
Abstract
Glioblastoma multiforme (GBM) is the most fatal of all brain cancers, and the standard care protocol for GBM patients is surgical tumor resection followed by radiotherapy and temozolomide (TMZ)-mediated chemotherapy. However, tumor recurrence frequently occurs, and recurrent GBM exhibits more malignancy and less sensitivity in response to chemotherapy. The malignancy and drug resistance primarily reflect the small population of glioma stem-like cells (GSC). Therefore, understanding the mechanism that controls GSC enrichment is important to benefit the prognosis of GBM patients. Nucleolin (NCL), which is responsible for ribosome biogenesis and RNA maturation, is overexpressed in gliomas. However, the role of NCL in GSC development and drug resistance is still unclear. In this study, we demonstrate that NCL attenuated GSC enrichment to enhance the sensitivity of GBM cells in response to TMZ. In GSC enrichment, NCL was significantly reduced at the protein level as a result of decreased protein stability. In particular, the inhibition of HDAC activity by suberoylanilide hydroxamic acid rescued NCL acetylation accompanied by the loss of mouse double minute 2 homolog (MDM2)-mediated ubiquitination. In addition, we found that NCL ubiquitination resulted from the activation of STAT3- and JNK-mediated signaling in GSC. Moreover, NCL inhibited the formation of stem-like spheres by attenuating the expression of Sox2, Oct4, and Bmi1. Furthermore, NCL sensitized the response of GBM cells to TMZ. Based on these findings, NCL expression is a potential indicator to predict chemotherapeutic efficiency in GBM patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Repeated recurrence mediated by drug and radioresistance after therapy is the primary reason for the death of cancer patients [1, 2]. Glioblastoma multiforme (GBM) is the most malignant of brain tumors and consistently develops resistance in response to chemotherapy, leading to recurrence. The standard care protocol of GBM is surgical resection following chemotherapy and radiotherapy. Temozolomide (TMZ), the major chemotherapeutic drug for GBM, does not benefit all patients, and its effects are limited to a short time window [3,4,5]. In particular, TMZ slightly extends the median survival from 12.1 to 14.6 months in GBM patients who received radiotherapy [4]. After resection and therapy, the recurrent tumors are more aggressive and less sensitive to therapy. A small subpopulation of cells, termed GBM stem-like cells (GSC), has strong self-renewal potential and contributes to tumorigenic initiation, progression, and resistance [6]. Radio- and chemotherapy enrich the population of GSC, characterized by the expression of CD133, Oct4, Sox2, and Bmi1, thereby increasing the difficulty of treatment [6,7,8].
Compared with normal tumor cells, cancer stem cells exhibit more resistance to radio- and chemotherapy [9, 10], and TMZ treatment ultimately expands the GSC population [11, 12]. In addition, inhibition of Oct4 and Sox2, both of which are markers of GSC, significantly improved TMZ-mediated therapy in the orthotopic GBM mouse model [13]. In particular, the master regulators of GSC, such as Sox2, Oct4, and Bmi1, were shown to mediate chemoresistance [14,15,16]. These results highlight the importance of the GSC population in glioma development, and the identification of therapeutic strategies targeting GSC enrichment is needed to cure glioma patients.
Recent studies have indicated that nucleolin (NCL) is overexpressed in glioma and that glycosylated NCL serves as a putative marker of glioma [17]. In addition to ribosome biogenesis, NCL extensively regulates several cellular functions, including transcription, RNA turnover, translation, and viral entry into the cellular membrane [18]. The understanding of NCL in stem cell biology is still limited. In embryonic stem cell maintenance, NCL suppresses the p53 pathway and affects Oct4-mediated transcription for self-renewal [19, 20]. In addition, NCL is essential to maintain hematopoietic progenitor cells [21]. However, the role of NCL in GSC enrichment and drug sensitivity remains unclear. In the present study, we found that NCL expression is decreased in GSC compared with normal GBM. The decreased NCL in GSC was caused by the ubiquitination-mediated protein destabilization controlled by mouse double minute 2 homolog (MDM2). Specifically, HDAC2-mediated deacetylation was required for NCL ubiquitination. Moreover, the downregulation of NCL contributes to the development of stemness and TMZ resistance.
Materials and Methods
Cell Culture and Chemicals
The glioma cell lines U87MG, U373MG, and A172 were purchased from ATCC (Manassas, VA, USA) and cultured in Dulbecco’s modified Eagle’s medium (DMEM, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (GE Healthcare Life Sciences, South Logan, UT, USA), 100 μg/ml penicillin, and 100 μg/ml streptomycin (Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C in a 5% CO2 incubator. Both FTY720 and SP600125 were purchased from Selleckchem (Houston, TX, USA).
Human Specimens
The use of human specimens was approved by the Clinical Research Ethics Committee at Taipei Medical University Hospital. The human GBM cells, pt#11, were isolated from the tumor tissue of a male glioma patient. Before radio- and chemotherapy, the GBM tissue was excised from the patient at the first surgery, and tissue was freshly homogenized by collagenase type IV and DNase I for purifying pt#11 cells. Pt#11 cells were maintained in the DMEM containing 10% FBS.
Bioinformatics
The messenger RNA (mRNA) level of NCL in primary and recurrent GBM was retrieved from the GSE4271 database of NCBI [22, 23]. The comparison of NCL in these two groups was analyzed by Student’s t test.
The Establishment of TMZ-Resistant Cells
The concentration of TMZ in the serum of patient which received TMZ-mediated chemotherapy is approximately 100 μM [24, 25], which is sufficient to induce apoptosis in GBM cells in in vitro studies [26, 27]. In addition, IC50 of U87MG cells in response to TMZ is approximately 100–172 μM [28, 29]. Therefore, we attempted to establish the cells which exhibit resistance to 100–200 μM TMZ. After treatment with TMZ (Sigma-Aldrich, St. Louis, MO, USA) for 24 h, the U87MG or pt#11 cells were seeded onto a 96-well plate (1 cell/well) and cultured in growth media containing 100 μM TMZ for an additional 21 days. On day 22, cells were maintained in the media containing 150 μM TMZ until day 42; on day 43, cells were maintained in the media containing 200 μM TMZ for 21 days. The surviving cells were gradually expanded into 12-well plates and 6- and 10-cm dishes with 200 μM TMZ treatment. The healthy and TMZ-resistant cells were maintained in the media containing 200 μM TMZ. The resistance characteristics were confirmed using a colony formation assay.
Colony Formation Assay
One hundred U87MG and TMZ-resistant U87MG cells were seeded onto 6-cm dishes and incubated for 3 days. On day 4, cells were treated with the indicated dose of TMZ for additional 9 days, and the media containing TMZ was renewed every 2 days. On day 12, cell colonies were stained with 0.05% crystal violet (Sigma-Aldrich) overnight. After washing by tap water, cell colonies were photographed by a scanner, and the number of colony was quantified by the Image J software (National Institute of Mental Health, Bethesda, MD, USA).
Glioma Sphere Culture and Treatment
Using a previously established protocol [30, 31], the cells (1.6 × 105) were suspended in F12/DMEM (Thermo Fisher Scientific) containing 20 ng/ml FGF (Sigma-Aldrich), 20 ng/ml EGF (Sigma-Aldrich), and N2 supplement (Thermo Fisher Scientific); seeded onto 6-cm dishes coated with poly(2-hydroxyethyl methacrylate) (Santa Cruz Biotechnology, Inc., Dallas, TX, USA); and incubated at 37 °C and 5% CO2 for the indicated time. The number of spheres was quantified using ImageJ software (National Institute of Mental Health).
Transfection
The cells were transfected with the indicated plasmids using Polyjet reagent (SignaGen Laboratories, Rockville, MD, USA) according to the manufacturer’s instructions. The plasmid, pEGFP-C2-NCL (GFP-NCL), was constructed as previously described [32]. For transfection of GFP-NCL, 2 × 105 cells were seeded onto 6-well plates and incubated overnight. Two micrograms of GFP-NCL was diluted in a 1.5-ml Eppendorf tube containing 100 μl serum-free media; 6 μl of Polyjet reagent was diluted in a 1.5-ml Eppendorf tube containing 100 μl serum-free media. After mixing and spinning down, serum-free media containing Polyjet reagent was transferred into a 1.5-ml Eppendorf tube containing serum-free media and GFP-NCL plasmid. After incubating for 15 min at room temperature, the mixture containing the plasmid and Polyjet reagent was transferred into the well containing 2 × 105 cells and 1 ml growth media. To overexpress GFP-NCL in spheroid U87MG, cells were transfected with the GFP-NCL plasmid overnight and subsequently subjected to sphere culture methods.
Lentivirus-Mediated Gene Knockdown
The shRNA-expressing lentiviruses targeting scramble (Clone ID: ASN0000000001; oligo sequence: CCGGAGTTCAGTTACGATATCATGTCTCGAGACATTCGCGAGTAACTGAACTTTTTT; target sequence: none), NCL (Clone ID: TRCN0000062284; oligo sequence: CCGGCGGTGAAATTGATGGAAATAACTCGAGTTATTTCCATCAATTTCACCGTTTTTG;target sequence: CGGTGAAATTGATGGAAATAA), or MDM2 (Clone ID: TRCN0000355725; oligo sequence: CCGGATTATCTGGTGAACGACAAAGCTCGAGCTTTGTCGTTCACCAGATAATTTTTTG; target sequence: ATTATCTGGTGAACGACAAAG) were constructed at the RNAi Core Facility of Academic Sinica (Taipei, Taiwan) and were used according to the provider’s instructions.
Western Blotting
Cellular proteins were lysed by RIPA lysis buffer (EMD Millipore, Billerica, MA, USA) containing protease inhibitors (EMD Millipore), and protein concentration was determined by BCA assay (Thermo Fisher Scientific). Twenty micrograms of proteins were subjected to western blotting. After protein electrophoresis and transfer, the blocked PVDF membrane was incubated with primary antibody, including anti-NCL (1:2000; Santa Cruz Biotechnology, Inc.), anti-CD133 (1:1000; Proteintech Group, Chicago, IL, USA), anti-Sox2 (1:1000; GeneTex, Inc., Irvine, CA, USA), anti-Bmi1 (1:1000; GeneTex, Inc.), anti-Oct4 (1:1000; Cell Signaling Technology, Danvers, MA, USA), anti-MDM2 (1:1000; Santa Cruz Biotechnology, Inc.), anti-ubiquitin (1:2000; GeneTex, Inc.), anti-acetyl-lysine (1:2000; Cell Signaling Technology), anti-p300 (1:2000; GeneTex, Inc.), anti-Nanog (1:1000; GeneTex, Inc.), anti-HDAC2 (1:1000; EMD Millipore), anti-pSTAT3 (1:1000; Cell Signaling Technology), anti-STAT3 (1:3000; Santa Cruz Biotechnology, Inc.), anti-pJNK (1:1000; Cell Signaling Technology), anti-JNK (1:3000; Cell Signaling Technology), anti-pERK (1:2000; Cell Signaling Technology), anti-ERK (1:5000; EMD Millipore), anti-pAkt (1:1000; Cell Signaling Technology), anti-Akt (1:3000; Santa Cruz Biotechnology, Inc.), anti-pp38 (1:1000; Cell Signaling Technology), anti-p38 (1:3000; Cell Signaling Technology), anti-PARP (1:1000; Cell Signaling Technology), anti-caspase 3 (1:1000; Cell Signaling Technology), anti-GAPDH (1:20,000; Proteintech Group), or anti-tubulin (1:20,000; Sigma-Aldrich), at 4 °C overnight. After incubation with secondary antibody conjugated with HRP (1:20,000; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA), the signals amplified using enhanced chemiluminescence reagent (ECL, GE Healthcare Life Sciences) were captured using the ChemiDoc™ Touch Imaging System (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Protein Stability Analysis
The cells were treated with 10 μM cycloheximide (CHX), and 20 μg of the lysates was collected at the indicated times for western blot using the anti-NCL antibody.
Immunoprecipitation
Five hundred micrograms of proteins were incubated with 2 μg of the anti-NCL antibody or IgG (Santa Cruz Biotechnology, Inc.) for 1 h at 4 °C, and the cell lysates containing the antibody were mixed with 50 μl of protein A/G PLUS agarose (Santa Cruz Biotechnology, Inc.) for 1 h at 4 °C. After centrifugation at 3000 rpm for 5 min at 4 °C, the immune-complex was collected and analyzed using western blot.
RT-qPCR
TRIsure reagent (SignaGen Laboratories) was used to prepare total RNA according to the manufacturer’s instructions. The cDNA was synthesized using the SensiFAST™ cDNA Synthesis Kit (Bioline USA, Inc., Taunton, CA, USA) and was subjected to real-time PCR using the SensiFAST™ SYBR® Hi-ROX Kit (Bioline USA Inc.) in a StepOnePlus™ Real-Time PCR System (Thermo Fisher Scientific). The following primers were used for qPCR: Sox2, F-AAATGGGAGGGGTGCAAAAGAGGAG, R-CAGCTGTCATTTGCTGTGGGTGAT G; Bmi1, F-TGGAGAAGGAATGGTCCACTT C, R-GTGAGGAAACTGTGGATGAGGA; Oct4, F-CTTGCTGCAGAAGTGGGTGGAGGAA, R-CTGCAGTGTGGGTTTCGGGCA; NCL, F-ATGGTGAAGCTCGCGAAGGC, R-ATCCTCCTCTTCATCACTGT; and GAPDH, F-CCATCACCATCTTCCAGGAG, R-CCTGCTTCACCACCTTCTTG.
Statistical Analysis
Student’s t test was used to compare the differences between the groups. Protein stability was compared using a two-way analysis of variance (ANOVA). P <0.05 was considered statistically significant.
Results
NCL Downregulation in Recurrent and TMZ-Resistant Gliomas
Because the role of NCL in the occurrence of drug resistance is unknown, we compared NCL levels in gliomas with or without TMZ resistance. In Fig. 1a, NCL mRNA was significantly decreased in recurrent GBM according to GSE4271, suggesting that NCL downregulation is involved in drug resistance. In addition, glioma stem-like cells potentially contribute to TMZ resistance. Therefore, we established TMZ-resistant GBM cells (Fig. 1b) and found that NCL was significantly decreased concomitant with the significant upregulation of stemness markers, including CD133, Sox2, Bmi1, and Oct4, in TMZ-resistant U87MG and pt#11 cells (Fig. 1c, d). These results suggest that the decreased expression of NCL contributes to the TMZ resistance of the expanding GSC population.

Reduction of NCL expression in recurrent and TMZ-resistant glioma. a The mRNA level of NCL was retrieved from the GSE4271 database. The comparison was performed using Student’s t test (*P < 0.05). b The sensitivity of U87MG and U87MG-R to TMZ was evaluated based on colony formation and was shown as the quantitative result. After seeding cells for 3 days, cells were treated with TMZ for additional 9 days and stained with crystal violet. Independent experiments were performed three times in triplicate. The difference between two groups was analyzed by Student’s t test. P value was indicated. c The cell lysates were subjected to western blotting using the antibody recognizing NCL, CD133, Oct4, Bmi1, or tubulin. d Quantitated results of western blotting in c. Experiments were performed three times independently, and the difference was analyzed by Student’s t test (*P < 0.05, **P < 0.01)
The Reduction of NCL Expression in Glioma Stem-Like Spheres
Based on the reduction of NCL in TMZ-resistant cells which exhibit GSC stemness, we attempted to evaluate the expression of NCL in GSC. Therefore, compared with adherent glioma cells, we evaluated the NCL expression in GSC-like spheres of U87MG, U373MG, A172, and pt#11 cells (Fig. 2A). As the spheres developed stemness, characterized by CD133, Sox2, Oct4, and Bmi1, NCL expression was significantly decreased in spheroid U87MG, U373MG, A172, and pt#11 cells (Fig. 2B). In particular, the spheroid formation of U87MG/GSC is more significant than that of A172, U373, and pt#11 cells (data not shown). These findings suggest that NCL is a negative regulator of GSC enrichment.

The decrease of NCL in GSC-like spheres compared with normal glioma. Spheres were induced and cultured as described in the “Materials and Methods.” A Representative images of adherent and spheroid glioma cells. The formation of GSC sphere was induced and cultured according to the “Materials and Methods” section. Magnification ×10; scale bar 100 μm. B The lysates of adherent and spheroid glioma, including U87MG, U373MG, A172, and pt#11 cells, were collected on the indicated day and were subjected to western blotting. The protein level of NCL was quantified and normalized to tubulin. a adhered cells. Experiments were performed three times independently and difference between two groups was analyzed by Student’s t test (P* < 0.05, P** < 0.01, P*** < 0.001)
MDM2-Mediated Ubiquitination Degrades NCL in GSC
To characterize the mechanism underlying NCL downregulation in GSC, we analyzed NCL mRNA in U87MG and U373MG cells, both of which were cultured in adherent type and spheroid type of GSC. In Fig. 3a, NCL mRNA expression remained stable, whereas the mRNA level of stemness markers was significantly increased as the spheres developed, suggesting that transcription is not involved in NCL alterations in GSC. Therefore, we tested NCL protein stability, as shown in Fig. 3b, and found that NCL protein was significantly more unstable for degradation in GSC. Protein stability is highly modulated by posttranslational modification, particularly ubiquitination. Accordingly, NCL ubiquitination was identified in spheroid U87MG/GSC, but not in adherent U87MG cells (Fig. 3c). Because NCL was shown to interact with MDM2 [33, 34], we postulated that MDM2 is the E3 ligase for NCL ubiquitination; therefore, this interaction in GSC was identified (Fig. 3c). Interestingly, NCL acetylation was lost in GSC (Fig. 3c), in which NCL interacted with HDAC2 (Fig. 3c) but not with HDAC1 or HDAC3 (data not shown). To demonstrate whether MDM2 influences NCL for GSC enrichment, MDM2-knocked down cells were induced to develop stem-like spheres. MDM2 knockdown not only impaired sphere formation but also increased NCL protein levels (Fig. 3d, e). Furthermore, MDM2 knockdown significantly abolished NCL ubiquitination in GSC to rescue NCL degradation (Fig. 3e). All together, these findings suggest that MDM2-mediated NCL ubiquitination is important for GSC enrichment.

MDM2 degrades NCL through inducing ubiquitination in GSC. a The mRNA levels of NCL and stemness markers. b Adherent and spheroid U87MG cells were treated with 10 μM CHX, and the cell lysates were harvested at the indicated times for western blotting. Lower panel, the stability of NCL protein in adherent and spheroid U87MG cells was compared using two-way ANOVA. Experiments were performed three times independently. P value is indicated. The protein level of NCL at the indicated treatment time of CHX in adherent and spheroid U87MG was compared using Student’s t test (*P < 0.05, **P < 0.01, ***P < 0.001). c Five hundred of cell lysates from adherent (a) and spheroid (s) U87MG cells were immunoprecipitated using the anti-NCL antibody, and the immune-complex was analyzed by western blotting using the anti-ubiquitin, anti-MDM2, anti-NCL, anti-acetyl-lysine (Ac-K), anti-p300, anti-HDAC2, or anti-Nanog antibody. Experiments were performed three times independently. d After MDM2 knockdown, U87MG cells were induced to form GSC spheres for 6 days, and the number of spheres was counted (P** < 0.01). Magnification ×2.5; scale bar 25 μm. e Left panel, after MDM2 knockdown, the cell lysates were analyzed by western blotting using the anti-MDM2 or anti-NCL antibody. Right panel, quantitated results. Experiments were performed three times independently, and the difference was analyzed by Student’s t test (*P < 0.05). f Left panel, the lysates of spheroid U87MG with or without MDM2 knockdown were immunoprecipitated using the anti-NCL antibody, and the precipitates were analyzed by western blotting using the anti-ubiquitin or anti-MDM2 antibody. Right panel, the ubiquitination of NCL was quantified and analyzed by Student’s t test (*P < 0.05)
Inhibition of HDAC Prevents NCL Ubiquitination and Impairs GSC Enrichment
Based on the acetylation loss of NCL in GSC, we attempted to determine whether HDAC-mediated NCL deacetylation is important for GSC. As shown in Fig. 4a, following treatment with suberoylanilide hydroxamic acid (SAHA), a potent HDAC inhibitor, spheroid formation was significantly inhibited. In addition, SAHA increased NCL expression accompanied by Sox2 downregulation and apoptosis, as evidenced by PARP and caspase 3 cleavage (Fig. 4b). To determine whether SAHA increases transcription of NCL promoter resulting in the upregulation of the NCL protein, we analyzed NCL mRNA expression and found that SAHA did not affect NCL mRNA expression (Supplementary Fig. S1), and we speculated that acetylation is involved in NCL protein stability. In particular, SAHA significantly extended the half-life of NCL protein in GSC (Fig. 4c). Furthermore, SAHA rescued acetylation through abolishing the interaction with HDAC2 and prevented ubiquitination by inhibiting the interaction with MDM2 (Fig. 4d), leading to NCL accumulation in GSC. These results suggest that NCL is deacetylated by HDAC2, followed by MDM2-mediated ubiquitination in GSC. The restoration of acetylation by inhibiting HDAC successfully prevents NCL ubiquitination and impairs GSC enrichment.

Effect of SAHA on GSC formation and NCL expression. a After treatment with SAHA for 48 h, the GSC spheres on day 6 were photographed. Magnification ×10; scale bar 100 μm. b GSC spheres were collected for protein preparation and analyzed by western blotting. Lower panel, the quantitative results of NCL and Sox2. Experiments were performed three times independently, and the difference was analyzed by Student’s t test (*P < 0.05, **P < 0.01). c Upper panel, in the presence or absence of SAHA, the GSC spheres on day 6 were treated with CHX for the indicated times. The NCL expression was analyzed by western blotting, and the stability was compared using two-way ANOVA (lower panel). Experiments were performed three times independently. P value is indicated. The protein level of NCL at the indicated treatment time point of CHX was compared using Student’s t test (*P < 0.05). d Upper panel, the immune-complex precipitated using the anti-NCL antibody was analyzed by western blotting using the anti-acetyl-lysine, anti-HDAC2, anti-ubiquitin, or anti-MDM2 antibody. Lower panel, both acetylation and ubiquitination of NCL were quantified. a adherent, s spheroid. Experiments were performed three times independently, and the difference was analyzed by Student’s t test (*P < 0.05, **P < 0.01)
Activation of STAT3 and JNK-Mediated Signaling Contributes to NCL Reduction and GSC-Like Spheres Formation
To investigate the mechanism contributing to NCL reduction in GSC, we analyzed the phosphorylation of several kinases in adherent GBM and GSC-like spheres. As shown in Fig. 5a, compared with adherent U87MG and pt#11 cells, the phosphorylation of STAT3 and JNK1 was significantly increased in both spheroid U87MG and pt#11. Spheroid pt#11, not spheroid U87MG cells, exhibited the significantly increased levels of phosphorylated Akt and ERK (Fig. 5a). Accordingly, we proposed that the reduction of NCL in GSC is caused by the activation of STAT3 and JNK, and subsequent experiments showed that the blockade of JNK or STAT3 by SP600125 or FTY720, respectively, is sufficient to rescue NCL reduction in spheroid U87MG (Fig. 5b, c) and pt#11 cells (Supplementary Fig. S2). All together, these findings indicate that STAT3- and JNK-mediated signaling promotes GSC enrichment by downregulating NCL expression.

Kinase phosphorylation in adherent and spheroid glioma cells. a Cell lysates were prepared and subjected to western blotting using the indicated antibody. Adh adherent, Sph spheroid. Experiments were performed three times independently, and the difference was analyzed by Student’s t test (*P < 0.05, **P < 0.01). b, c After treatment with SP600125 (upper panel) or FTY720 (lower panel) for 48 h, the day 6 GSC spheres were collected to prepare the protein lysates and subjected to western blotting. Experiments were performed three times independently, and the difference was analyzed by Student’s t test (*P < 0.05, **P < 0.01)
NCL Inhibits GSC Enrichment and Sensitizes Glioma to TMZ Treatment
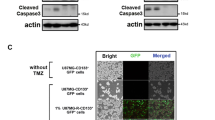
To confirm whether NCL impairs GSC enrichment, we subjected U87MG-expressing GFP-NCL to sphere culture. As shown in Fig. 6A, GFP-NCL not only reduced the size but also significantly decreased the number and size of spheroid U87MG cells (Fig. 6A). Furthermore, the stem-like characteristics, as evidenced by Sox2, Oct4, and Bmi1 expression, were significantly decreased by NCL (Fig. 6B). Additionally, the inhibitory effect of NCL on Sox2 and Oct4 was due to transcription inhibition and mRNA destabilization (Supplementary Fig. S3). Therefore, we speculated that NCL prevents TMZ resistance as a result of its inhibitory effect on GSC enrichment. In Fig. 6C, spheroid U87MG cells exhibited resistance to 200 μM TMZ, whereas GFP-NCL-expressing spheres were destroyed in response to treatment with 100 μM TMZ. In addition, TMZ exhibited a more potent effect in NCL-expressing spheres on inducing apoptosis, as characterized by caspase 3 cleavage (Fig. 6C). We also examined the effect of NCL on TMZ sensitivity in adherent U87MG cells and found that NCL overexpression enhanced TMZ-decreased cell survival (Supplementary Fig. S4). In contrast, NCL knockdown surprisingly induced the formation of spheroid U87MG cells and increased Sox2 expression (Fig. 7a, b). Moreover, NCL knockdown significantly attenuated TMZ-induced apoptosis, as evidenced by cleaved PARP and caspase 3 (Fig. 7c, d). Based on these results, in TMZ-sensitive glioma, acetylated NCL constitutively inhibits Sox2, Oct4, and Bmi1 to prevent GSC expansion, and in TMZ-resistant cells, HDAC2-mediated deacetylation and MDM2-mediated ubiquitination induce NCL degradation to de-repress Sox2, Oct4, and Bmi1, leading to GSC enrichment and TMZ resistance (Fig. 8).

Effect of NCL overexpression on GSC enrichment and GBM sensitivity to TMZ. A After transfection with GFP-NCL, U87MG cells were induced to form GSC spheres for 7 days, which were photographed (left) and quantified (right). Magnification ×2.5; scale bar 25 μm; magnification ×10; scale bar 100 μm. Experiments were performed three times independently, and the difference was analyzed by Student’s t test (*P < 0.05). B The cell lysates of day 7 spheroid U87MG cells were analyzed by western blotting (left), and the expression of stemness markers was quantified (right panel). Experiments were performed three times independently, and the difference was analyzed by Student’s t test (*P < 0.05). C After GFP-NCL overexpression, the spheroid U87MG cells on day 5 were treated with TMZ for 48 h. The GSC spheres were photographed (a) and counted (b). Magnification ×2.5; scale bar 25 μm; magnification ×10; scale bar 100 μm. Experiments were performed three times independently, and the difference was analyzed by Student’s t test (**P < 0.01, ***P < 0.001). c Subsequently, the spheres were homogenized to prepare protein lysates, and the lysates were analyzed by western blotting. d Quantitative results. Experiments were performed three times independently, and the difference was analyzed by Student’s t test (P value is indicated)

Effect of NCL silence on GSC enrichment and GBM sensitivity to TMZ. a After infection with shNCL-expressing lentivirus for 5 days, the U87MG cells were photographed (left panel) and counted (right panel). Magnification ×2.5; scale bar 25 μm; magnification ×10; scale bar 100 μm. b Subsequently, the cells were harvested for protein preparation and analyzed by western blotting. Experiments were performed three times independently, and the difference was analyzed by Student’s t test (*P < 0.05, **P < 0.01). c After NCL knockdown, the U87MG cells were treated with TMZ for 48 h and lysates were collected for western blotting using the anti-NCL, PARP, or caspase 3 antibodies. d Quantitated results. Two groups at the same dose of TMZ were compared using Student’s t test (*P < 0.05, **P < 0.01, ***P < 0.001)

The present study demonstrates that NCL is acetylated, and NCL inhibits stemness development in TMZ-sensitive glioma. In contrast, in STAT3- and JNK-induced GSC spheres, NCL protein was degraded through HDAC2-mediated deacetylation and MDM2-mediated ubiquitination, thus leading to the enhancement of stemness and TMZ resistance
Discussion
In this study, we demonstrated that NCL downregulation in GSC contributed to TMZ resistance. Lower expression of NCL was accompanied by the upregulation of markers of GSC, including CD133, Oct4, Bmi1, and Sox2. We further demonstrated that the protein level of NCL was decreased in GSC stem-like spheres. In particular, both HDAC2-mediated deacetylation and MDM2-mediated ubiquitination contributed to protein instability of NCL in GSC, leading to protein degradation. Furthermore, p-STAT3 and p-JNK, both of which are required for GSC-like spheres enrichment [35, 36], caused NCL downregulation. Moreover, NCL enhanced TMZ-induced apoptosis, characterized by detecting caspase 3 and PARP cleavages. Accordingly, NCL sensitizes the response of GBM to TMZ through inhibiting GSC formation.
Over the past decade, NCL has been identified as an oncogenic protein that regulates proliferation, metastasis, and angiogenesis [18]. In addition to expression, NCL localization is essential for tumor development. Membrane NCL cooperates with surface receptors, such as EGFR, ErbB2, and TGFβ receptor, to promote tumorigenicity [37,38,39]. Strategies targeting surface NCL have been identified [40], thus implicating a critical role for NCL in tumor development. However, the role of NCL in cancer stem cell enrichment, which contributes to chemoresistance, remains unclear. The results of the present study provide a novel insight into how NCL suppresses GSC to prevent the occurrence of TMZ resistance.
Although we clarified that NCL was decreased in recurrent glioma and GSC (Figs. 1 and 2), it remains unclear whether NCL is a biomarker to predict the response of patients to TMZ administration. However, additional studies are needed to confirm whether patients with low NCL expression exhibit a poor response to TMZ. The NCL downregulation in GSC is different from that in both breast cancer stem cells and hematopoietic progenitor cells whose NCL is upregulated [21, 41]. The discrepancy between these controversial results remains unknown. In addition to regulating GSC expansion, NCL affects drug sensitivity by regulating the DNA repair system. The deletion of NCL/Nsr1 reduces the sensitivity to camptothecin by affecting topoisomerase I localization [42]. NCL also inhibits nucleotide excision repair, which antagonizes TMZ treatment [43]. To confirm whether NCL affects TMZ sensitivity in GSC-independent pathways, we showed that TMZ exhibited a stronger effect on inhibiting cell survival in adherent glioma cells overexpressing GFP-NCL (Supplementary Fig. S4). The underlying mechanism remains unknown.
Studies on NCL acetylation have not been well established. In the present study, we indicated that NCL acetylation confers to enhanced protein stability, which has never been reported. In addition, because NCL ubiquitination was identified, the E3 ligase for this modification should be determined. Previously, NCL was shown to associate with MDM2 and antagonize the function of this protein [33], and MDM2 has been implicated in the stemness development of GBM [44]. Therefore, we examined whether MDM2 interacts with NCL in GSC and found that acetylation blocked MDM2-mediated NCL ubiquitination in GSC (Fig. 4), suggesting that acetylation competes with ubiquitination through an unknown mechanism. Other studies have indicated that NCL acetylation at the K88 residue is involved in mRNA maturation and splicing [45], suggesting the importance of acetylation for NCL stability and function. Proteins such as drosha, COMMD1, and epithelial sodium channel have been shown to require acetylation to maintain protein stability and prevent ubiquitination [46,47,48].
Although NCL has been shown to enhance leukemia stemness [21], the effect of NCL on the stemness markers responsible for self-renewal remains unclear. In Fig. 5, NCL significantly decreased the expression of Sox2, Oct4, and Bmi1 at the protein level, whereas CD133 was not affected (Fig. 5b). However, NCL increases CD133 in leukemia progenitor cells [21]. The discrepancy between these differences may be attributed to different cancer types, but this question needs further elucidation. In terms of NCL containing RNA-binding domains [18, 49], we confirmed that NCL decreased Sox2 and Oct4 mRNA stability (Supplementary Fig. S3B). However, although NCL has been implicated as an RNA stabilizer, these data are not consistent with this theory. In addition, NCL, which lacks zinc-finger domains for binding to DNA, was also shown to regulate gene transcription [18, 49]. The results indicating that NCL decreased Sox2 and Oct4 promoter activity (Supplementary Fig. S3A) are distinct from most studies claiming that NCL exerts a positive effect on transcription. However, NCL was reported to inhibit c-myc promoter activity [50], and c-myc expression is involved in glioma progression and recurrence in parallel with Oct4 expression [51]. These findings suggest that NCL may attenuate GSC expansion through the inhibition of c-myc-mediated GSC self-renewal.
According to these findings, NCL plays a negative role in GSC development and sensitizes the response of glioma to TMZ. The reduction of NCL, resulting from HDAC2-mediated deacetylation and MDM2-mediated ubiquitination, is required for GSC expansion. The role of NCL in GSC is different from that in normal glioma. However, it remains unclear whether NCL expression is an independent predictor of chemotherapeutic efficiency in glioma patients, and additional clinical studies are needed to confirm this hypothesis.
References
Kim SH, Ezhilarasan R, Phillips E, Gallego-Perez D, Sparks A, Taylor D, Ladner K, Furuta T et al (2016) Serine/threonine kinase MLK4 determines mesenchymal identity in glioma stem cells in an NF-kappaB-dependent manner. Cancer Cell 29(2):201–213
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD et al (2016) The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131(6):803–820
Ramirez YP, Weatherbee JL, Wheelhouse RT, Ross AH (2013) Glioblastoma multiforme therapy and mechanisms of resistance. Pharmaceuticals (Basel) 6(12):1475–1506
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA et al (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352(10):987–996
Wen PY, Kesari S (2008) Malignant gliomas in adults. N Engl J Med 359(5):492–507
Jackson M, Hassiotou F, Nowak A (2015) Glioblastoma stem-like cells: at the root of tumor recurrence and a therapeutic target. Carcinogenesis 36(2):177–185
Beier D, Schulz JB, Beier CP (2011) Chemoresistance of glioblastoma cancer stem cells—much more complex than expected. Mol Cancer 10:128
Zhou W, Cheng L, Shi Y, Ke SQ, Huang Z, Fang X, Chu CW, Xie Q et al (2015) Arsenic trioxide disrupts glioma stem cells via promoting PML degradation to inhibit tumor growth. Oncotarget 6(35):37300–37315
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD et al (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444(7120):756–760
Cheng L, Wu Q, Huang Z, Guryanova OA, Huang Q, Shou W, Rich JN, Bao S (2011) L1CAM regulates DNA damage checkpoint response of glioblastoma stem cells through NBS1. EMBO J 30(5):800–813
Auffinger B, Tobias AL, Han Y, Lee G, Guo D, Dey M, Lesniak MS, Ahmed AU (2014) Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ 21(7):1119–1131
Bleau AM, Hambardzumyan D, Ozawa T, Fomchenko EI, Huse JT, Brennan CW, Holland EC (2009) PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell 4(3):226–235
Yang YP, Chien Y, Chiou GY, Cherng JY, Wang ML, Lo WL, Chang YL, Huang PI et al (2012) Inhibition of cancer stem cell-like properties and reduced chemoradioresistance of glioblastoma using microRNA145 with cationic polyurethane-short branch PEI. Biomaterials 33(5):1462–1476
Facchino S, Abdouh M, Chatoo W, Bernier G (2010) BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery. J Neurosci 30(30):10096–10111
Lee Y, Kim KH, Kim DG, Cho HJ, Kim Y, Rheey J, Shin K, Seo YJ et al (2015) FoxM1 promotes stemness and radio-resistance of glioblastoma by regulating the master stem cell regulator Sox2. PLoS One 10(10):e0137703
Lopez-Bertoni H, Lal B, Li A, Caplan M, Guerrero-Cazares H, Eberhart CG, Quinones-Hinojosa A, Glas M et al (2015) DNMT-dependent suppression of microRNA regulates the induction of GBM tumor-propagating phenotype by Oct4 and Sox2. Oncogene 34(30):3994–4004
Galzio R, Rosati F, Benedetti E, Cristiano L, Aldi S, Mei S, D’Angelo B, Gentile R et al (2012) Glycosilated nucleolin as marker for human gliomas. J Cell Biochem 113(2):571–579
Abdelmohsen K, Gorospe M (2012) RNA-binding protein nucleolin in disease. RNA Biol 9(6):799–808
Johansson H, Svensson F, Runnberg R, Simonsson T, Simonsson S (2010) Phosphorylated nucleolin interacts with translationally controlled tumor protein during mitosis and with Oct4 during interphase in ES cells. PLoS One 5(10):e13678
Yang A, Shi G, Zhou C, Lu R, Li H, Sun L, Jin Y (2011) Nucleolin maintains embryonic stem cell self-renewal by suppression of p53 protein-dependent pathway. J Biol Chem 286(50):43370–43382
Bhatia S, Reister S, Mahotka C, Meisel R, Borkhardt A, Grinstein E (2015) Control of AC133/CD133 and impact on human hematopoietic progenitor cells through nucleolin. Leukemia 29(11):2208–2220
Costa BM, Smith JS, Chen Y, Chen J, Phillips HS, Aldape KD, Zardo G, Nigro J et al (2010) Reversing HOXA9 oncogene activation by PI3K inhibition: epigenetic mechanism and prognostic significance in human glioblastoma. Cancer Res 70(2):453–462
Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM et al (2006) Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9(3):157–173
Hammond LA, Eckardt JR, Kuhn JG, Gerson SL, Johnson T, Smith L, Drengler RL, Campbell E et al (2004) A randomized phase I and pharmacological trial of sequences of 1,3-bis(2-chloroethyl)-1-nitrosourea and temozolomide in patients with advanced solid neoplasms. Clin Cancer Res 10(5):1645–1656
Tomicic MT, Meise R, Aasland D, Berte N, Kitzinger R, Kramer OH, Kaina B, Christmann M (2015) Apoptosis induced by temozolomide and nimustine in glioblastoma cells is supported by JNK/c-Jun-mediated induction of the BH3-only protein BIM. Oncotarget 6(32):33755–33768
Das A, Banik NL, Patel SJ, Ray SK (2004) Dexamethasone protected human glioblastoma U87MG cells from temozolomide induced apoptosis by maintaining Bax:Bcl-2 ratio and preventing proteolytic activities. Mol Cancer 3(1):36
Roos WP, Batista LF, Naumann SC, Wick W, Weller M, Menck CF, Kaina B (2007) Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene 26(2):186–197
Baer JC, Freeman AA, Newlands ES, Watson AJ, Rafferty JA, Margison GP (1993) Depletion of O6-alkylguanine-DNA alkyltransferase correlates with potentiation of temozolomide and CCNU toxicity in human tumour cells. Br J Cancer 67(6):1299–1302
Kanzawa T, Germano IM, Kondo Y, Ito H, Kyo S, Kondo S (2003) Inhibition of telomerase activity in malignant glioma cells correlates with their sensitivity to temozolomide. Br J Cancer 89(5):922–929
Balasubramaniyan V, Vaillant B, Wang S, Gumin J, Butalid ME, Sai K, Mukheef F, Kim SH et al (2015) Aberrant mesenchymal differentiation of glioma stem-like cells: implications for therapeutic targeting. Oncotarget 6(31):31007–31017
Sharma VP, Anderson NT, Geusz ME (2014) Circadian properties of cancer stem cells in glioma cell cultures and tumorspheres. Cancer Lett 345(1):65–74
Wang SA, Li HY, Hsu TI, Chen SH, Wu CJ, Chang WC, Hung JJ (2011) Heat shock protein 90 stabilizes nucleolin to increase mRNA stability in mitosis. J Biol Chem 286(51):43816–43829
Bhatt P, d’Avout C, Kane NS, Borowiec JA, Saxena A (2012) Specific domains of nucleolin interact with Hdm2 and antagonize Hdm2-mediated p53 ubiquitination. FEBS J 279(3):370–383
Saxena A, Rorie CJ, Dimitrova D, Daniely Y, Borowiec JA (2006) Nucleolin inhibits Hdm2 by multiple pathways leading to p53 stabilization. Oncogene 25(55):7274–7288
Villalva C, Martin-Lanneree S, Cortes U, Dkhissi F, Wager M, Le Corf A, Tourani JM, Dusanter-Fourt I et al (2011) STAT3 is essential for the maintenance of neurosphere-initiating tumor cells in patients with glioblastomas: a potential for targeted therapy? Int J Cancer 128(4):826–838
Yoon CH, Kim MJ, Kim RK, Lim EJ, Choi KS, An S, Hwang SG, Kang SG et al (2012) c-Jun N-terminal kinase has a pivotal role in the maintenance of self-renewal and tumorigenicity in glioma stem-like cells. Oncogene 31(44):4655–4666
Goldshmit Y, Trangle SS, Kloog Y, Pinkas-Kramarski R (2014) Interfering with the interaction between ErbB1, nucleolin and Ras as a potential treatment for glioblastoma. Oncotarget 5(18):8602–8613
Lv S, Zhang J, Han M, Wang W, Zhang Y, Zhuang D, Shi R, Bian R et al (2015) Nucleolin promotes TGF-beta signaling initiation via TGF-beta receptor I in glioblastoma. J Mol Neurosci 55(1):1–6
Wolfson E, Goldenberg M, Solomon S, Frishberg A, Pinkas-Kramarski R (2016) Nucleolin-binding by ErbB2 enhances tumorigenicity of ErbB2-positive breast cancer. Oncotarget 7:65320–65334
Palmieri D, Richmond T, Piovan C, Sheetz T, Zanesi N, Troise F, James C, Wernicke D et al (2015) Human anti-nucleolin recombinant immunoagent for cancer therapy. Proc Natl Acad Sci U S A 112(30):9418–9423
Fonseca NA, Rodrigues AS, Rodrigues-Santos P, Alves V, Gregorio AC, Valerio-Fernandes A, Gomes-da-Silva LC, Rosa MS et al (2015) Nucleolin overexpression in breast cancer cell sub-populations with different stem-like phenotype enables targeted intracellular delivery of synergistic drug combination. Biomaterials 69:76–88
Edwards TK, Saleem A, Shaman JA, Dennis T, Gerigk C, Oliveros E, Gartenberg MR, Rubin EH (2000) Role for nucleolin/Nsr1 in the cellular localization of topoisomerase I. J Biol Chem 275(46):36181–36188
Yang C, Kim MS, Chakravarty D, Indig FE, Carrier F (2009) Nucleolin binds to the proliferating cell nuclear antigen and inhibits nucleotide excision repair. Mol Cell Pharmacol 1(3):130–137
Daniele S, Costa B, Zappelli E, Da Pozzo E, Sestito S, Nesi G, Campiglia P, Marinelli L et al (2015) Combined inhibition of AKT/mTOR and MDM2 enhances glioblastoma multiforme cell apoptosis and differentiation of cancer stem cells. Sci Rep 5:9956
Das S, Cong R, Shandilya J, Senapati P, Moindrot B, Monier K, Delage H, Mongelard F et al (2013) Characterization of nucleolin K88 acetylation defines a new pool of nucleolin colocalizing with pre-mRNA splicing factors. FEBS Lett 587(5):417–424
Butler PL, Staruschenko A, Snyder PM (2015) Acetylation stimulates the epithelial sodium channel by reducing its ubiquitination and degradation. J Biol Chem 290(20):12497–12503
O’Hara A, Simpson J, Morin P, Loveridge CJ, Williams AC, Novo SM, Stark LA (2014) p300-mediated acetylation of COMMD1 regulates its stability, and the ubiquitylation and nucleolar translocation of the RelA NF-kappaB subunit. J Cell Sci 127(Pt 17):3659–3665
Tang X, Wen S, Zheng D, Tucker L, Cao L, Pantazatos D, Moss SF, Ramratnam B (2013) Acetylation of drosha on the N-terminus inhibits its degradation by ubiquitination. PLoS One 8(8):e72503
Tajrishi MM, Tuteja R, Tuteja N (2011) Nucleolin: the most abundant multifunctional phosphoprotein of nucleolus. Commun Integr Biol 4(3):267–275
Gonzalez V, Hurley LH (2010) The C-terminus of nucleolin promotes the formation of the c-MYC G-quadruplex and inhibits c-MYC promoter activity. Biochemistry 49(45):9706–9714
Hattermann K, Fluh C, Engel D, Mehdorn HM, Synowitz M, Mentlein R, Held-Feindt J (2016) Stem cell markers in glioma progression and recurrence. Int J Oncol 49(5):1899–1910
Acknowledgements
We thank the support from the Ministry of Science and Technology of Taiwan (MOST 103-2320-B-038-046-MY3 and 105-2320-B-038-062). Grants MOHW 105-TDU-B-212-134001 and 106-TDU-B-212-144001 from the Ministry of Health and Welfare and the Health and Welfare surcharge of tobacco products were also appreciated.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Electronic supplementary material
ESM 1
(DOCX 722 kb)
Rights and permissions
About this article

Cite this article
Ko, CY., Lin, CH., Chuang, JY. et al. MDM2 Degrades Deacetylated Nucleolin Through Ubiquitination to Promote Glioma Stem-Like Cell Enrichment for Chemotherapeutic Resistance. Mol Neurobiol 55, 3211–3223 (2018). https://doi.org/10.1007/s12035-017-0569-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0569-4