
Abstract
The drastic increase in the consumption of fructose encouraged the research to focus on its effects on brain physio-pathology. Although young and adults differ largely by their metabolic and physiological profiles, most of the previous studies investigated brain disturbances induced by long-term fructose feeding in adults. Therefore, we investigated whether a short-term consumption of fructose (2 weeks) produces early increase in specific markers of inflammation and oxidative stress in the hippocampus of young and adult rats. After the high-fructose diet, plasma lipopolysaccharide and tumour necrosis factor (TNF)-alpha were found significantly increased in parallel with hippocampus inflammation, evidenced by a significant rise in TNF-alpha and glial fibrillar acidic protein concentrations in both the young and adult groups. The fructose-induced inflammatory condition was associated with brain oxidative stress, as increased levels of lipid peroxidation and nitro-tyrosine were detected in the hippocampus. The degree of activation of the protein kinase B, extracellular signal-regulated kinase 1/2, and insulin receptor substrate 1 pathways found in the hippocampus after fructose feeding indicates that the detrimental effects of the fructose-rich diet might largely depend on age. Mitochondrial function in the hippocampus, together with peroxisome proliferator-activated receptor gamma coactivator 1-alpha content, was found significantly decreased in fructose-treated adult rats. In vitro studies with BV-2 microglial cells confirmed that fructose treatment induces TNF-alpha production as well as oxidative stress. In conclusion, these results suggest that unbalanced diet, rich in fructose, may be highly deleterious in young people as in adults and must be strongly discouraged for the prevention of diet-associated neuroinflammation and neurological diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The consumption of fructose has increased drastically over the past two centuries [1], mainly because of consumption of sucrose or high-fructose corn syrup in industrial foods and beverages. Fructose may have a role in the development of obesity by contributing, together with other nutrients, to an overall excessive energy intake [2]. In addition, a high-fructose intake also increases hepatic de novo lipogenesis, very low-density lipoprotein triglyceride secretion, and intrahepatic fat concentrations [2]. The fructose-fed rat represents a commonly used animal model for studying diet-induced metabolic disturbances [3]. Using this experimental animal model, we previously showed that long-term treatment with a fructose-rich diet elicits in adult rats the development of obesity, insulin resistance, and hepatic steatosis [4–6]. We also demonstrated that fructose worsened the deleterious effects of a high-fat diet after just 2 weeks of treatment [7, 8], thus suggesting that even short periods of high-fructose intake may induce metabolic perturbations.
In the last decade, attention was focused on the harmful effects of fructose intake on the brain as well. Indeed, long-term high-fructose intake was shown to induce a reduction of hippocampal neurogenesis [9], accumulation of advanced glycation end products, widespread reactive gliosis, and altered mitochondrial activity in the hippocampus [10]. In addition, high-fructose diet was associated with hypothalamic astrogliosis and neuroinflammation [11] and with high extent of brain oxidative stress [10, 12, 13], which are both implicated in the pathogenesis of neurodegenerative diseases [14].
Widespread overconsumption of added sugars, especially fructose-sweetened beverages, which are the hallmark of modern lifestyles, is particularly prominent in young populations [15]. Although young and adults differ largely by their metabolic and physiological profiles, most of the previous studies investigated fructose-induced metabolic disturbances in adults. In addition, no data are available about the effect of high-fructose diet on the hippocampus of rats of different ages, although aging is associated with increased susceptibility to disease and represents the dominant non-genetic risk factor for neurodegenerative disorders such as Alzheimer’s disease (AD) [16]. Hence, we investigated the effect of fructose feeding on specific markers of inflammation and oxidative stress, as well as on mitochondrial function, in the hippocampus of young and adult rats. This brain region, crucial for learning and memory, is considered one of the most vulnerable sites in AD and other neurodegenerative disease development [17] and is particularly susceptible to disruption by dietary factors. Since most studies on hippocampal dysfunction due to fructose administration addressed the impact of long-term feeding, for more than 4 weeks, the present work focused on the early alterations in this brain region after a short-term consumption of fructose of 2 weeks.
Methods
Materials
Bovine serum albumin fraction V (BSA), goat anti-rabbit horseradish peroxidase-conjugated IgG (GAR-HRP), mouse anti-β actin IgG, goat anti-mouse horseradish peroxidase-conjugated IgG (GAM-HRP), o-phenylenediamine, MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide], hexadecyltrimethylammoniumbromide (HTAB), 3-amino-1,2,4 triazole, and salts and buffers were purchased from Sigma-Aldrich (St. Louis, MO, USA). Tetramethylbenzidine (TMB) was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Rabbit anti-N-Tyr of Covalab was purchased from Vinci-Biochem (Vinci, Italy). Goat anti-rabbit alkaline phosphatase-conjugated secondary antibody (GAR-AP) was purchased from Promega (WI, USA). The dye reagent for protein titration and PVDF membranes were from Bio-Rad (Bio-Rad, Hercules, CA). Polystyrene 96-well ELISA MaxiSorp plates, with high affinity to proteins with mixed hydrophilic/hydrophobic domains, were purchased from Nunc (Roskilde, Denmark). Cell culture medium and components were from Gibco (Life Technologies Italy, Monza, Italy). Cell culture flasks (75 cm2) and 12- and 96-well cell culture plates of Sarstedt (Verona, Italy) were used.
Animals and Treatments
Male Sprague-Dawley rats (Charles River, Italy) of 25 (young) or 90 (adult) days of age were caged singly in a temperature-controlled room (23 ± 1 °C) with a 12-h light/dark cycle (06.30–18.30). Treatment, housing, and euthanasia of animals met the guidelines set by the Italian Health Ministry. All experimental procedures involving animals were approved by “Comitato Etico-Scientifico per la Sperimentazione Animale” of the University of Naples Federico II.
The young and adult rats were divided into two groups and were fed a fructose-rich or control diet for 2 weeks. The two diets were obtained by adding the components reported in Table 1 to 50 g of standard chow (4RF21, Mucedola, Italy). The rats were pair-fed for the whole experimental period, by giving them the same amount of diet, both as weight and as caloric content, and each rat consumed the full portion of the diet. During the treatment, body weight, food, and water intake were monitored daily. At the end of the experimental period, the rats were euthanized by decapitation and blood was collected for subsequent analyses.
The brain was quickly removed and the hippocampus was dissected on ice. Samples were snap-frozen in liquid nitrogen immediately and stored at −80 °C for subsequent analyses.
Cell Culture
The mouse microglial cell line BV-2 was purchased from IRCCS Azienda Ospedaliera Universitaria San Martino-Istituto Nazionale per la Ricerca sul Cancro (Genova, Italy). Cells (1.5 × 106) were seeded in 250-ml tissue culture flasks (75-cm2 surface) and grown in RPMI supplemented with 10% FBS, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (complete medium) at 37 °C under humidified atmosphere of 5% CO2 in air. The medium was changed twice a week, and the cells were sub-cultivated when confluent.
In order to evaluate the effect of fructose on survival of BV-2, cells were cultured into a 96-well plate (20,000 cells/well) for 20 h in complete medium, rinsed with RPMI, and finally incubated (24 h, 37 °C) in serum-free RPMI containing different amounts of fructose (0, 0.005, 0.05, 0.1, 0.5, 1.0, 1.5, 2.5, 5, 7.5, or 10 mM). The medium was then removed, and cell survival was evaluated by MTT reduction assay [18]. The data were expressed as viability percentage, assuming the absorbance value from untreated cells as 100%.
In order to study the influence of fructose on cell inflammatory and oxidative status, as well as on mitochondrial biogenesis, BV-2 cells were cultured for 20 h into a 12-well plate (250,000 cells/well) and, after medium removal and rinsing with RPMI, were incubated (24 h, 37 °C) in RPMI containing different amounts of fructose (0, 2.5, 5, or 10 mM). At the end of incubation, media samples were collected, supplemented with Tissue Protease Inhibitor Cocktail (Sigma-Aldrich, 1:500, v/v), cleared of any cellular debris by centrifugation (400×g, 10 min), and then used for titrating tumour necrosis factor (TNF)-alpha concentration by ELISA sandwich, as described below.
In order to titrate nitro-tyrosine (N-Tyr) or peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), the cells were extensively washed with RPMI, detached by treatment (5 min, 37 °C) with 250 μl of trypsin (TrypLE Express, Gibco), and finally lysed with 125 μl of RIPA buffer (150 mM NaCl, 50 mM Tris-HCl, 0.5% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, pH 8) containing the protease inhibitor cocktail (1:500, v/v). The lysates were centrifuged (12,000×g, 30 min, 4 °C) and analysed for total protein concentration [19] and for N-Tyr by ELISA or PGC-1α content by western blotting as reported below.
In order to assess thiobarbituric acid-reactive substance (TBARS) concentration, BV-2 cells, after trypsin treatment, were centrifuged at 500×g for 5 min and pellets used for TBARS measurement as described below.
In several experiments, after incubation with fructose, cells were resuspended in RPMI medium and used for measurement of oxygen consumption as reported below.
Plasma LPS Levels
Plasma lipopolysaccharide (LPS) determinations were performed using a kit based upon a Limulus amaebocyte lysate (LAL) extract (Lonza, Switzerland). Briefly, samples were mixed with the LAL reagent and chromogenic substrate reagent for 16 min, and absorbance readings were performed on a plate reader at 405 nm.
TNF-Alpha Levels in Plasma, Hippocampus, and BV-2 Cell Supernatants
TNF-alpha concentrations in plasma and hippocampus were determined using a rat-specific enzyme-linked immunosorbent assay (ELISA) (R&D Systems, MN, USA) according to the manufacturer’s instruction. Briefly, the wells of a microtitre plate were coated with 100 μl of mouse anti-rat TNF-alpha (4 μg/ml) in PBS (137 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, 1.5 mM KH2PO4, pH 7.4) and incubated overnight at room temperature. The antibody excess was then removed by washing with wash buffer (containing 0.05% (v/v) Tween 20 in PBS, pH 7.4), and the remaining sites on the plate were blocked with a reagent diluent (PBS containing 1% BSA; 1 h, room temperature). After extensive washing, 100 μl of plasma (1:10 dilution in a reagent diluent) were added to the wells and incubated for 2 h at room temperature. After further washing, the wells were incubated with biotinylated goat anti-rat TNF-alpha (225 ng/ml in a reagent diluent) followed by treatment with streptavidin-HRP (1:200 dilution; 1 h, room temperature). Peroxidase-catalysed colour development from o-phenylenediamine was measured at 492 nm. Data were reported as picograms of TNF-alpha per millilitre of plasma.
For quantification of TNF-alpha in the hippocampus, proteins were extracted by homogenizing frozen tissues as previously reported [18]. Hippocampal homogenates were centrifuged at 18,000×g for 45 min at 4 °C, and the protein concentration of soluble samples was measured according to a published procedure [19]. TNF-alpha concentration was titrated by ELISA as described for plasma samples. Each hippocampus homogenate was diluted (1:5, 1:10, 1:30) in the assay. Data were reported as picograms of TNF-alpha per milligram of proteins.
TNF-alpha secretion by the BV-2 cell line was evaluated in cell culture supernatants using the mouse TNF-alpha ELISA MAX kit (BioLegend, distributed by Aurogene, Rome, Italy), essentially according to the manufacturer’s instructions. Each supernatant was diluted (1: 10, 1:30) in the assay. Data were reported as picograms of TNF-alpha per milligram of cell proteins.
MPO Activity in the Hippocampus
The determination of myeloperoxidase (MPO) activity can be used as a surrogate marker of inflammation, since it has been shown that the activity of MPO solubilized from inflamed tissue is directly proportional to the number of neutrophils seen in histologic sections [20]. MPO activity was therefore assessed in the hippocampus using a modification of the method reported by Kim et al. [21]. Briefly, tissue samples were homogenized in HTAB buffer (0.5% HTAB in 50 mM phosphate buffer, pH 6.0) and centrifuged at 15,000×g for 40 min at 4 °C. Then, supernatants were sonicated for 2 min and thereafter incubated for 1 h at 37 °C with 20 mM 3-amino-1,2,4 triazole (catalase inhibitor). Aliquots of the treated samples (containing about 87 μg of tissue) were combined with 200 μl of TMB-hydrogen peroxide (0.26 g/l). The change in absorbance at 450 nm was measured, and 1 unit of MPO activity was defined as that degrading 1 μmol of peroxide per minute at 25 °C.
Lipid Peroxidation in Rat Hippocampus or BV-2 Lysates
Lipid peroxidation was determined according to Fernandes et al. [22], by measuring TBARS, using the thiobarbituric acid assay. Aliquots of hippocampal homogenates prepared in KCl medium were added to 0.3 ml of ice-cold 40% trichloroacetic acid. Then, 1 ml of 0.67% of aqueous thiobarbituric acid containing 0.01% of 2,6-di-tert-butyl-p-cresol was added. The mixtures were heated at 90 °C for 15 min, then cooled in ice for 10 min, and centrifuged at 850×g for 10 min. The supernatant fractions were collected, and lipid peroxidation was estimated spectrophotometrically at 530 nm. The amount of TBARS formed was calculated using a molar extinction coefficient of 1.56 × 105/M/cm and expressed as nanomoles of TBARS per gram of tissue.
Pellets from BV-2 cells were mixed with 0.5 ml of ice-cold 40% trichloroacetic acid and 0.5 ml of 0.67% of aqueous thiobarbituric acid containing 0.01% of 2,6-di-tert-butyl-p-cresol. The mixture was then processed as above, and the results were expressed as nanomoles of TBARS per milligram of proteins.
Titration of N-Tyr in Rat Hippocampus or BV-2 Lysates
For N-Tyr titration, proteins were extracted from the hippocampus by homogenizing frozen tissues (−80 °C) as previously described [18]. N-Tyr in individual hippocampal homogenates or in BV-2 lysates was quantified by ELISA. Samples were diluted (1:500, 1:1500, 1:3000, and 1:6000) with coating buffer, and aliquots (50 μl) were then incubated in the wells of a microtitre plate (overnight, 4 °C). After four washes with T-TBS (130 mM NaCl, 20 mM Tris-HCl, 0.05% Tween 20, pH 7.4) and four washes with high-salt TBS (500 mM NaCl, 20 mM Tris-HCl at pH 7.4), the wells were blocked with TBS containing 0.5% BSA (1 h, 37 °C). After washing, the wells were incubated (2 h, 37 °C) with 50 μl of rabbit anti-N-Tyr (1:600 dilution in T-TBS containing 0.25% BSA) followed by 60 μl of GAR-HRP IgG (1:3500 dilution; 1 h, 37 °C). Peroxidase-catalysed colour development from o-phenylenediamine was measured at 492 nm. Data were reported as OD per milligram of proteins.
Mitochondrial Analyses in the Hippocampus and BV-2 Cells
Tissue samples were homogenized in a KCl medium containing 100 mM KCl, 50 mM HEPES, 5 mM MgCl2, 1 mM EDTA, 5 mM EGTA, and 1 mM ATP, pH 7.0. Aliquots of homogenates were then incubated for 30 min at 0 °C after the addition of 1.5 mg/ml Lubrol.
Samples of tissue homogenates were transferred into 2-ml calibrated Oxygraph-2k (O2k, Oroboros Instruments, Innsbruck, Austria) chambers. Oxygen polarography was performed at 37 ± 0.001 °C (electronic Peltier regulation) in the O2k chambers, and oxygen concentration (μM) as well as oxygen flux per tissue mass (pmol O2/s/mg) were recorded real-time using DatLab software (Oroboros Instruments, Innsbruck, Austria). The substrate-uncoupler-inhibitor titration protocol was used for measurement of the single step of cytochrome oxidase (COX) activity in a medium containing 75 mM HEPES, 30 μM cytochrome c, 10 mM malonate, 4 μM rotenone, 0.5 mM dinitrophenol, 4 mM ascorbate, and 0.3 mM N,N,N′,N′-tetramethyl-P-phenylenediamine [23]. Chemical background oxygen consumption induced by auto-oxidation of TMPD and ascorbate was assessed after inhibition of CIV by sodium azide (100 mM) and reoxygenation with gaseous oxygen. Respiratory fluxes were corrected automatically for instrumental background by DatLab taking into account oxygen consumption of the oxygen sensor and oxygen diffusion out of or into the oxygraph chamber measured at experimental conditions in incubation medium without a biological sample.
Citrate synthase (CS) activity was measured according to Srere [24] by following the appearance of the free SH group of the released CoASH at 412 nm by using 5,5′-dithiobis-(2-nitrobenzoate).
BV-2 cells (75 × 103) suspended in RPMI medium were incubated in the same medium at 37 °C in the Oxygraph-2k for polarographic measurement of oxygen consumption.
Western Blotting
Proteins were extracted from the hippocampus by homogenizing frozen tissues (−80 °C) in ten volumes (w/v) of cold RIPA buffer containing Tissue Protease Inhibitor Cocktail (Sigma-Aldrich, 1:500, v/v). Homogenates were then centrifuged (14,000×g, 45 min, 4 °C), and the protein concentration of supernatants was measured according to a published procedure [19].
The supernatants were then used to evaluate the content of several proteins, namely extracellular signal-regulated kinases (Erk) 1 and 2, p-Erk 1 and 2, glial fibrillar acidic protein (GFAP), PGC-1α, p-insulin receptor substrate (IRS) 1, protein kinase B (Akt), and p-Akt.
Aliquots (40 μg) of supernatants were fractionated by electrophoresis on 12% (in order to quantify p-Akt/Akt) or 10% (in order to quantify p-Erk/Erk, GFAP, and PGC-1α) or 7.5% (in order to quantify p-IRS) polyacrylamide gel, under denaturing and reducing conditions [25]. After electrophoresis, proteins were blotted onto PVDF membranes. The membranes for detecting p-Erk/Erk, GFAP, and PGC-1α were blocked with T-TBS containing 5% non-fat milk (60 min, 37 °C), while the membrane for detecting p-IRS was blocked with TBS containing 0.1% Tween 20 and 3% BSA. p-Erk 1 and 2 were revealed by incubation (overnight, 4 °C) with anti-p-Erk IgG (Cell Signaling, MA, USA; 1:2000 dilution in T-TBS containing 5% BSA), followed by GAR-HRP IgG (1:4000 dilution in T-TBS containing 0.25% non-fat milk; 1 h, 37 °C). After p-Erk detection, the membrane was extensively washed with T-TBS and submerged in stripping buffer (100 mM β-mercaptoethanol, 2% SDS, 62.5 mM Tris-HCl, pH 6.7; 45 min, 50 °C). Erk1/2 was revealed by incubation (overnight, 4 °C) with rabbit anti-Erk1/2 (Cell Signaling, MA, USA; 1:1000 in T-TBS containing 5% BSA) followed by RAG-HRP IgG (1: 4000 dilution in T-TBS containing 0.25% non-fat milk; 1 h, 37 °C).
PGC-1α was revealed by incubation (overnight, 4 °C) with rabbit anti-human PGC-1α IgG (Millipore, MA, USA; 1:2000 dilution in T-TBS containing 0.25% non-fat milk), followed by GAR-HRP IgG (1:15,000 dilution; 1 h, 37 °C). GFAP was revealed by incubation (overnight, 4 °C) with rabbit anti-human GFAP IgG (Cell Signaling, MA, USA; 1:1000 dilution in T-TBS containing 3% BSA), followed by GAR-HRP IgG (1:8000 dilution T-TBS containing 5% non-fat milk; 1 h, 37 °C). p-IRS 1 was revealed by incubation (overnight, 4 °C) with mouse anti-p-IRS1 (Millipore; 1:1000 dilution in blocking buffer), followed by GAM-HRP IgG (1:20,000 dilution in blocking buffer; 1 h, 37 °C).
Loading control was carried out by incubating the membranes (overnight, 4 °C) with mouse anti-β-actin IgG (1:1000 dilution in T-TBS containing 0.25% non-fat milk) followed by GAM-HRP IgG (1:10,000 dilution). All the above immunocomplexes were detected by the ECL detection system.
The membranes for detecting p-Akt/Akt were preblocked in blocking buffer (PBS, 5% milk powder, 0.5% Tween 20) for 1 h and then incubated overnight at 4 °C with polyclonal antibody for p-Akt (Cell Signaling, MA, USA; diluted 1:1000 in blocking buffer). The membranes were washed three times for 15 min in PBS/0.5% Tween 20 and three times for 15 min in PBS and then incubated for 1 h at room temperature with GAR-AP (diluted 1:5000 in blocking buffer) (Promega, WI, USA). The membranes were washed as described above, rinsed with distilled water, and incubated at room temperature with a chemiluminescent substrate, CDP-Star (Sigma-Aldrich, MO, USA). Akt was detected with polyclonal antibody (Cell Signaling, MA, USA; diluted 1:1000 in blocking buffer), followed by GAR-AP (diluted 1:7000 in blocking buffer).
Quantification of signals was carried out by UN-SCAN-IT gel software (Silk Scientific, UT, USA).
Statistical Analysis
Data were expressed as mean values ± SEM. The program GraphPad Prism 6 (GraphPad Software, San Diego, CA) was used to perform one-way or two-way ANOVA, followed by the Tukey post hoc test. Linear regression analysis was used to analyse the association between variables. P < 0.05 was considered significant.
Results
Animal Data
During the 2-week period of dietary treatment, no significant variation was evident in body weight, epididymal fat weight, body weight gain, and food intake of the rats (Table 2).
Plasma Levels of Inflammation Markers
Plasma concentrations of LPS and TNF-alpha were detected as markers of systemic inflammation, and the results show that after 2 weeks of dietary treatment with a fructose-rich diet, LPS and TNF-alpha were significantly increased in both young and adult rats (Fig. 1a, b). These results show that a short-time feeding of a fructose-rich diet is associated with a systemic inflammatory condition. In addition, an age-dependent increase in inflammatory profile was found in the control and fructose-fed rats (Fig. 1).

Plasma inflammatory markers. Concentrations of lipopolysaccharide (LPS) (a) and tumour necrosis factor (TNF)-alpha (b) were measured in plasma samples from young or adult rats fed a control or fructose-rich diet for 2 weeks. Data reported as means ± SEM of six rats/group. *p < 0.05, **p < 0.01, ****p < 0.0001 compared to control rats, §p < 0.05, §§p < 0.01, §§§§p < 0.0001 compared to young rats (two-way ANOVA followed by the Tukey post-test)
Analysis of Inflammation Markers in Rat Hippocampus
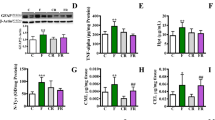
TNF-alpha was titrated in hippocampus homogenates, and significant diet-related changes of its level were observed. In particular, TNF-alpha concentration was significantly higher in the fructose-fed rats than in the control rats, in both the young (about 2.3-fold; p < 0.0001) and adult (about 1.5-fold; p < 0.01) groups (Fig. 2a), thus suggesting that just 2 weeks of high-fructose feeding might induce brain inflammation. GFAP was evaluated as a marker of astrogliosis, and its amount was found significantly higher in the fructose-fed rats in both young (about 2.6-fold; p < 0.001) and adults (about 1.4-fold; p < 0.05) (Fig. 2b). Overall, these results suggest an early reactive astrocytic response of the hippocampus to the fructose diet. Further, age-related changes of TNF-alpha and GFAP concentrations were observed in the control rats (Fig. 2a, b). In particular, we found that the concentration of this cytokine was about 1.7-fold higher (p < 0.05) in adult than in young rats (Fig. 2a). These results show that aging, in rats receiving a standard diet, is associated with the increase in cerebral inflammation markers, in agreement with the age-associated shift toward an inflammatory profile previously described in the brain [26].

Inflammatory markers in rat hippocampus. Tumour necrosis factor (TNF)-alpha was titrated by sandwich ELISA (a), glial fibrillary acidic protein (GFAP) was quantified by western blot (b), while myeloperoxidase (MPO) activity was assessed spectrophotometrically (c) in the hippocampus of young or adult rats fed a control or fructose-rich diet for 2 weeks. Data are reported as means ± SEM of six rats/group. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to control rats, §p < 0.05, §§p < 0.01 compared to young rats (two-way ANOVA followed by the Tukey post-test)
The degree of hippocampus inflammation was also evaluated by measuring the activity of MPO (an enzyme produced in leukocytes and whose activity is linearly related to neutrophil infiltration of the tissues) as an index of inflammatory response under pathological conditions [20] (Fig. 2c). Increasing age or high-fructose intake did not have any effect on neutrophil infiltration in this brain region.
Oxidative Damage in the Hippocampus
High-fructose diet was previously reported to induce oxidative stress in the liver and skeletal muscle of rats [4, 5]. More recently, acute fructose administration and long-term fructose diet feeding were shown to induce oxidative stress in rat brain [13, 27, 28]. Therefore, we investigated whether short-term fructose diet affects brain oxidative status. In order to verify this hypothesis, we analysed lipid peroxidation and N-Tyr levels in the hippocampus of control and fructose-fed rats as a marker of oxidative damage to lipids and proteins, respectively.
Lipid peroxidation was significantly higher in the young and adult rats fed a fructose-rich diet (Fig. 3a). In addition, the N-Tyr level was found more elevated in the hippocampus of the fructose-fed than in the control diet-fed rats in both young (Fig. 3c) (about 1.5-fold; p < 0.05) and adult (about 1.7-fold; p < 0.0001) rats. These results suggest that fructose feeding can induce oxidative damage to lipids and proteins in the brain. Further, we observed age-dependent differences of N-Tyr amount only in the fructose-fed rats. Indeed, the N-Tyr level was about 1.3-fold higher (p < 0.05) in the adult than in the young rats after fructose-rich diet (Fig. 3c), thus suggesting that adult rats are more prone to cerebral protein nitration when fed a fructose-rich diet.

Markers of oxidative damage in rat hippocampus. Lipid peroxidation (a) was measured in the hippocampus of young or adult rats fed a control or fructose-rich diet for 2 weeks by assessing the amount of thiobarbituric acid-reactive substances (TBARS). N-Tyr amount (c) was measured by ELISA in the hippocampus of young or adult rats fed a control or fructose-rich diet for 2 weeks. Values are reported as means ± SEM of six rats/group. *p < 0.05 compared to control rats;****p < 0.0001 compared to control rats; §p < 0.05 compared to young rats (two-way ANOVA followed by the Tukey post-test). Correlation between TNF-alpha and lipid peroxidation (b) or TNF-alpha and N-Tyr content (d) in rat hippocampus was evaluated by linear regression analysis
Interestingly, a positive correlation between the hippocampal level of TNF-alpha and TBARS (r 2 = 0.2330; p = 0.0197; Fig. 3b) or N-Tyr amount (r 2 = 0.4262; p = 0.0005 Fig. 3d) was observed. This result suggests that inflammatory status is associated with a higher extent of lipid peroxidation and protein nitration in the brain, in agreement with the previously described strong association between oxidative stress and inflammation [29].
Fructose Effect on p-Akt, p-Erk, and p-IRS1 Protein Levels in the Hippocampus
The activation of the Akt pathway, namely the ratio p-Akt/Akt, was evaluated by western blot. This ratio was found significantly decreased by high-fructose diet in young rats, while the opposite effect, i.e. a significant increase, was found in adult rats (Fig. 4). p-IRS1 was found significantly lower in both young and adult rats (Fig. 4).

Fructose-induced changes in the activation of kinase Akt and p-IRS1 in the hippocampus. The degree of activation of protein kinase B (Akt) and insulin receptor substrate (IRS) 1 pathways was evaluated by measuring p-Akt/Akt and p-IRS1/actin ratio, respectively, by western blot on protein extracts from the hippocampus of young or adult rats fed a control or fructose-rich diet for 2 weeks. Data (with representative western blots shown below) are reported as means ± SEM of six (p-Akt) or four (p-IRS) rats/group. *p < 0.05, **p < 0.01 compared to control rats (two-way ANOVA followed by the Tukey post-test)
The activation of the Erk 1 and 2 pathways was also evaluated, and a different effect of fructose feeding in young and adult rats was found (Fig. 5). In fact, the p-Erk1/Erk1 ratio was found significantly increased in young and adult rats after fructose-rich diet (Fig. 5c), due to a decrease in Erk1 in young rats (Fig. 5b) or to an increase in the degree of phosphorylation of Erk1 in adult rats (Fig. 5a). As for Erk2, again young and adult rats displayed a different response. In fact, no variation was found in young rats, while a significant increase in the p-Erk2/Erk2 ratio was evident in adult rats (Fig. 5f), due to an increase in the degree of phosphorylation of Erk2 (Fig. 5d). Finally, the degree of phosphorylation of Erk1 and 2 significantly decreased with increasing age, independent of dietary treatment (Fig. 5c, f).

a–f Fructose-induced changes in the Erk1 and Erk2 pathways in the hippocampus. The degree of activation of extracellular signal-regulated kinases (Erk) 1 and 2 was evaluated by measuring the p-Erk/Erk ratio by western blot on protein extracts from the hippocampus of young or adult rats fed a control or fructose-rich diet for 2 weeks. Data (with representative western blots shown below) are reported as means ± SEM of six rats/group. *p < 0.05, **p < 0.01 compared to control rats; §p < 0.05 compared to young rats (two-way ANOVA followed by the Tukey post-test)
Mitochondrial Function in Rat Hippocampus
Mitochondrial function in the hippocampus was evaluated on whole tissue homogenates. CS activity is a commonly used functional marker of the content of intact mitochondria representing a single-enzyme marker of the mitochondrial matrix, while COX activity is a functional marker of the electron transport chain [30]. We found a significant age-induced increase in COX activity in the control groups and a significant decrease induced by the fructose-rich diet only in the adult rats (Fig. 6a). To discriminate whether the above change could be the result of changes in the activity of the enzyme or in the mitochondrial mass, we also assessed the behavior of the enzyme CS, which is representative of the Krebs cycle (Fig. 6b). The very similar magnitude of the decrease found here for the two different enzymes seems to be indicative of a decrease in mitochondrial mass. The analysis of PGC-1α was performed as a marker of mitogenesis, and we found a significantly lower level in the fructose-fed adult rats with respect to the control rats (Fig. 6c) in line with the results obtained from COX and CS activity measurement.

Markers of mitochondrial activity and biogenesis in rat hippocampus. Cytochrome oxidase activity was assessed polarographycally (a), citrate synthase activity was measured spectrophotometrically (b), and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) was quantified by western blot (c) in hippocampus homogenates from young or adult rats fed a control or fructose-rich diet for 2 weeks. Values are reported as means ± SEM of six rats/group. *p < 0.05 compared to control rats, §p < 0.05, §§p < 0.01 compared to young rats (two-way ANOVA followed by the Tukey post-test)
Fructose Effect on TNF-Alpha Production and Mitochondrial Function in BV-2 Microglial Cells
As reported above, our results demonstrate that a 2-week feeding with fructose-rich diet is associated with a significant increase in hippocampal TNF-alpha. The induction of brain inflammation might be the consequence of systemic inflammation or might be generated by fructose entry into the brain by Glut-5 [10, 31], thus inducing a local event of gliosis and inflammation. In order to further investigate this last possibility, we studied the effect of fructose treatment on BV-2 microglial cells.
Preliminarily, the effect of this compound on cell survival was evaluated by MTT assay, in order to choose concentrations not affecting cell viability (data not shown).
BV-2 cells were incubated (24 h, 37 °C) in RPMI containing different amounts of fructose (0, 2.5, 5, or 10 mM). After each treatment, cell culture supernatants were analysed by sandwich ELISA for measuring TNF-alpha concentration. As shown in Fig. 7b, we found that fructose treatment stimulates TNF-alpha secretion by BV-2 cells by about 1.3-fold (p < 0.05) at any concentration used. These results suggest that fructose might directly act as a pro-inflammatory stimulus on microglial cells, in agreement with previous data showing a higher level of TNF-alpha mRNA level in fructose-treated BV-2 cells [32]. No mitochondrial impairment was evident with all the tested concentrations, since oxygen consumption of the cells after the treatment was not affected by fructose (Fig. 7a). In addition, no significant change in the level of PGC-1α was found at any concentration used (Fig. 7c).

Effect of fructose on TNF-alpha secretion and mitochondrial activity and biogenesis in BV-2 cells. BV-2 cells were incubated into a 12-well plate (24 h; 250,000 cells/well) in RPMI containing different amounts of fructose (0, 2.5, 5, or 10 mM). a Mitochondrial was measured spectrophotometrically in RPMI medium at 37 °C, and data are expressed as nanomoles of O2 per minute x milligrams of cell protein. bZ At the end of incubation, media samples were collected, and TNF-alpha was measured, in individual samples, by sandwich ELISA. Data were expressed as picograms per milligram of cell protein. c Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) was quantified by western blot and normalized to actin signal. Values are reported as means from three independent experiments ± SEM.*p < 0.05, **p < 0.01, ****p < 0.0001 compared to control cells cultured in the absence of fructose (one-way ANOVA followed by the Tukey post-test)
Fructose Effect on Oxidative Status of BV-2 Microglial Cells
Our data demonstrate that fructose feeding, just for 2 weeks, is associated with increased lipid and protein oxidative damage in rat hippocampus. We therefore investigated whether this result can be ascribed to an increase in microglial cell oxidative modifications as well. To this aim, BV-2 cells were incubated, as described above, with different amounts of fructose (0, 2.5, 5, or 10 mM), and at the end of treatment, cells were lysed and analysed for lipid peroxidation and N-Tyr content. A pro-oxidative effect of fructose on lipids was found starting from a 5-mM concentration (Fig. 8a). In addition, we found that fructose treatment induces an increase in the cell content of N-Tyr, at any concentration used (p < 0.05; Fig. 8b).

Effect of fructose on markers of oxidative damage in BV-2 lysates. BV-2 cells were incubated into a 12-well plate (24 h; 250,000 cells/well) in RPMI containing different amounts of fructose (0, 2.5, 5, or 10 mM). At the end of incubation, cells were lysed with RIPA buffer. Lipid peroxidation (a) was measured by assessing the amount of thiobarbituric acid-reactive substances (TBARS). N-Tyr level (b) was measured by ELISA. Data are reported as means from three independent experiments ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to control cells cultured in the absence of fructose (one-way ANOVA followed by the Tukey post-test)
Discussion
The deleterious effects of unbalanced nutrition on brain function have been extensively investigated in the last decade, and a high-fructose intake was recently proposed as a risk factor for neurodegenerative diseases [33–35]. Since there is no information about early modifications that occur during high intake of fructose in the hippocampus, the primary goal of this work was to study biochemical changes in this particular region of short-term fructose-fed rats. It is important to mention that the diet used in this work substantially reflects western habits of consuming diets containing high-fructose syrup and therefore is a good model to study metabolic alterations due to high-fructose consumption.
High-fructose intake for 2 weeks did not affect either body weight gain or epididymal fat mass, taken as an index of whole body adiposity. Nevertheless, key markers of systemic inflammation, namely plasma LPS and TNF-alpha, were significantly increased in fructose-fed rats. In addition, an age-dependent increase in systemic inflammation was evident, in both control and fructose-fed rats, in agreement with the notion of “inflammaging” theory [36]. Our present data indicate that inflammation elicited by a fructose-rich diet is an early event that takes place before the onset of obesity. We have previously shown that after 8 weeks of fructose feeding in adult rats, a remodeling of intestinal microbiota takes place, with a concomitant increase in systemic plasma concentrations of LPS and TNF-alpha [37]. Although the dietary treatment in the present study is shorter than that performed in the previous study [37], the similarity of the plasma inflammatory profile suggests that microbiota remodeling is already present in this condition.
Another relevant finding of the present study is that the concentration of TNF-alpha, as well as of GFAP, was higher in the hippocampus of fructose-fed rats, in both young and adult rats, thus indicating that the fructose-rich diet is also associated with neuroinflammation. The fructose effects on the hippocampus could be due to its entry into the brain, since the fructose transporter Glut-5 is expressed in the choroid plexus and hippocampal microglial cells [10, 31]. Fructose feeding was also reported to induce an increased expression of both Glut5 mRNA and protein in the brain of young (35-day-old) and adult rats [38]. Further, it was shown that rat neocortex cells, as well as mice hippocampal cells, are able to take up and metabolize fructose [10, 31].
Oxidative stress and inflammation were suggested to play a central role in fructose-induced damage to the brain [10, 32], as inflammation is associated with the release of reactive oxygen and nitrogen species [39] and with the activation of the inducible nitric oxide synthase (iNOS) gene in astrocytes and microglia [40, 41]. Therefore, we evaluated whether the fructose-induced inflammation would be associated with oxidative brain injury. We titrated N-Tyr to evaluate the extent of oxidative damage to protein, as they are the footprint of peroxynitrite, a potent oxidant which has been implicated in neuronal cell death, together with the amount of TBARS, as markers of oxidative damage to lipids. We report here that short-term fructose feeding is associated with a higher extent of brain oxidative damage, as N-Tyr and TBARS levels were significantly higher in the hippocampus of fructose-fed rats, in both the young and adult groups. Further, we detected an age-dependent increase in cerebral protein nitration in fructose-fed rats, thus suggesting that aging might exacerbate the oxidative condition induced by this diet. This result is of relevance, since protein nitration was shown to play a role in the pathogenesis and progression of AD [42–45]. Our results also support the hypothesis that the fructose-induced inflammatory condition could contribute to the increased hippocampal oxidative damage, as TNF-alpha concentration correlates with both N-Tyr and T-BARS amount.
Intriguingly, we found that high-fructose diet induces a significant decrease in p-Akt in the hippocampus of young rats, while an increase in Akt phosphorylation is found in adult rats. Kinase Akt is at crossroad of several signaling pathways. To better characterize the early effects of fructose feeding on both metabolic and survival signaling, we analysed additional markers, namely p-IRS1 and p-Erk. The decrease in p-IRS found in young and adult rats suggests an impairment in insulin signaling (Fig. 4), in agreement with systemic insulin resistance found at the whole body level in these rats (data not shown). On the other hand, the analysis of Erk1 and Erk2 pathways suggests that the severity of the stress caused by fructose feeding may largely depend on age. Indeed, adult rats seem to be more affected by fructose feeding than young ones, as evidenced by the marked increase in the activation of both Erk1 and Erk2 pathways. The activation of the Erk pathway is suggestive of the onset of hippocampal damage. In fact, Erk1/2 activation seems to play an active role in several models of neuronal death [46]. For example, Erk1/2 activation is implicated in hyperglycemia-mediated cerebral damage [47] as well as in β-amyloid-induced neuronal cell death [48]. Finally, an anti-inflammatory mechanism of resveratrol involving the inactivation of the Erk1/2 pathway has been evidenced in the hippocampus [49]. In line with previous similar results, we also found an age-related decrease in Erk1 and Erk2 activation. Overall, the above results seem to suggest that the reduced p-Akt found in young rats is mainly the result of a decreased insulin signaling, while the increased p-Akt found in adult rats, similarly to Erk activation, mainly reflects a more detrimental effect of fructose feeding. Interestingly, some papers reported increased, rather than decreased, accumulation of active phosphorylated Akt in models of neurodegeneration [50–52].
In the brain, the energy requirements are so high that any defect in mitochondria could be potentially the cause of cognitive impairments [53]. Therefore, we focused on the functioning of mitochondrial compartment in response to fructose-rich diet. The parallel decrease found in COX and CS activity in the hippocampus from adult, but not young, rats is suggestive of a decrease in the mass of these organelles in response to a fructose-rich diet. This hypothesis is supported by the reduction in the expression of PGC-1α. Accordingly, suppression of mitochondrial biogenesis, leading to reduced mitochondrial mass and impaired mitochondrial respiratory capacity, has been found in preparations from postmortem AD cortex compared to controls [54]. The reduced mitochondrial mass implies a decrease in the cellular capacity to produce ATP, which in turn may result in the loss of synapses and synaptic function leading to cognitive decline [55]. In addition, increased mitochondrial COX and CS activity found in adults compared to young rats could be related to the increasing energy needs required for the energy metabolism of a fully developed, neurologically competent mammalian brain. Accordingly, increased respiration rates were found in 14-month-old rats as compared with 1-month-old rats [56], and a significant increase in the expression of mitochondrial proteins has been found in adult mice compared to young ones [57].
It is generally assumed that oxidative stress is a consequence of mitochondrial respiratory chain dysfunction. However, other cellular sites of ROS production are present in the cells, such as NADPH oxidase and NOS [58, 59]. In particular, there is evidence for the presence of oxidative stress and the involvement of NADPH oxidase in neurodegeneration [60, 61]. In addition, an increased oxidative stress could also arise from decreased antioxidant defenses. In agreement, a significant decrease in the activity of the antioxidant enzyme superoxide dismutase has been found in the liver [4], skeletal muscle [5], and brain [10].
The ability of fructose to induce TNF-alpha secretion and affect oxidative status was confirmed in a culture of microglial cells, which are known to be at the forefront of inflammation. In fact, microglia respond to and propagate inflammatory signals initiated at the periphery, thus producing pro-inflammatory cytokines [62] and secondary inflammatory mediators [63, 64]. Therefore, we supposed that the diet-dependent effects on inflammatory and oxidative status in the hippocampus might be likely due to the activation of resident microglia. This hypothesis was supported by the finding of an increase in TNF-alpha concentration, N-Tyr amount, and TBARS level following BV-2 microglia cell incubation with fructose, in the absence of any deleterious effect on mitochondrial function or PGC-1α expression.
In conclusion, this work provides novel evidence of the early biochemical alterations caused by short-term fructose feeding in the hippocampus of rats of different ages, which might determine cognitive impairment and neurodegenerative disease susceptibility after longer exposure to such a nutritional imbalance. Western diet, rich in fructose, may be highly deleterious in young people and must be strongly discouraged for the prevention of diet-associated neuroinflammation and neurological diseases.
References
Campos VC, Tappy L (2016) Physiological handling of dietary fructose-containing sugars: implications for health. Int J Obes Suppl 1:S6–11. doi:10.1038/ijo.2016.8
Stanhope KL (2016) Sugar consumption, metabolic disease and obesity: the state of the controversy. Crit Rev Clin Lab Sci 53:52–67. doi:10.3109/10408363.2015.1084990
Tran LT, Yuen VG, McNeill JH (2009) The fructose-fed rat: a review on the mechanisms of fructose-induced insulin resistance and hypertension. Mol Cell Biochem 332:145–159
Crescenzo R, Bianco F, Falcone I, Coppola P, Liverini G, Iossa S (2013) Increased hepatic de novo lipogenesis and mitochondrial efficiency in a model of obesity induced by diets rich in fructose. Eur J Nutr 52:537–545. doi:10.1007/s00394-012-0356-y
Crescenzo R, Bianco F, Coppola P, Mazzoli A, Cigliano L, Liverini G, Iossa S (2013) Increased skeletal muscle mitochondrial efficiency in rats with fructose-induced alteration in glucose tolerance. Br J Nutr 110:1996–2003. doi:10.1017/S0007114513001566
Crescenzo R, Bianco F, Coppola P, Mazzoli A, Valiante S, Liverini G, Iossa S (2014) Adipose tissue remodeling in rats exhibiting fructose-induced obesity. Eur J Nutr 53:413–419. doi:10.1007/s00394-013-0538-2
Crescenzo R, Bianco F, Coppola P, Mazzoli A, Tussellino M, Carotenuto R, Liverini G, Iossa S (2014) Fructose supplementation worsens the deleterious effects of short-term high-fat feeding on hepatic steatosis and lipid metabolism in adult rats. Exp Physiol 99:1203–1213. doi:10.1113/expphysiol.2014.079632
Crescenzo R, Bianco F, Coppola P, Mazzoli A, Cigliano L, Liverini G, Iossa S (2015) The effect of high-fat--high-fructose diet on skeletal muscle mitochondrial energetics in adult rats. Eur J Nutr 54:183–192. doi:10.1007/s00394-014-0699-7
Van der Borght K, Köhnke R, Göransson N, Deierborg T, Brundin P, Erlanson-Albertsson C, Lindqvist A (2011) Reduced neurogenesis in the rat hippocampus following high fructose consumption. Regul Pept 167:26–30. doi:10.1016/j.regpep.2010.11.002
Mastrocola R, Nigro D, Cento AS, Chiazza F, Collino M, Aragno M (2016) High-fructose intake as risk factor for neurodegeneration: key role for carboxymethyllysine accumulation in mice hippocampal neurons. Neurobiol Dis 89:65–75. doi:10.1016/j.nbd.2016.02.005
Li JM, Ge CX, Xu MX, Wang W, Yu R, Fan CY, Kong LD (2015) Betaine recovers hypothalamic neural injury by inhibiting astrogliosis and inflammation in fructose-fed rats. Mol Nutr Food Res 59:189–202. doi:10.1002/mnfr.201400307
Yin Q, Ma Y, Hong Y, Hou X, Chen J, Shen C, Sun M, Shang Y et al (2014) Lycopene attenuates insulin signaling deficits, oxidative stress, neuroinflammation, and cognitive impairment in fructose-drinking insulin resistant rats. Neuropharmacology 86:389–396. doi:10.1016/j.neuropharm.2014.07.020
Lopes A, Vilela TC, Taschetto L, Vuolo F, Petronilho F, Dal-Pizzol F, Streck EL, Ferreira GC et al (2014) Evaluation of the effects of fructose on oxidative stress and inflammatory parameters in rat brain. Mol Neurobiol 50:1124–1130. doi:10.1007/s12035-014-8676-y
Freeman LR, Haley-Zitlin V, Rosenberger DS, Granholm AC (2014) Damaging effects of a high-fat diet to the brain and cognition: a review of proposed mechanisms. Nutr Neurosci 17:241–251. doi:10.1179/1476830513Y.0000000092
Ford CN, Slining MM, Popkin BM (2013) Trends in dietary intake among US 2- to 6-year-old children, 1989-2008. J Acad Nutr Diet 113:35–42. doi:10.1016/j.jand.2012.08.022
Cummings JL, Cole G (2002) Alzheimer disease. JAMA 287:2335–2338
Raskin J, Cummings J, Hardy J, Schuh K, Dean RA (2015) Neurobiology of Alzheimer’s disease: integrated molecular, physiological, anatomical, biomarker, and cognitive dimensions. Curr Alzheimer Res 12(8):712–722
Spagnuolo MS, Mollica MP, Maresca B, Cavaliere G, Cefaliello C, Trinchese G, Scudiero R, Crispino M et al (2015) High fat diet and inflammation—modulation of haptoglobin level in rat brain. Front Cell Neurosci 9:479. doi:10.3389/fncel.2015.00479
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Krawisz JE, Sharon P, Stenson WF (1984) Quantitative assay for acute intestinal inflammation based on myeloperoxidase activity. Assessment of inflammation in rat and hamster models Gastroenterol 87:1344–1350
Kim JJ, Shajib MS, Manocha MM, Khan WI (2012) Investigating intestinal inflammation in DSS-induced model of IBD. J Vis Exp 60:3678. doi:10.3791/3678
Fernandes MA, Custódio JB, Santos MS, Moreno AJ, Vicente JA (2006) Tetrandrine concentrations not affecting oxidative phosphorylation protect rat liver mitochondria from oxidative stress. Mitochondrion 6:176–185
Barré H, Bailly L, Rouanet JL (1987) Increased oxidative capacity in skeletal muscle from cold-acclimated ducklings: a comparison with rats. Comp Biochem Physiol 88B:5119–5522
Srere PA (1969) Citrate synthase. Meth Enzymol 13:3–11
Spagnuolo MS, Maresca B, Mollica MP, Cavaliere G, Cefaliello C, Trinchese G, Esposito MG, Scudiero R et al (2014) Haptoglobin increases with age in rat hippocampus and modulates apolipoprotein E mediated cholesterol trafficking in neuroblastoma cell lines. Front Cell Neurosci 8:212. doi:10.3389/fncel.2014.00212
Miller DB, O’Callaghan JP (2005) Aging, stress and the hippocampus. Ageing Res Rev 4:123–140
Lin S, Yang Z, Liu H, Tang L, Cai Z (2011) Beyond glucose: metabolic shifts in responses to the effects of the oral glucose tolerance test and the high-fructose diet in rats. Mol BioSyst 7:1537–1548. doi:10.1039/c0mb00246a
Madani Z, Malaisse WJ, Ait-Yahia D (2015) A comparison between the impact of two types of dietary protein on brain glucose concentrations and oxidative stress in high fructose-induced metabolic syndrome rats. Biomed Rep 3:731–735. doi:10.3390/nu3110987
Roberts RA, Smith RA, Safe S, Szabo C, Tjalkens RB, Robertson FM (2010) Toxicological and pathophysiological roles of reactive oxygen and nitrogen species. Toxicology 276:85–94. doi:10.1016/j.tox.2010.07.009
Holloszy JO, Oscai LB, Don IJ, Molé PA (1970) Mitochondrial citric acid cycle and related enzymes: adaptive response to exercise. Biochem Biophys Res Commun 40(6):1368–1373
Hassel B, Elsais A, Froland AS, Tauboll E, Gjerstad L, Quan Y, Dingledine R, Rise F (2015) Uptake and metabolism of fructose by rat neocortical cells in vivo and by isolated nerve terminals in vitro. J Neurochem 133:572–581. doi:10.1111/jnc.13079
Xu MX, Yu R, Shao LF, Zhang YX, Ge CX, Liu XM, Wu WY, Li JM et al (2016) Up-regulated fractalkine (FKN) and its receptor CX3CR1 are involved in fructose-induced neuroinflammation: suppression by curcumin. Brain Behav Immun 58:69–81. doi:10.1016/j.bbi.2016.01.001
Stephan BC, Wells JC, Brayne C, Albanese E, Siervo M (2010) Increased fructose intake as a risk factor for dementia. J Gerontol A Biol Sci Med Sci 65:809–814. doi:10.1093/gerona/glq079
Hipkiss AR (2014) Aging risk factors and Parkinson’s disease: contrasting roles of common dietary constituents. Neurobiol Aging 35:1469–1472. doi:10.1016/j.neurobiolaging.2013.11.032
Hsu TM, Konanur VR, Taing L, Usui R, Kayser BD, Goran MI, Kanoski SE (2015) Effects of sucrose and high fructose corn syrup consumption on spatial memory function and hippocampal neuroinflammation in adolescent rats. Hippocampus 25:227–239. doi:10.1002/hipo.22368
Fougère B, Boulanger E, Nourhashémi F, Guyonnet S, Cesari M (2016) Chronic inflammation: accelerator of biological aging. J Gerontol A Biol Sci Med Sci. doi:10.1093/gerona/glw240
Di Luccia B, Crescenzo R, Mazzoli A, Cigliano L, Venditti P, Walser JC, Widmer A, Baccigalupi L et al (2015) Rescue of fructose-induced metabolic syndrome by antibiotics or faecal transplantation in a rat model of obesity. PLoS One 10:e0134893. doi:10.1371/journal.pone.0134893
Shu HJ, Isenberg K, Cormier RJ, Benz A, Zorumski CF (2006) Expression of fructose sensitive glucose transporter in the brains of fructose-fed rats. Neuroscience 140:889–895
Urrutia PJ, Mena NP, Núñez MT (2014) The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders. Front Pharmacol 5:38 . doi:10.3389/fphar.2014.00038 eCollection 2014
Possel H, Noack H, Putzke J, Wolf G, Sies H (2000) Selective upregulation of inducible nitric oxide synthase (iNOS) by lipopolysaccharide (LPS) and cytokines in microglia: in vitro and in vivo studies. Glia 32:51–59
Hewett JA, Hewett SJ (2012) Induction of nitric oxide synthase-2 expression and measurement of nitric oxide production in enriched primary cortical astrocyte cultures. Methods Mol Biol 814:251–263. doi:10.1007/978-1-61779-452-0_17
Chung KK, David KK (2010) Emerging roles of nitric oxide in neurodegeneration. Nitric Oxide 22:290–295. doi:10.1016/j.niox.2010.02.002
Malinski T (2007) Nitric oxide and nitroxidative stress in Alzheimer’s disease. J Alzheimers Dis 11:207–218
Butterfield DA, Reed TT, Perluigi M, De Marco C, Coccia R, Keller JN, Markesbery WR, Sultana R (2007) Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: implications for the role of nitration in the progression of Alzheimer’s disease. Brain Res 1148:243–248
Butterfield DA, Reed T, Sultana R (2011) Roles of 3-nitrotyrosine- and 4-hydroxynonenal-modified brain proteins in the progression and pathogenesis of Alzheimer’s disease. Free Radic Res 45:59–72. doi:10.3109/10715762.2010.520014
Subramaniam S, Unsicker K (2010) ERK and cell death: ERK1/2 in neuronal death. FEBS J 277:22–29. doi:10.1111/j.1742-4658.2009.07367.x
Zhang JZ, Jing L, Guo FY, Ma Y, Wang YL (2007) Inhibitory effect of ketamine on phosphorylation of the extracellular signal-regulated kinase 1/2 following brain ischemia and reperfusion in rats with hyperglycemia. Exp Toxicol Pathol 59:227–235
Frasca G, Carbonaro V, Merlo S, Copani A, Sortino MA (2008) Integrins mediate beta-amyloid-induced cell-cycle activation and neuronal death. J Neurosci Res 86:350–355
Bellaver B, Souza DG, Bobermin LD, Souza DO, Gonçalves CA, Quincozes-Santos A (2015) Resveratrol protects hippocampal astrocytes against LPS-induced neurotoxicity through HO-1, p38 and ERK pathways. Neurochem Res 40:1600–1608. doi:10.1007/s11064-015-1636-8
Pei JJ, Khatoon S, An WL, Nordlinder M, Tanaka T, Braak H, Tsujio I, Takeda M et al (2003) Role of protein kinase B in Alzheimer’s neurofibrillary pathology. Acta Neuropathol (Berl) 105:381–392
Rickle A, Bogdanovic N, Volkman I, Winblad B, Ravid R, Cowburn RF (2004) Akt activity in Alzheimer’s disease and other neurodegenerative disorders. Neuroreport 15:955–959
Griffin RJ, Moloney A, Kelliher M, Johnston JA, Ravid R, Dockery P, O’Connor R, O’Neill C (2005) Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer’s disease pathology. J Neurochem 93:105–117. doi:10.1111/j.1471-4159.2004.02949.x
Lee J, Boo JH, Ryu H (2009) The failure of mitochondria leads to neurodegeneration: do mitochondria need a jump start? Adv Drug Deliv Rev 61:1316–1323. doi:10.1016/j.addr.2009.07.016
Young-Collier KJ, McArdle M, Bennett JP (2012) The dying of the light: mitochondrial failure in Alzheimer’s disease. J Alzheimers Dis 28:771–781. doi:10.3233/JAD-2011-111487
Butterfield DA, Perluigi M, Sultana R (2006) Oxidative stress in Alzheimer’s disease brain: new insights from redox proteomics. Eur J Pharmacol 545:39–50
Bustamante J, Czerniczyniec A, Cymeryng C, Lores-Arnaiz S (2008) Age related changes from youth to adulthood in rat brain cortex: nitric oxide synthase and mitochondrial respiratory function. Neurochem Res 7:1216–1223
Manczak M, Jung Y, Park BS, Partovi D, Reddy PH (2005) Time-course of mitochondrial gene expressions in mice brains: implications for mitochondrial dysfunction, oxidative damage, and cytochrome c in aging. J Neurochem 92:494–504. doi:10.1111/j.1471-4159.2004.02884.x
Li J, O W, Li W, Jiang ZG, Ghanbari HA (2013) Oxidative stress and neurodegenerative disorders. Int J Mol Sci 14:24438–24475. doi:10.3390/ijms141224438
Gandhi S, Abramov AY (2012) Mechanism of oxidative stress in neurodegeneration. Oxidative Med Cell Longev 2012:428010. doi:10.1155/2012/428010
Shimohama S, Tanino H, Kawakami N, Okamura N, Kodama H, Yamaguchi T, Hayakawa T, Nunomura A et al (2000) Activation of NADPH oxidase in Alzheimer’s disease brains. Biochem Biophys Res Commun 273:5–9
De La Monte SM, Wands JR (2006) Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J Alzheim Dis 9:167–181
Hanisch UK, Kettenmann H (2007) Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 10:1387–1394
Konsman JP, Kelley K, Dantzer R (1999) Temporal and spatial relationships between lipopolysaccharide-induced expression of Fos, interleukin-1beta and inducible nitric oxide synthase in rat brain. Neuroscience 89:535–548
Marty V, El Hachmane M, Amedee T (2008) Dual modulation of synaptic transmission in the nucleus tractus solitarius by prostaglandin E2 synthesized downstream of IL-1beta. Eur J Neurosci 27:3132–3150. doi:10.1111/j.1460-9568.2008.06296.x
Acknowledgements
This work was supported by a grant from the University of Naples Federico II—Ricerca Dip 2015—and by a FIRB-Futuro in Ricerca grant (RBFR12QW4I_004) from the Italian Ministry of Education, University and Research (MIUR). The authors wish to thank Dr. Emilia de Santis for the skillful management of the animal house.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Treatment, housing, and euthanasia of animals met the guidelines set by the Italian Health Ministry. All experimental procedures involving animals were approved by “Comitato Etico-Scientifico per la Sperimentazione Animale” of the University of Naples Federico II.
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Cigliano, L., Spagnuolo, M.S., Crescenzo, R. et al. Short-Term Fructose Feeding Induces Inflammation and Oxidative Stress in the Hippocampus of Young and Adult Rats. Mol Neurobiol 55, 2869–2883 (2018). https://doi.org/10.1007/s12035-017-0518-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0518-2