated for each sample as the ratio between the mean fluorescence value in the channel of the probe-labeled cells versus the unstained ones (negative).
To quantify oxidative stress and mitochondria integrity, cells were stained with DHE (superoxide), H2DCF-DA (H2O2) and MitoTracker Red. Cells were quickly scraped on ice, washed twice in cold PBS, and re-suspended in 5 μM DHE (20′, 37 °C in the dark, in PBS), 5 μM H2DCF-DA (30′, 37 °C in the dark, in HBSS) or 100 nM MitoTracker Red (20′, 37 °C in the dark, in PBS). After a final wash in PBS, cells were immediately transferred into a tube on ice for flow cytofluorometric analysis [15].
DNA Methylation Analysis
DNA methylation analysis was performed by bisulfite conversion and pyrosequencing, as detailed in Consales et al. [48]. Briefly, DNA was extracted by a commercial kit (Zymo Research, Irvine, CA, USA); 500 ng of DNA were bisulfite-converted using the EZ DNA Methylation-Gold™ Kit (Zymo research) according to the manufacturer’s protocol. Bisulfite-treated DNA (50 ng) was amplified in a 50 μL reaction mixture containing 25 μL of PyroMark PCR master mix (New England Biolabs, Ipswich, MA, USA) and 0.2 μM of the primers specific for the human miR-34b/c promoter region (Online Resource 1).
The level of methylation at human miR-34b/c amplified promoter was evaluated by pyrosequencing (PyroMark ID Q24, Qiagen, Hilden, Germany), according to manufacturer’s instructions. Methylation level quantification was performed using the provided software. The percentage of methylation (% of 5-methyl Cytosine, 5-mC) was expressed as the number of 5-mC divided by the sum of methylated and un-methylated cytosines. One investigator, blind to all the information regarding the samples except their code, performed the pyrosequencing analyses. Different internal controls were included in every pyrosequencing run to ensure the completion of bisulfite modification, the specificity of PCR amplification, and the success of pyrosequencing reactions. A universal methylated DNA standard (Zymo Research,) was used as a positive control for bisulfite conversion [48].
Fluorescence Microscopy for Quantification of α-Synuclein Aggregates
SH-SY5Y cells were cultured on cover-slips, fixed in 4% paraformaldehyde in PBS for 30 min, then permeabilized by addition of 0.25% Triton X-100 in PBS for 10 min. After blocking (5% normal goat serum), cells were incubated with the anti-α-synuclein antibody (Cell Signaling Technologies, 1:50 in 5% normal goat serum, over-night at +4 °C), washed with PBS and incubated with Alexa Fluor 488-conjugated anti-rabbit IgG (Invitrogen) at room temperature for 1 h. For nuclear staining, cells were incubated with 1 μg/mL DAPI. Cells were then washed with PBS and analyzed under a fluorescence microscope (Nikon Eclipse 80-I). For quantification of aggregates, 4 microscopic fields containing at least 100 cells per group were randomly selected, and the average number of aggregates per 100 cells was calculated.
Statistical Analysis
The variations of samples values are reported as Mean ± S.D. calculated in N ≥ 3 replicates; the precise number of experimental replicates has been detailed in each figure legend. The statistical differences were analyzed trough the KailedaGraph program (Synergy Software, Reading PA, USA) by applying the two-sided Student’s t test; the experimental groups that have been compared and statistically analyzed are detailed in each figure legend. P values < 0.05 were considered statistically significant and indicated as follows: *P < 0.05; **P < 0.01; ***P < 0.001, n.d.: variation not statistically different.
Results
Fifty-Hertz Magnetic Field Drives miR-34b and miR-34c Impairment in Neuronal Cells
The expression level of miR-133b and miR-34b/c cluster was first assessed in proliferating SH-SY5Y human neuroblastoma cells following either Sham or continuous exposure to 50 Hz (1 mT) magnetic fields for 48 h. As reported in Fig. 1a, no change in miR-133b level was detected, whereas both miR-34b and miR-34c expression underwent a significant decrement in ELF-MFs-exposed cells compared to Sham counterparts. In order to determine the kinetics of the ELF-MFs-driven deregulation in miR-34 pattern, miR-34b and 34c expression was screened from 6 to 72 h of continuous exposure (Fig. 1b). The level of both micro-RNAs was gradually impaired as early as 24 h, and was maintained decreased up to 72 h of continuous exposure, with a higher effect reported for the 34b mature molecule compared to the 34c counterpart.

Fifty-Hertz MFs affect miR-34b and miR-34c expression in neuronal cells. Evaluation of miR-133b, miR-34b and miR-34c expression levels by qRT-PCR in (a, b) proliferating, (c, d) RA-PMA differentiated (DAergic) human SH-SY5Y cells, and (e) mouse PCNs, in response to ELF-MFs and Sham exposure. In RA + PMA-treated cells (c), the expression level of the dat gene has been also evaluated. Values are the means ± S.D. (N = 5 for SH-SY5Y cells; N = 3 for PCNs). *P < 0.05, **P < 0.01, **P < 0.001, n.d. = no statistical difference, calculated in (a, b, d, e) ELF-MF vs sham-exposed cells, and in (c) RA + PMA-treated vs untreated cells
To test the ability of ELF-MFs to affect miR-34b/c expression also in post-mitotic neurons, SH-SY5Y cells were incubated with RA in combination with PMA to acquire a DAergic phenotype prior ELF-MFs exposure [15, 49]. Differentiation of SH-SY5Y cells stimulated the expression of both dat (a DAergic-specific gene) and miR-34b/c level (Fig. 1c). Once ELF-MFs were applied, miR-34b and 34c expression was not induced if compared to Sham-exposed cells, with this effect being maintained all over the exposure (72 h; see Fig. 1d). Neuronal response to ELF-MFs was further assessed in mouse PCNs. Also in this case, miR-34b and miR-34c expression was inhibited by continuous exposure to radiations, thus indicating a general ability of the 50 Hz-MF to specifically impair miR-34b/c cluster expression in both highly proliferating cancer cells and post-mitotic neurons.
Fifty-Hertz Magnetic Field Regulates miR-34b/c Expression at Transcriptional Level Via Promoter DNA Methylation
According to gene structure, mature miR-34b and 34c arise from a common pri-miR transcript, set under the control of both p53 transcription factor and CpG island methylation (Online Resource 2a) [37, 39]. As shown in Fig. 2a, continuous ELF-MFs exposure affected pri-miR-34b/c regulation at transcriptional level. In all the neuronal models analyzed, the pri-miR-34b/c expression was significantly reduced, with the early effective decrement preceding those observed in the mature miR form (Fig. 1). At longer exposure times, pri-miR-34 expression was progressively recovered, the difference between ELF and Sham being no more observed (Fig. 2a). We then analyzed the expression of Btg4 in response to ELF-MFs, as it has already been demonstrated to undergo epigenetic regulation and to share the same regulatory sequence on human genome with the miR-34b/c (Online Resource 2a) [43]. Although expressed at very low level in SH-SY5Y cells, Btg4 transcript displayed a decrement trend upon 50 Hz (1 mT) radiation exposure that overlapped the pri-miR-34b expression (Online Resource 2b). No statistically significant difference was instead observed in the ELF-MFs-exposed mouse PCNs compared to Sham in terms of Btg4 expression (Online Resource 2b).

ELF-MF-driven miR-34b/c modulation does not depend on p53. a Evaluation of the pri-miR-34b/c expression level carried out by qRT-PCR in the indicated neuronal cells. Data are reported as relative transcript expression in the ELF-MFs vs Sham-exposed cells. b Western blot analysis of p53 protein expression evaluated in response to the ELF-MFs. Each panel reports the blot image as well as the densitometry of p53 normalized vs GAPDH expression. Treatment with the Doxorubicin (Doxo, 2 μM, 24 h) has been included as positive control for p53 induction. c p53 and pri-miR-34b/c expression analysis carried out by Western blot and qRT-PCR, respectively, in DAergic SH-SY5Y cells in response to ELF-MFs ± Nutlin-3a (50 μM, 6 h). Values are the means ± S.D. (N = 3); n.d. = no statistical difference; *P < 0.05, **P < 0.01, **P < 0.001 calculated in ELF-MFs vs Sham-exposed cells. Immunoblots are representative of three different experiments giving similar results
To verify the involvement of p53 in pri-miR-34 deregulation induced by ELF-MFs, we evaluated p53 mRNA and protein level (Online Resource 2c, Fig. 2b). No statistical difference in p53 transcript was detected both in SH-SY5Y cells and in mouse PCNs (Online Resource 2c). At protein level, a slight p53 decrement was observed later on in the exposure kinetics (Fig. 2b). However, this phenomenon was significantly delayed if compared to the early pri-miR inhibition triggered by ELF-MFs, indicating that a p53-independent mechanism of miR-34b/c regulation was likely induced under our experimental conditions. Consistently, p53 stabilization, achieved via the administration of the MDM2-inhibitor Nutlin-3a [50], resulted in no significant change of pri-miR-34b/c level (Fig. 2c), strengthening the hypothesis that miR-34b/c transcriptional regulation depends on regulatory mechanisms unrelated to p53.
We next tested the hypothesis that ELF-MF might affect the miR-34b/c level by other mechanisms, such as by DNA methylation. We thus evaluated the miR-34b/c promoter methylation content via DNA bisulphite conversion and pyrosequencing of the 7 elements located in a fragment of the human CpG island (CpG 79 region, Fig. 3a). The average methylation level, measured in DAergic neuronal cells, showed a significant increase upon ELF-MFs exposure, whose extent was significant already after 4 h and, mostly, consistent with the timing of pri-miR-34 silencing (Fig. 3b). Coherently, incubations with the demethylating agent 5-aza-2-deoxycytidine (DAC) efficiently reverted ELF-MFs-induced pri-miR-34 silencing and Btg4 expression release (Fig. 3c).

Fifty-Hertz MFs trigger the hyper-methylation of the miR-34b/c promoter. a Genome structure of the 5′ flanking region of the human miR-34b/c gene cluster obtained from the ucsc genome browser (https://genome.ucsc.edu). Nucleotide sequence of a portion of CpG island 79 has been reported, with the indication of the seven CG dinucleotides and the position of the forward (Fw), reverse (Rev) and sequencing (Pyro) primers used for bisulfite conversion, PCR amplification and pyrosequencing. b Percentage of cytosine methylation carried out in the DAergic SH-SY5Y cells in response to 4- to 48 h of continuous exposure to 50-Hz MFs and Sham. c Analysis of the Btg4 and pri-miR-34b/c expression level performed in the DAergic SH-SY5Y cells exposed to Sham, ELF-MFs and ELF-MFs + DAC (10 μM, 6 h). Values are the means ± S.D. (N = 4); *P < 0.05, calculated in (b) ELF-MF vs sham, and in (c) ELF-MF vs ELF-MF + DAC exposed cells
miR-34b/c Deregulation Does Not Depend on the ELF-Driven Oxidative Stress, but Triggers Mitochondria and Oxidative Imbalance
We recently reported that ELF-MFs trigger oxidative stress and protein carbonylation in both proliferating and DAergic SH-SY5Y cells [15]. The correlation potentially occurring between the ELF-driven redox imbalance and the miR-34b/c impairment was thus investigated.
Incubations with the thiol antioxidant compounds GSHest or NAC [15] did not restore miR-34b and miR-34c expression, suggesting that the ELF-MFs trigger miR deregulation independently of ROS generation and thiol impairment (Online Resource 3a). Coherently, the inhibition of the GSH de novo synthesis by BSO, as well as the induction of a direct oxidative damage by H2O2, did not affect the miR-34b/c expression level in Sham-exposed cells (Online Resource 3b). A significant stimulation of the miR-34b/c levels could be exclusively detected at higher H2O2 concentration (25 μM) (Online Resource 3b), in response to a massive increase in ROS generation that significantly exceeds those measured upon ELF exposure (Online Resource 3c) [15].
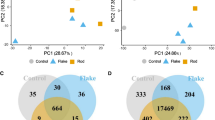
In order to assess whether miR-34 deregulation might contribute to the ELF-MFs-induced redox imbalance, either miR-34b or miR-34c expression was selectively increased by exogenous administration of mimic miR molecules (Online Resource 3d) [45]. Under these conditions, oxidative stress was partially restored, with the extent of ROS (both superoxide and H2O2 species) being attenuated at 48 h following ELF-MFs exposure (Fig. 4a, b). Coherently, anti-miR-mediated silencing of miR-34b and miR-34c (Online Resource 3e) [45] produced a time-dependent increase of ROS in Sham cells (Online Resource 3f), clearly suggesting an inverse functional correlation between miRs-34 and oxidative stress. Further assessment of mitochondrial function by MitoTracker staining indicated the occurrence of an early impairment of the mitochondria integrity upon ELF-MFs exposure, that could be partially reverted by the administration of miR-34b mimic molecule (Fig. 4c), suggesting that, at least in part, ROS were generated at the mitochondrial level. By comparing in silico datasets of human miR-34b or miR-34c, obtained from the screening of different algorithms of microRNAs target prediction [45], we identified a set of putative genes involved in the control of mitochondrial function and redox balance, including a cluster of 14 target genes shared by both miRs-34, e.g., the mitochondrial Superoxidase Dismutase 2 and many mitochondrial ribosomal proteins (Fig. 4d) that might account for the miR-34-mediated effects on oxidative stress elicited by the ELF-MFs. Each single microRNA also displays specific target sites (Online Resources 4 and 5), such as the Nox family members, which can suggest the way these miRs-34 can individually control mitochondrial function and redox imbalance.

miR-34b/c deregulation is involved in mitochondria and oxidative stress. a, b Flow cytometric analysis of superoxide and H2O2 carried out by DHE and H2-DCFDA, respectively, in cells loaded with the reported miR mimic molecules and exposed to either Sham or ELF-MF. Data are expressed as the ratio of the mean fluorescence intensity (MFI) calculated in ELF-MFs vs Sham-exposed cells. Scr: scramble; 34b: miR-34b mimic; 34c: miR-34c mimic, (-): no mimic molecule. Values are the means ± S.D. (N = 3). *P < 0.05, n.d. = no statistical difference, calculated in Scrambled vs miR-34 Mimic molecules. c Evaluation of the mitochondria integrity performed by Mito Tracker Red in cells loaded with the reported miR mimic molecules and exposed to either Sham or ELF-MFs. Data are expressed as the ratio of the mean fluorescence intensity (MFI) calculated in ELF-MFs vs Sham-exposed cells. Values are the means ± S.D. (N = 3). *P < 0.05, **P < 0.01, n.d. = no statistical difference, calculated in ELF-MF vs Sham-exposed cells (left histogram) and in Scrambled vs miR-34 mimic molecule (right panel). FACS plots reported in each panels are from one experiment representative of three which gave similar results. d Venn diagrams representing miR-34b and 34c putative targets selected in the context of genes controlling mitochondrial homeostasis and oxidative stress pathways; the 14 targets displayed at the intersection represent those shared by both miRs
Fifty-Hertz MFs Exposure Stimulates SNCA Expression Via miR-34
Besides controlling mitochondrial function and redox balance, miR-34b/c target prediction highlighted a consistent set of transcripts specifically involved in neuronal functions (Fig. 5a and Online Resource 6), including the human Snca /Park1. The 3’UTR of the Snca mRNA displays different regions for 34b/c putative binding sites (Online Resource 7a), some recently identified as miR-34b/c targets in neuroblastoma cells [32], strongly suggesting it may be regulated by this cluster in response to specific stimuli. Continuous exposure of human neuronal cells to 50-Hz MFs up-regulated SNCA at both mRNA (Online Resource 7b) and protein level (Fig. 5b). Moreover, a slight but significant (P = 0.049) increase in the intracellular α-synuclein aggregation was observed in response to 72 h of continuous ELF-MF if compared to Sham-exposed cells (Fig. 5c).

ELF-MFs exposure induces SNCA via miR-34b and oxidative stress. a Venn diagrams showing set of specific neuronal targets recognized by miR-34b and 34c; the genes shared by both miRs are listed in the table below. b Western blot analysis of SNCA expression level carried out in the DAergic SH-SY5Y cells after 4, 24 and 48 h of ELF-MFs or Sham exposure. The histogram represents blot densitometry. Values are the means ± S.D. (N = 3). **P < 0.01, n.d. = no statistical difference, calculated in the ELF-MF vs Sham-exposed cells. c Percentage of alpha-synuclein intracellular aggregates quantified by fluorescence microscopy at 72 h of continuous exposure to either ELF-MF or Sham. Values are the means ± S.D. (N = 3). **P < 0.05, calculated in the ELF-MF vs Sham-exposed cells. d Evaluation of Snca mRNA (qRT-PCR, upper histograms) and SNCA protein levels (Western blot, bottom panels) performed in response to ELF-MFs or Sham exposure in combination with the indicated miR mimic or anti-miR agents. Values are the means ± S.D. (N = 3). *P < 0.05, **P < 0.01, n.d. = no statistical difference, calculated in the miR-34 vs Scramble molecules. e Western blot analysis of the SNCA protein levels carried out in mouse PCNs in response to 48 h of ELF-MFs or Sham exposure. f Evaluation of Snca mRNA expression performed by qRT-PCR in response to the indicated combined treatments. Values are the means ± S.D. (N = 5). *P < 0.05, **P < 0.01, ***P < 0.001, calculated in Sham vs ELF exposure, and in ELF + NAC/34b Mimic/34b Mimic + NAC vs ELF exposure
In line with the in silico prediction, SNCA transcript and protein levels were reduced in ELF-MF-exposed cells by the addition of miR-34b mimic and increased in Sham cells upon treatment with anti-miR-34b (Fig. 5d), whereas it coherently but not significantly changed upon miR-34c modulation (Fig. 5d). These results identified miR-34b, more than miR-34c, as major epigenetic determinant in the SNCA stimulation upon ELF-MFs. Surprisingly, ELF-MFs exposure induced SNCA expression also in mouse PCNs (Fig. 5e), even though the 3’UTR of mouse Snca mRNA does not contain any miR-34b/c putative binding site. This unexpected result prompted us to hypothesize that some additional mechanism(s) can affect SNCA expression in response to ELF-MFs stimulation, such as oxidative stress, which is known to stimulate SNCA [51]. The administration of NAC actually inhibited the ELF-MFs-driven SNCA increase, with the combination NAC/miR-34b mimic molecule accomplishing the highest inhibition of SNCA expression (Fig. 5f), thus disclosing a much more complex network of regulation elicited by ELF-MFs in neuronal cells, which could predispose to oxidative damage and degenerative phenotype (sketched in Fig. 6).

Graphical Abstract representing the proposed mechanisms underlying the interaction between the ELF-MF (50 Hz, 1 mT) and the neuronal cell system. Pathway (1): ELF-MF triggers redox imbalance by increasing ROS generation, depleting the thiols pool and stimulating protein carbonylation [ref. 15]. Pathway (2): ELF-MF stimulates cytosine hyper-methylation in the miR-34b/c cluster gene promoter thus inhibiting miR expression; miR-34 impairment drives SNCA stimulation and affects the redox balance likely through the regulation the genes controlling mitochondria function. The stimulation of the SNCA expression also depends on the ELF-driven oxidative stress. Solid lines represent what demonstrated in the paper by experimental data; dashed lines show potential molecular pathways to be further experimentally validated.
Discussion
Over the last years, epidemiological studies have been suggesting a potential neurodegenerative property of ELF-MFs exposure, mainly based on a significant association with AD and ASL [4,5,6,7,8]. Despite these lines of evidence, still a limited set of experimental data have attempted to identify the cellular and molecular pathways underlying the possible pathogenic role of ELF-MFs in brain; besides, most data are based on in vitro findings. In accordance with previous data reporting a pro-oxidant ability of the ELF-MFs in different experimental models [7, 10,11,12,13,14], we recently demonstrated that 50-Hz MFs directly trigger redox imbalance in both proliferating and DAergic SH-SY5Y neuroblastoma cells, which is characterized by ROS generation, thiol content depletion and protein carbonylation, and sensitizes neurons to the PD-neurotoxin MPP+ [15]. Oxidative and nitrosative stress, as well as mitochondria dysfunction, are major responsible for brain aging and neurodegeneration [52, 53]. The sustained oxidative stress triggered by ELF-MFs exposure, along with the resulting damage to cellular bio-molecules and mitochondria [47], might thus help shifting neurons toward a degenerative phenotype, and disturb the intracellular environment in a way that sensitizes them to a second damaging stressor (such as the MPP+) [15, 54].
Besides oxidative stress, the aetiopathogenesis of the neurodegenerative diseases, including PD, recently encountered the involvement of the epigenetic machinery, in terms of altered DNA methylation pattern and histone modifications, as well as dysregulated microRNA expression, mainly including miR-133, miR-7 and miR-34b/c [16,17,18,19,20,21,22,23,24,25,26, 30, 31]. The epigenetic level of gene expression control specifically tunes the neuronal response to different environmental stressors, including ionizing radiations and ELF-MFs [55,56,57,58,59,60,61]. Based on this evidence, we here assessed the expression level of some PD-related microRNAs and demonstrated that 50-Hz MFs strongly decrease the expression of both miR-34b and miR-34c mature molecules in proliferating SH-SY5H and post-mitotic neuronal cells. Although based on in vitro experimental evidences, these data are particularly relevant, as the miR-34b/c locus is a well characterized tumor suppressor gene [33, 34] and the ability of the ELF-MFs to switch-off their expression in a neuronal context might open a new scenario in the comprehension of those mechanisms potentially underlying cancer transformation by ELFs. Different groups have indeed documented the ability of the 50-Hz MF to alter the proliferative status of neuroblastoma cells, especially in response to long (up to 15 days) exposure to the radiation, supporting the hypothesis that ELF-MF might trigger a shift toward a more invasive phenotype [47, 62,63,64,65]. Our findings add a small piece to the puzzle by involving the epigenetic control via miR-34; many different targets devoted to proliferation control and cancer progression have been identified as both direct and indirect miR-34 targets, such as c-Myc and N-Myc, the YY1 transcription factor, the Notch signaling, Sox-2, and PDGFRA [45, 66,67,68,69,70,71]; they might account for the pro-proliferative effect elicited by the long-term exposure to ELF-MF. Concordantly, the differentiation processes is known to stimulate the expression of all miR-34 family members in neurons [30, 72], in line with what we observe in SH-SY5Y cells upon RA/PMA administration. This is consistent with the idea that the sustained expression of miR-34 family helps in maintaining the mature neurons in a non-proliferative stage; those stimuli, such as the ELF-MFs, that reduce miR-34 expression might lead to a growth advantage and more aggressive behavior that typically characterizes cancer lesions. The relevant role for miR-34b/c in neuronal physiology is confirmed by data reporting their dysregulation in major neurodegenerative diseases, such as AD, PD and Huntington’s Disease. In particular, miR-34b/c reduced expression has been associated with both memory impairment and neuronal survival, providing a potential common molecular mechanism contributing to dementia [73, 74]. Moreover, miRs-34 have been linked with aging in Drosophila, where loss of these miRNAs leads to brain degeneration and decline in flies survival [31].
In our in vitro experimental models, ELF-driven reduction of miR-34b and miR-34c mature species relies on the dysregulation of their common pri-miR transcript. At the transcriptional level, all miR-34 family members are the most prevalent p53-induced miRNAs. However, surprisingly, we did not observe any p53 involvement in miR-34b/c decrement induced by ELF-MFs exposure, at least in the early (effective) phases of this phenomenon, which is actually attributable to the hypermethylation of the CpG island controlling the miR-34 gene cluster. Specifically, we found out that ELF-MFs increase the percentage of the methylated cytosines in the human CpG 79 island, where we analyzed seven CG sites. These cytosines are the best characterized across the miR-34 CpG island and their methylation status has been associated with miR-34b/c knock-down in many different cancer histotypes and metabolic diseases, this suggesting an interesting interplay between two components of the epigenetic machinery, i.e., the DNA methylation and the miRoma [39,40,41,42,43, 75]. According to the genome sequence, the CpG 79 island contains other putative methylation sites that we have not pyrosequenced yet and that might further account for radiation-induced effects. In support to this hypothesis, we provided evidence that the inhibition of DNA methyltransferase by DAC results in the recovery of miR-34, as well as Btg4 expression level, which is set under the same regulatory control. Future investigation might add a further step in the comprehension of such transcriptionl regulation; it might occur that specific repressors take part and help coordinate the repression of the downstream miR sequence upon ELF exposure. P73 and ΔNp73 for instance share the same regulatory sequences with p53 on many different gene promoters likely including miR-34 [76] and are specifically involved in gene transcription in brain where they control physiological and pathological pathways [77, 78]. ΔNp73α in particular works as an antagonist of p53/p73 functions in growth suppression and/or apoptosis and might be involved in miR-34 inhibition in response to ELF.
miR-34 is a miRNA family emerging from recent radiobiology studies, mainly as responsive to conventional tumor radiotherapy [79], thus suggesting that it could represent a key checkpoint controlling different downstream pathways in response to both ionizing and non-ionizing radiations through epigenetic control. Moreover, the ability of ELF-MFs to affect the cellular epigenetic layout is coming out in different experimental models, mainly in terms of microRNAs expression change [58, 60, 61, 80,81,82]. Together with previous findings, our in vitro data can help shading light on a new set of pathways underlying the biological effects of non-ionizing radiations and their potential impact on human health. The epigenetic machinery may act as an interface between the environment and the genome, thus influencing both aging and different degenerative phenotypes, such as PD, which imply an epigenetic deregulation, as well as oxidative stress and damage among the main molecular determinants underlying their insurgence.
Oxidative stress and microRNA deregulation can actively cooperate to accelerate neurodegeneration [83, 84]. Minones-Moyano et al. highlighted this synergy by demonstrating that miR-34b/c depletion in DAergic SH-SY5Y cells triggered an altered mitochondria function and dynamics, which resulted in oxidative stress [30]. However, this study, similarly to many others published in this field, failed so far to provide a reasonable mechanism linking radiations and oxidative stress, taking for granted that ROS are produced downstream of any still undefined effects produced by electromagnetic fields. In this study, we provide evidence supporting the hypothesis in which oxidative stress is a consequence of the ELF-induced miR-34b/c decrease. Our data show that miR-34b/c decrement contributes to redox imbalance, as the administration of exogenous miR mimic molecules helped recovering oxidative imbalance and mitochondria dysfunction caused by ELF-MFs exposure. To give strength to this new idea, in silico prediction of putative miR-34b/c shared targets identified a mitochondrial/redox signature that might explain such an involvement in ROS levels maintenance. Interestingly, we found out that miR-34b might bind to and positively regulate the Nox-1 mRNA, encoding the NADPH oxidase (NOX) 1, an enzyme belonging to the NOXs family that specifically generates superoxide anion. According to this hypothesis, the ROS-generating ability elicited by ELF-MFs in neurons may be attributable to the stimulation of NOX-1 via miR-34b silencing. There is a growing evidence that NOX members can be upregulated by a variety of neurodegenerative factors and that pharmacological inhibition of NADPH oxidase enzymes are neuroprotective [85]. Along this line of reasoning, ELF-MFs could represent a detrimental agent through their ability to stimulate the NOX family. It has been reported that administration of diphenyleneiodonium (DPI), a specific inhibitor for the NADPH oxidases, prevents MF-driven pro-oxidant effect in monocytes and human amniotic epithelial cells, this strongly suggesting that ELF-MFs exposure might induce ROS production via the activation of the NADH oxidase [86, 87].
In silico screening of human miR-34b/c putative targets also highlighted the enrichment in many different genes underlying neuronal functions, including Bdnf (Brain-derived neutrophic factor), Nos1 (neuronal nitric oxide synthase 1), Park2, and Park1/Snca, the last being already demonstrated as a miR-34b/c target in SH-SY5Y neuroblastoma cells [32]. Results shown in this paper provide useful and novel information in terms of biological functions of miR-34/snca association in response to non-ionizing radiations. SNCA is a protein implicated in the aetiopathogenesis of PD and inhibits neurotransmitter release when overexpressed [29, 88]. Surprisingly, although mouse 5’UTR of the Snca mRNA does not contain any miR-34b/c putative binding site, we found that its expression was stimulated by the ELF-MF exposure in PCNs. Therefore, the stimulation of SNCA expression by 50-Hz MF implies both a redox and an epigenetic component in our in vitro experimental models. The exposure to the ELF-MF also drives a slight but significant change in the percentage of intracytoplasmic SNCA aggregates—a typical biomarker in PD phenotype—thus indicating that ELF exposure can stimulate SNCA protein increase and promote its aggregation in vitro. Future studies might help identify whether other Park genes are deregulated by ELF exposure, to investigate the ability of the 50-Hz MFs to drive neurons toward a PD-like phenotype by other mechanisms. Most park genes display a direct or indirect impact on the oxidative stress, mitochondria integrity and function [89, 90] and might be thus additional mediators of the ELF-driven biological effect in neuronal cells.
Overall, our data support the hypothesis that the prolonged exposure to the ELF-MFs in SH-SY5Y and in primary mouse neuronal cultures, although not able to commit per se neuronal cells to death, can promote a set of molecular events (e.g., tune the intracellular biochemical and epigenetic balance) that might progressively boost neurons toward a degenerative phenotype. The molecular alterations induced by 50-Hz-MFs resemble some typical PD molecular damages, such as oxidative imbalance, thiols depletion and protein carbonylation [15], as much as the miR-34 epigenetic deregulation and SNCA stimulation (Fig. 6), all these suggesting a pathogenic role of ELF-MFs in PD predisposition and onset. Albeit our findings identified a redox-epigenetic axis in response to ELF toward neuro-degeneration, they are all performed on in vitro experimental models; they can therefore help shade light on the pathways tuned by radiations and open the way to future validation in animal models where the neuro-degenerative potential of the 50-Hz MF can be assessed in the central nervous system.
Abbreviations
- AD:
-
Alzheimer’s disease
- ALS:
-
Amyotrophic lateral sclerosis
- B field:
-
Magnetic field
- BSO:
-
Buthionine-s,r-sulfoximine
- BTG4:
-
B cell translocation gene 4
- 5-mC:
-
5-Methyl cytosine
- DAergic:
-
Dopaminergic
- DAC:
-
5-Aza-2′-deoxycytidine
- DHE:
-
Dihydroethidium
- E field:
-
Electric field
- ELF-MF:
-
Extremely low-frequency magnetic field
- GSH:
-
Reduced glutathione
- GSHest:
-
Reduced glutathione ethyl ester
- H2-DCFDA:
-
2′,7′-Dichlorofluorescin diacetate
- MicroRNAs/MiRs:
-
MicroRNAs
- MF:
-
Magnetic field
- MFI:
-
Mean fluorescence intensity
- MPP+ :
-
1-Methyl-4-phenylpyridinium
- NAC:
-
N-Acetyl-l-cysteine
- PCNs:
-
Primary cortical neurons
- PD:
-
Parkinson’s disease
- PI:
-
Propidium iodide
- PMA:
-
Phorbol 12-myristate 13-acetate
- RA:
-
Retinoic acid
- RMS:
-
Root-mean-square
- ROS:
-
Reactive oxygen species
- SNCA:
-
α-Synuclein
- sncRNAs:
-
Small non-coding RNAs
- 3’UTR:
-
3′-Untranslated region
References
WHO-World Health Organization (2007) Extremely low frequency fields. Environmental Health Criteria, vol 238. World Health Organization, Geneva
IARC, Non Ionizing Radiation Part 1: Static and Extremely Low Frequency (ELF) Electric and Magnetic Fields. IARC monographs on the evaluation of the carcinogenic risks to Human, Vol. 80, 2002
IARC, Non Ionizing Radiation Part 2: Radiofrequency Electromagnetic Fields. IARC monographs on the evaluation of the carcinogenic risks to Human, Vol. 102, 2013
Savitz DA, Checkoway H, Loomis DP (1998) Magnetic field exposure and neurodegenerative disease mortality among electric utility workers. Epidemiology 4:398–404
Davanipour Z, Tseng CC, Lee PJ, Sobel E (2007) A case-control study of occupational magnetic field exposure and Alzheimer’s disease: results from the California Alzheimer's Disease Diagnosis and Treatment Centers. BMC Neurol 7:13. https://doi.org/10.1186/1471-2377-7-13
Li CY, Sung FC (2003) Association between occupational exposure to power frequency electromagnetic fields and amyotrophic lateral sclerosis: a review. Am J Ind Med 43(2):212–220. https://doi.org/10.1002/ajim.10148
Consales C, Merla C, Marino C, Benassi B (2012) Electromagnetic fields, oxidative stress, and neurodegeneration. Int J Cell Biol 2012:683897. https://doi.org/10.1155/2012/683897
Wechsler LS, Checkoway H, Franklin GM, Costa LG (1991) A pilot study of occupational and environmental risk factors for Parkinson's disease. Neurotoxicology 12(3):387–392. https://doi.org/10.1002/mds.870050212
Röösli M (2008) Commentary: epidemiological research on extremely low frequency magnetic fields and Alzheimer's disease—biased or informative? Int J Epidemiol 37(2):341–343. https://doi.org/10.1093/ije/dyn024
KJ B, Masters CL, Bush AI (2004) Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov 3(3):205–214. https://doi.org/10.1038/nrd1330
Mattsson MO, Simkó M (2014) Grouping of experimental conditions as an approach to evaluate effects of extremely low-frequency magnetic fields on oxidative response in in vitro studies. Front Public Health 2:132. https://doi.org/10.3389/fpubh.2014.00132
Adair RK (1999) Effects of very weak magnetic fields on radical pair reformation. Bioelectromagnetics 20(4):255–263
Cho SI, Nam YS, Chu LY, Lee JH, Bang JS, Kim HR et al (2012) Extremely low-frequency magnetic fields modulate nitric oxide signaling in rat brain. Bioelectromagnetics 33(7):568–574. https://doi.org/10.1002/bem.21715
Falone S, Mirabilio A, Carbone MC, Zimmitti V, Di Loreto S, Mariggiò MA et al (2008) Chronic exposure to 50Hz magnetic fields causes a significant weakening of antioxidant defence systems in aged rat brain. Int J Biochem Cell Biol 40(12):2762–2770. https://doi.org/10.1016/j.biocel.2008.05.022
Benassi B, Filomeni G, Montagna C, Merla C, Lopresto V, Pinto R et al (2016) Extremely low frequency magnetic field (ELF-MF) exposure sensitizes SH-SY5Y cells to the pro-Parkinson’s disease toxin MPP+. Mol Neurobiol 53(6):4247–4260. https://doi.org/10.1007/s12035-015-9354-4
Coppedè F (2012) Genetics and epigenetics of Parkinson's disease. Sci World J 2012:489830. https://doi.org/10.1100/2012/489830
Farrer MJ (2006) Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet 7(4):306–318. https://doi.org/10.1038/nrg1831
Goodall EF, Heath PR, Bandmann O, Kirby J, Shaw PJ (2013) Neuronal dark matter: the emerging role of microRNAs in neurodegeneration. Front Cell Neurosci 7:178. https://doi.org/10.3389/fncel.2013.00178
Feng Y, Jankovic J, Wu YC (2015) Epigenetic mechanisms in Parkinson’s disease 349(1–2):3–9. Review. doi:https://doi.org/10.1016/j.jns.2014.12.017
Tan L, Yu JT, Tan L (2015) Causes and consequences of MicroRNA dysregulation in neurodegenerative diseases. Mol Neurobiol 51(3):1249–1262. Review. https://doi.org/10.1007/s12035-014-8803-9
Heman-Ackah SM, Hallegger M, Rao MS, Wood MJ (2013) RISC in PD: the impact of microRNAs in Parkinson's disease cellular and molecular pathogenesis. Front Mol Neurosci 6:40. https://doi.org/10.3389/fnmol.2013.00040
Masliah E, Dumaop W, Galasko D, Desplats P (2013) Distinctive patterns of DNA methylation associated with Parkinson disease: identification of concordant epigenetic changes in brain and peripheral blood leukocytes. Epigenetics 8(10):1030–1038. https://doi.org/10.4161/epi.25865
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116(2):281–297
Kim J, Inoue K, Ishii J, Vanti WB, Voronov SV, Murchison E et al (2007) A MicroRNA feedback circuit in midbrain dopamine neurons. Science 317(5842):1220–1224. https://doi.org/10.1126/science.1140481
Junn E, Lee KW, Jeong BS, Chan TW, Im JY, Mouradian MM (2009) Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc Natl Acad Sci U S A 106(31):13052–13057. https://doi.org/10.1073/pnas.0906277106
Hoss AG, Labadorf A, Beach TG, Latourelle JC, Myers RH (2016) MicroRNA profiles in Parkinson’s disease prefrontal cortex. Front Aging Neurosci 8:36. https://doi.org/10.3389/fnagi.2016.00036
Ammal Kaidery N, Tarannum S, Thomas B (2013) Epigenetic landscape of Parkinson's disease: emerging role in disease mechanisms and therapeutic modalities. Neurotherapeutics 10(4):698–708. https://doi.org/10.1007/s13311-013-0211-8
Schlaudraff F, Gründemann J, Fauler M, Dragicevic E, Hardy J, Liss B (2014) Orchestrated increase of dopamine and PARK mRNAs but not miR-133b in dopamine neurons in Parkinson's disease. Neurobiol Aging 35(10):2302–2315. https://doi.org/10.1016/j.neurobiolaging.2014.03.016
Gründemann J, Schlaudraff F, Haeckel O, Liss B (2008) Elevated alpha-synuclein mRNA levels in individual UV-laser-microdissected dopaminergic substantia nigra neurons in idiopathic Parkinson’s disease. Nucleic Acids Res 36(7):e38. https://doi.org/10.1093/nar/gkn084
Miñones-Moyano E, Porta S, Escaramís G, Rabionet R et al (2011) MicroRNA profiling of Parkinson's disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum Mol Genet 20(15):3067–3078. https://doi.org/10.1093/hmg/ddr210
Liu N, Landreh M, Cao K, Abe M, Hendriks GJ, Kennerdell JR et al (2012) The microRNA miR-34 modulates ageing and neurodegeneration in Drosophila. Nature 482(7386):519–523. https://doi.org/10.1038/nature10810
Kabaria S, Choi DC, Chaudhuri AD, Mouradian MM, Junn E (2015) Inhibition of miR-34b and miR-34c enhances α-synuclein expression in Parkinson's disease. FEBS Lett 589(3):319–325. https://doi.org/10.1016/j.febslet.2014.12.014
He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y et al (2007) A microRNA component of the p53 tumour suppressor network. Nature 447(7148):1130–1134. https://doi.org/10.1038/nature05939
Maroof H, Salajegheh A, Smith RA, Lam AK (2014) MicroRNA-34 family, mechanisms of action in cancer: a review. Curr Cancer Drug Targets 14(8):737–751 Review
Zhang DG, Zheng JN, Pei DS (2014) P53/microRNA-34-induced metabolic regulation: new opportunities in anticancer therapy. Mol Cancer 3:115. Review. https://doi.org/10.1186/1476-4598-13-115
Hiroki E, Suzuki F, Akahira J, Nagase S, Ito K, Sugawara J et al (2012) MicroRNA-34b functions as a potential tumor suppressor in endometrial serous adenocarcinoma. Int J Cancer 131(4):E395–E404. https://doi.org/10.1002/ijc.27345
Corney DC, Flesken-Nikitin A, Godwin AK, Wang W, Nikitin AY (2007) MicroRNA-34b and MicroRNA-34c are targets of p53 and cooperate in control of cell proliferation and adhesion-independent growth. Cancer Res 67(18):8433–8438. https://doi.org/10.1158/0008-5472.CAN-07-1585
Wong MY, Yu Y, Walsh WR, Yang JL (2011) microRNA-34 family and treatment of cancers with mutant or wild-type p53. Int J Oncol 38(5):1189–1195. Review. https://doi.org/10.3892/ijo.2011.970
Deneberg S, Kanduri M, Ali D, Bengtzen S, Karimi M, Qu Y et al (2014) microRNA-34b/c on chromosome 11q23 is aberrantly methylated in chronic lymphocytic leukemia. Epigenetics 9(6):910–917. https://doi.org/10.4161/epi.28603
Nadal E, Chen G, Gallegos M, Lin L, Ferrer-Torres D, Truini A et al (2013) Epigenetic inactivation of microRNA-34b/c predicts poor disease-free survival in early-stage lung adenocarcinoma. Clin Cancer Res 19(24):6842–6852. https://doi.org/10.1158/1078-0432.CCR-13-0736
Wang Z, Chen Z, Gao Y, Li N, Li B, Tan F et al (2011) DNA hypermethylation of microRNA-34b/c has prognostic value for stage non-small cell lung cancer. Cancer Biol Ther 11(5):490–496. https://doi.org/10.4161/cbt.11.5.14550
Xu Y, Liu L, Liu J, Zhang Y, Zhu J, Chen J et al (2011) A potentially functional polymorphism in the promoter region of miR-34b/c is associated with an increased risk for primary hepatocellular carcinoma. Int J Cancer 128(2):412–417. https://doi.org/10.1002/ijc.25342
Toyota M, Suzuki H, Sasaki Y, Maruyama R, Imai K, Shinomura Y et al (2008) Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res 68(11):4123–4132. https://doi.org/10.1158/0008-5472.CAN-08-0325
Rizza S, Cirotti C, Montagna C, Cardaci S, Consales C, Cozzolino M et al (2015) S-nitrosoglutathione reductase plays opposite roles in SH-SY5Y models of parkinson's disease and amyotrophic lateral sclerosis. Mediat Inflamm 2015:536238. https://doi.org/10.1155/2015/536238
Benassi B, Flavin R, Marchionni L, Zanata S, Pan Y, Chowdhury D et al MYC is activated by USP2a-mediated modulation of microRNAs in prostate cancer. Cancer Discov 2(3):236–247. https://doi.org/10.1158/2159-8290.CD-11-0219
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C[T]) normalized to glyceraldehyde-3-phosphate dehydrogenase levels. qRT-PCR was method. Methods 25:402–408. https://doi.org/10.1006/meth.2001.1262
Falone S, Santini S Jr, di Loreto S, Cordone V, Grannonico M, Cesare P, Cacchio M, Amicarelli F (2016) Improved mitochondrial and methylglyoxal-related metabolisms support hyperproliferation induced by 50 Hz magnetic field in neuroblastoma cells. J Cell Physiol 231(9):2014–2025. https://doi.org/10.1002/jcp.25310
Consales C, Leter G, Bonde JP, Toft G, Eleuteri P, Moccia T et al (2014) Indices of methylation in sperm DNA from fertile men differ between distinct geographical regions. Hum Reprod 29(9):2065–2072. https://doi.org/10.1093/humrep/deu176
Presgraves SP, Ahmed T, Borwege S, Joyce JN (2004) Terminally differentiated SH-SY5Y cells provide a model system for studying neuroprotective effects of dopamine agonists. Neurotox Res 5(8):579–598
Vassilev LT, BT V, Graves B, Carvajal D, Podlaski F, Filipovic Z et al (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303(5659):844–848. https://doi.org/10.1126/science.1092472
Esteves AR, Arduíno DM, Swerdlow RH, Oliveira CR, Cardoso SM (2009) Oxidative stress involvement in alpha-synuclein oligomerization in Parkinson's disease cybrids. Antioxid Redox Signal 11(3):439–448. https://doi.org/10.1089/ARS.2008.2247
Manoharan S, Guillemin GJ, Abiramasundari RS, Essa MM, Akbar M, Akbar MD (2016) The role of reactive oxygen species in the pathogenesis of Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease: a mini review. Oxidative Med Cell Longev 2016:8590578. https://doi.org/10.1155/2016/8590578
Angelova PR, Abramov AY (2016) Alpha-synuclein and beta-amyloid - different targets, same players: calcium, free radicals and mitochondria in the mechanism of neurodegeneration. Biochem Biophys Res Commun. Review. https://doi.org/10.1016/j.bbrc.2016.07.103
Luukkonen J, Liimatainen A, Höytö A, Juutilainen J, Naarala J (2011) Pre-exposure to 50 Hz magnetic fields modifies menadione-induced genotoxic effects in human SH-SY5Y neuroblastoma cells. PLoS One 6(3):e18021. https://doi.org/10.1371/journal.pone.0018021
Hollins SL, Cairns MJ (2016) MicroRNA: Small RNA mediators of the brains genomic response to environmental stress. Prog Neurobiol 43:61–81. https://doi.org/10.1016/j.pneurobio.2016.06.005
Impey S, Pelz C, Tafessu A, Marzulla T, Turker MS, Raber J (2016) Proton irradiation induces persistent and tissue-specific DNA methylation changes in the left ventricle and hippocampus. BMC Genomics 17:273. https://doi.org/10.1186/s12864-016-2581-x
Ji S, Tian Y, Lu Y, Sun R, Ji J, Zhang L et al (2014) Irradiation-induced hippocampal neurogenesis impairment is associated with epigenetic regulation of bdnf gene transcription. Brain Res 1577:77–88. https://doi.org/10.1016/j.brainres.2014.06.035
Leone L, Fusco S, Mastrodonato A, Piacentini R, Barbati SA, Zaffina S et al (2014) Epigenetic modulation of adult hippocampal neurogenesis by extremely low-frequency electromagnetic fields. Mol Neurobiol 49(3):1472–1486. https://doi.org/10.1007/s12035-014-8650-8
Trosko JE (2000) Human health consequences of environmentally-modulated gene expression: potential roles of ELF-EMF induced epigenetic versus mutagenic mechanisms of disease. Bioelectromagnetics 21(5):402–406 Review
Pasi F, Fassina L, Mognaschi ME, Lupo G, Corbella F, Nano R et al (2016) Pulsed electromagnetic field with temozolomide can elicit an epigenetic pro-apoptotic effect on glioblastoma T98G cells. Anticancer Res 36(11):5821–5826. 10.21873/anticanres.11166
Marchesi N, Osera C, Fassina L, Amadio M, Angeletti F, Morini M et al (2014) Autophagy is modulated in human neuroblastoma cells through direct exposition to low frequency electromagnetic fields. J Cell Physiol 229(11):1776–1786. https://doi.org/10.1002/jcp.24631
Sulpizio M, Falone S, Amicarelli F, Marchisio M, Di Giuseppe F, Eleuterio E, Di Ilio C, Angelucci S (2011) Molecular basis underlying the biological effects elicited by extremely low-frequency magnetic field (ELF-MF) on neuroblastoma cells. J Cell Biochem 112(12):3797–3806. https://doi.org/10.1002/jcb.23310
Pirozzoli MC, Marino C, Lovisolo GA, Laconi C, Mosiello L, Negroni A (2003) Effects of 50 Hz electromagnetic field exposure on apoptosis and differentiation in a neuroblastoma cell line. Bioelectromagnetics 24(7):510–516. https://doi.org/10.1002/bem.10130
Grassi C, D'Ascenzo M, Torsello A, Martinotti G, Wolf F, Cittadini A, Azzena GB (2004) Effects of 50 Hz electromagnetic fields on voltage-gated Ca2+ channels and their role in modulation of neuroendocrine cell proliferation and death. Cell Calcium 35(4):307–315. https://doi.org/10.1016/j.ceca.2003.09.001
Martínez MA, Úbeda A, Cid MA, Trillo MÁ (2012) The proliferative response of NB69 human neuroblastoma cells to a 50 Hz magnetic field is mediated by ERK1/2 signaling. Cell Physiol Biochem 29(5–6):675–686. https://doi.org/10.1159/000178457
Cannell IG, Bushell M (2010) Regulation of Myc by miR-34c: a mechanism to prevent genomic instability? Cell Cycle The MYCN oncogene is a direct target of miR-34a
Wei JS, Song YK, Durinck S, Chen QR, Cheuk AT, Tsang P, Zhang Q, Thiele CJ et al (2008) The MYCN oncogene is a direct target of miR-34a. Oncogene 27(39):5204–5213. https://doi.org/10.1038/onc.2008 15;9(14):2726-30
Wang AM, Huang TT, Hsu KW, Huang KH, Fang WL, Yang MH, Lo SS, Chi CW et al (2014) Yin Yang 1 is a target of microRNA-34 family and contributes to gastric carcinogenesis. Oncotarget 5(13):5002–5016. 10.18632/oncotarget.2073
Bae Y, Yang T, Zeng HC, Campeau PM, Chen Y, Bertin T, Dawson BC, Munivez E et al (2012) miRNA-34c regulates Notch signaling during bone development. Hum Mol Genet 21(13):2991–3000. https://doi.org/10.1093/hmg/dds129
Zou Y, Huang Y, Yang J, Wu J, Luo C (2017) miR-34a is downregulated in human osteosarcoma stem-like cells and promotes invasion, tumorigenic ability and self-renewal capacity. Mol Med Rep 15(4):1631–1637. https://doi.org/10.3892/mmr.2017.6187
Wang P, Xu J, Hou Z, Wang F, Song Y, Wang J, Zhu H, Jin H (2016) miRNA-34a promotes proliferation of human pulmonary artery smooth muscle cells by targeting PDGFRA. Cell Prolif 49(4):484–493. https://doi.org/10.1111/cpr.12265
Jauhari A, Singh T, Singh P, Parmar D, Yadav S (2017, 2017) Regulation of miR-34 Family in Neuronal Development. Mol Neurobiol. https://doi.org/10.1007/s12035-016-0359-4
Hernandez-Rapp J, Rainone S, Hébert SS (2017) MicroRNAs underlying memory deficits in neurodegenerative disorders. Prog Neuro-Psychopharmacol Biol Psychiatry 73:79–86. https://doi.org/10.1016/j.pnpbp.2016.04.011
Saito Y, Saito H (2012) MicroRNAs in cancers and neurodegenerative disorders. Front Genet 3:194. https://doi.org/10.3389/fgene.2012.00194
Poddar S, Kesharwani D, Datta M (2017, 2017) Interplay between the miRNome and the epigenetic machinery: implications in health and disease. J Cell Physiol. https://doi.org/10.1002/jcp.25819
Niklison-Chirou MV, Killick R, Knight RA, Nicotera P, Melino G, Agostini M (2016) How does p73 cause neuronal defects? Mol Neurobiol 53(7):4509–4520. https://doi.org/10.1007/s12035-015-9381-1
Agostini M, Melino G, Bernassola F (2017, 2017) The p53 family in brain disease, Antioxid Redox Signal. https://doi.org/10.1089/ars.2017.7302
Jacobs WB, Kaplan DR, Miller FD (2006) The p53 family in nervous system development and disease. J Neurochem 97(6):1571–1584. Review. https://doi.org/10.1111/j.1471-4159.2006.03980.x
Lacombe J, Zenhausern F (2017) Emergence of miR-34a in radiation therapy. Crit Rev Oncol Hematol 109:69–78. https://doi.org/10.1016/j.critrevonc.2016.11.017
Liu Y, Liu WB, Liu KJ, Ao L, Zhong JL, Cao J et al (2015) Effect of 50 Hz extremely low-frequency electromagnetic fields on the DNA methylation and DNA methyltransferases in mouse spermatocyte-derived cell line GC-2. Biomed Res Int 237183:2015. https://doi.org/10.1155/2015/237183
Liu Y, Liu WB, Liu KJ, Ao L, Cao J, Zhong JL et al (2015) Extremely low-frequency electromagnetic fields affect the miRNA-mediated regulation of signaling pathways in the GC-2 cell line. PLoS One 10(10):e0139949. https://doi.org/10.1371/journal.pone.0139949
Liu Y, Liu WB, Liu KJ, Ao L, Cao J, Zhong JL et al (2016) Overexpression of miR-26b-5p regulates the cell cycle by targeting CCND2 in GC-2 cells under exposure to extremely low frequency electromagnetic fields. Cell Cycle 15(3):357–367. https://doi.org/10.1080/15384101.2015.1120924
Prasad KN (2017) Oxidative stress, pro-inflammatory cytokines, and antioxidants regulate expression levels of MicroRNAs in Parkinson’s disease. Curr Aging Sci 2017 Jan 2
Xiong R, Wang Z, Zhao Z, Li H, Chen W, Zhang B et al (2014) MicroRNA-494 reduces DJ-1 expression and exacerbates neurodegeneration. Neurobiol Aging 35(3):705–714. https://doi.org/10.1016/j.neurobiolaging.2013.09.027
Ma MW, Wang J, Zhang Q, Wang R, Dhandapani KM, Vadlamudi RK et al (2017) NADPH oxidase in brain injury and neurodegenerative disorders. Mol Neurodegener 12(1):7. https://doi.org/10.1186/s13024-017-0150-7
Lupke M, Rollwitz J, Simkó M (2004) Cell activating capacity of 50 Hz magnetic fields to release reactive oxygen intermediates in human umbilical cord blood-derived monocytes and in Mono Mac 6 cells. Free Radic Res 38(9):985–993. https://doi.org/10.1080/10715760400000968
Feng B, Dai A, Chen L, Qiu L, Fu Y, Sun W (2016) NADPH oxidase-produced superoxide mediated a 50-Hz magnetic field-induced epidermal growth factor receptor clustering. Int J Radiat Biol 92(10):596–602. https://doi.org/10.1080/09553002.2016.1206227
Bendor JT, Logan TP, Edwards RH (2013) The function of alpha-synuclein. Neuron 79:1044–1066. https://doi.org/10.1016/j.neuron.2013.09.004
Dodson MW, Guo M (2007) Pink1, Parkin, DJ-1 and mitochondrial dysfunction in Parkinson's disease. Curr Opin Neurobiol 17(3):331–337. https://doi.org/10.1016/j.conb.2007.04.010 Review
Li JQ, Tan L, JT Y (2014) The role of the LRRK2 gene in Parkinsonism. Mol Neurodegener 9:47. https://doi.org/10.1186/1750-1326-9-47 Review
Funding
G.F. was supported by grants from the Danish Cancer Society (R72-A4647; R146-A9414); the Italian Association for Cancer Research, (AIRC-MFAG 2011 n.1145) and is part of the Center of Excellence in Autophagy, Recycling and Disease (CARD), funded by the Danish National Research Foundation.
We are very grateful to Francesca Pacchierotti for her helpful criticisms and scientific support.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Online Resource 1
(PPT 110 kb)
Online Resource 2
(PPT 356 kb)
Online Resource 3
(PPT 318 kb)
Online Resource 4
(PPT 896 kb)
Online Resource 5
(PPT 281 kb)
Online Resource 6
(PPT 283 kb)
Online Resource 7
(PPT 167 kb)
Rights and permissions
About this article

Cite this article
Consales, C., Cirotti, C., Filomeni, G. et al. Fifty-Hertz Magnetic Field Affects the Epigenetic Modulation of the miR-34b/c in Neuronal Cells. Mol Neurobiol 55, 5698–5714 (2018). https://doi.org/10.1007/s12035-017-0791-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0791-0