
Abstract
In a thromboembolic stroke model after reperfusion by recombinant tissue plasminogen activator (rt-PA), we aimed to determine whether therapeutic hypothermia (TH) and ethanol (EtOH) in combination with low concentration (60 %) of normobaric oxygen (NBO) enhanced neuroprotection, as compared to using each of these agents alone. We further aimed to elucidate a potential role of the NADPH oxidase (NOX), phosphorylated protein kinase B (Akt), and protein kinase C-δ (PKC-δ) pathway in oxidative stress and neuroprotection. In Sprague–Dawley rats, a focal middle cerebral artery (MCA) occlusion was induced by an autologous embolus in the following experimental groups: rt-PA treatment alone, rt-PA + NBO treatment, rt-PA + TH at 33 °C, rt-PA + EtOH, rt-PA + NBO + EtOH, rt-PA + NBO + TH, rt-PA + NOX inhibitor, rt-PA + EtOH + NOX inhibitor, or rt-PA + EtOH + Akt inhibitor. Control groups included sham-operated without stroke or stroke without treatment. Infarct volume and neurological deficit were assessed at 24 h after rt-PA-induced reperfusion with or without treatments. ROS levels, NOX activity, and the protein expression of NOX subunits p22phox, p47phox, p67phox, gp91phox, as well as PKC-δ and phosphorylated Akt were measured at 3 and 24 h after rt-PA-induced reperfusion. Following rt-PA in thromboembolic stroke rats, NBO combined with TH or EtOH more effectively decreased infarct volume and neurological deficit, as well as reactive oxygen species (ROS) production than with any of the used monotherapies. NOX activity and subunit expressions were downregulated and temporally associated with reduced PKC-δ and increased p-Akt expression. The present study demonstrated that combining NBO with either TH or EtOH conferred similar neuroprotection via modulation of NOX activation. The results suggest a role of Akt in NOX activation and implicate an upstream PKC-δ pathway in the Akt regulation of NOX. It is possible to substitute EtOH for TH, thus circumventing the difficulties in clinical application of TH through the comparatively easier usage of EtOH as a potential stroke management.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Stroke is a major cause of death and the leading cause of permanent disability worldwide [1–3]. In spite of debilitating effects due to stroke, research leading to therapies to improve the neurological outcome of patients has not made great clinical strides since the landmark FDA approval of recombinant tissue plasminogen activator (rt-PA) for ischemic stroke in 1996. However, after decades of rt-PA treatment, its clinical outcomes remain poor because of rt-PA’s short treatment widow, susceptibility to complications [4], and low frequency of use [5]. Even in patients where the clot is resolved and recanalization is achieved, extensive damage may have already occurred or be further exacerbated by subsequent reperfusion-related injury.
Normobaric oxygenation (NBO) is a simple and logical procedure in treating ischemic stress. However, higher levels of oxygenation have been associated with increased production of toxic reactive oxygen species (ROS), which can lead to further tissue injury [6]. In addition, hyperoxia following NBO treatment can cause vasoconstriction of the carotid and downstream cerebral arteries [7]. Currently, there is no convincing data to confirm that NBO monotherapy brings about significant benefits to patients after acute ischemic stroke [8]. In fact, the recent clinical trial for the use of 95 % NBO in acute ischemic stroke (NCT00414726) was terminated because of unclear therapeutic efficacy [9, 10]. This is likely due to the complexity of stroke and detrimental potential of oxygen-associated excessive generation of ROS. Furthermore, in a recent Indian trial that used 61 % (10 L/min for 12 h) NBO for anterior circulation ischemic strokes patients within 12 h after stroke, NBO did not improve their clinical outcome [11]. Therefore, failure of NBO as a neuroprotective agent in clinical trials has counter-indicated its use as an effective monotherapy in acute stroke [11, 12].
Perhaps the best studied neuroprotective treatment in ischemia models is the use of therapeutic hypothermia (TH), an approach that has been implemented in cardiac arrest protocols [13, 14]. Extensive data from ischemic animal models shows that TH with a body temperature target of 32–34 °C leads to decreased infarct volumes and improved outcomes in ischemic animal models [15–18]. Despite these findings, TH’s application in focal ischemic stroke has been limited by delayed cooling onset, prolonged duration, extensive medical and nursing efforts, and significant complications [19].
In recent studies, we have demonstrated a neuroprotective effect by ethanol (EtOH) after stroke due in part to EtOH’s stabilizing effects on cerebral metabolism in damaged brain tissue [20–22]. In a rat model of ischemic stroke, we further showed that a combination therapy of 95 % NBO with EtOH provided greater neuroprotection than either of these agents used alone [23, 24]. Because of the high translational degree of both NBO and EtOH (they are cost-effective, easy to manage, simple to administer, and readily available), we aimed at investigating their potential clinical application in a more clinically relevant animal model than those previously used. Here, we use an embolic model of stroke that closely mimics human thromboembolic stroke, where recanalization of an obstructed vessel can be re-established by administration of rt-PA. As such, this model is in concordance with the 1996 protocol for the treatment of ischemic stroke [25].
The current study builds on previous findings regarding the effects of NBO, TH, and EtOH as neuroprotective adjuncts to rt-PA. Because ROS production is a major concern with any oxygen treatment and in order to increase NBO’s feasibility in clinical use, here, we lowered the fraction of inspired oxygen (FiO2) to 60 % instead of the currently prescribed NBO protocol of 95 %. Due to the overproduction of deleterious ROS during ischemia, NADPH oxidase (NOX) is activated within brain cells and its superoxide-generating function leads to cell injury and death [26]. The primary substrate of NOX, NADPH, is created during reperfusion when the increased availability of glucose and oxygen leads to an increase in anaerobic metabolism by the hexose monophosphate shunt [27]. It is therefore critical to curb its activation in order to effectively provide neuroprotection [27].
Oxidative stress by increased oxygen-based free radical production in ischemia/reperfusion injury has been well documented [28]. Recent studies implicate Akt in oxidative stress and have suggested a role for the PI3K/Akt pathway in oxidative protection [29]. In addition, protein kinase C delta (PKC-δ) has been also proposed to mediate reperfusion injury via generation of ROS, apoptosis, and inflammation that contribute to the expansion of the ischemic infarct [30]. A major finding of the present study was that excessive ROS production in stroke could be reduced by PKC-Akt-NOX modulation through the use of NBO plus TH or EtOH.
Material and Methods
The experimental design and procedures used here were in accordance with the National Institute of Health and approved by the Animal Experimental Committee of Xuanwu Hospital at Capital Medical University. The procedures and data analysis were carried out in a blinded and randomized manner. A total of 174 Sprague–Dawley rats weighing 300 to 350 g (Beijing Vital River Laboratory Animal Co.) were divided before surgery into a sham group (animals underwent the entire surgical procedure except for embolization; n = 12) and ten stroke groups with different treatments. The stroke groups included the following: (1) control at normothermia with an injection of saline in place of rt-PA and other drugs (n = 18), (2) rt-PA alone (n = 18), (3) rt-PA + 60 % NBO (n = 18), (4) rt-PA + TH at 33 °C (n = 18), (5) rt-PA + EtOH at a dose of 1.0 g/kg (n = 18), (6) rt-PA+ NBO + TH (n = 18), (7) rt-PA + NBO + EtOH (n = 18), (8) rt-PA + apocynin (NOX inhibitor; n = 12), (9) rt-PA + EtOH + apocynin (n = 12), (10) rt-PA + EtOH + LY294002 (Akt inhibitor; n = 12). Animals in the eighth (subjected to apocynin), ninth (subjected to EtOH + apocynin), and tenth (subjected to LY294002) groups were further divided into two subgroups for analyses of neurologic deficits and infarct volume or biochemical assays at 24 h. The remaining groups were divided into three subgroups for data analyses of neurologic deficits and infarct volume at 24 h, or biochemical assays and protein expressions at 3 and 24 h after rt-PA treatment initiation.
Focal Cerebral Ischemia Induced by Autologous Embolus
Sprague–Dawley rats were intubated, and anesthesia was maintained with 1.5 % enflurane in 70 % nitrous oxide and 30 % oxygen through mechanical ventilation. Body temperature was monitored and kept at 37 ± 0.5 °C using a feedback-controlled heating blanket during the occlusion procedure. The right femoral artery was cannulated with a PE-50 catheter to monitor blood pressure and blood gases while the right femoral vein was cannulated for drug administration. The regional cerebral blood flow (rCBF) was monitored by laser-Doppler flowmetry (LDF) with the probe positioned 3 mm posterior and 5 mm lateral to bregma. The method to prepare an embolus was the same as described by Zhang et al. [25]. Briefly, femoral arterial blood was withdrawn into a section of PE-50 tubing and maintained in the tube for 2 h in a 37 °C water bath followed by 22 h at 4 °C. A section of the clot was cut and transferred to a PE-10 tube to wash with saline 10–15 times to flush out erythrocytes. The rats were injected with a 4 cm long, fibrin-rich clot at the origin of the MCA using a modified PE-50 catheter [25]. One hour after embolization, the rCBF was stable at ≤40 % of preischemic baseline.
rt-PA Treatment
rt-PA (Actilyse, Boehringer Ingelheim, Germany) at a total dose of 5 mg/kg body weight was administered through the right femoral vein [31]. Half of the dose was given as a bolus 1 h after injecting the thromboembolism (TE), and the rest was continuously infused over the next 30 min. Both sham and stroke groups received a saline injection instead of rt-PA.
NBO Treatment
Normobaric oxygen (60 %) was delivered by a ventilator for 0.5 h, commencing at 1 h after TE, after which the animal was placed in a sealed chamber and exposed to 60 % O2 for an additional 2.5 h. An oxygen probe assured a constant 60 % O2 in the chamber.
Hypothermia Treatment
TH was started at 1 h after TE and was sustained for 3 h. Rat whole body temperature of 33 °C was maintained by placing the ice under the body as described previously [32].
EtOH Treatment
EtOH at a dose of 1.0 g/kg was diluted in saline to 1.0 mL and administered at the end of rt-PA treatment which was delivered as a single bolus via the right femoral vein. In a previous study, the same dose of EtOH was found to have a mild neuroprotective effect [20].
Combination Therapy
In the NBO + EtOH group, NBO (60 %) was started simultaneously with rt-PA infusion at 1 h after TE (as described above) using a mechanical ventilator for the first 0.5 hour. The rats were then placed in a sealed chamber exposed to 60 % O2 for another 2.5 h. EtOH (1.0 g/kg) was administered at the end of rt-PA infusion as a bolus via the right femoral vein. In the NBO + TH group, both treatments were started simultaneously at 1 h after TE, with the oxygen delivered through ventilator. Sixty percent NBO and TH were maintained for a total of 3 h.
Akt Inhibition
In order to determine a potential role of Akt in EtOH-induced neuroprotection, the PI3K inhibitor LY294002 ([2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one], Sigma, St. Louis, MO, USA), previously shown to inhibit Akt phosphorylation in many studies [33–35], was used in EtOH treatment group. A microinjection was performed into the lateral ventricle ipsilateral to the ischemia over 10 min (from bregma: posterior, −1.0 mm; lateral, −1.5 mm; ventral, −4.0 mm), beginning at 1.5 h after TE. The dose of LY294002 at 30 μM (10 μL) was established previously [33].
NOX Inhibition
In order to determine a potential role of NOX in EtOH-induced neuroprotection, apocynin (4-hydroxy-3-methoxy-acetophenone; Sigma, St. Louis, MO, USA) was started at 0.5 h after TE as a bolus via the right femoral vein. It was dissolved in 10 % DMSO and 90 % saline and administered at 2.5 mg/kg body weight, as established previously [36].
Neurological Deficit
Neurological deficits were evaluated using a 12-point neurological scale [37] at 24 h after rt-PA administration. The higher scores were positively correlated with the more profound deficits observed.
Cerebral Infarct Volume
Infarct volume was evaluated at 24 h after rt-PA administration. As described before by us [23, 38], six coronal brain slices with a 2-mm thickness were cut for treatment with 2,3,5-triphenyltetrazolium chloride (TTC, Sigma, St. Louis, MO, USA) at 37 °C for 20 min and then were fixed in 10 % formalin solution. In order to minimize errors due to edema, an indirect percentage was used to calculate final infarct volume [20].
ROS Production
ROS generation was assessed by determining H2O2 levels by using the fluorescently labeled hydrogen peroxidase method, as previously shown [23]. An Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) was used. Based on protein concentration, homogenized brain samples were diluted to 10 mg/mL and 100 μg/ml of digitonin was added. After incubation for 30 min, the Amplex Red and Horse Radish Peroxidase were added and H2O2 levels in processed brain homogenates were detected at 37 °C for 30 min on a DTX-880 Multimode Detector.
NOX Activity
NADPH oxidase activity was determined by superoxide (O2·−) production from NADPH using the lucigenin-enhanced luminescence assay, as used previously [23]. Homogenized brain samples containing phenylmethylsulfonyl fluoride and a protease inhibitor cocktail (Thermo; 20 μL) and lucigenin were added to a 96-well luminescence plate and incubated in the dark for 10 min at 37 °C. NADPH (100 μmol/L) was added to start the reaction, and NOX activity was calculated by recording the change in luminescence over time using a DTX-880 Multimode Detector.
Protein Expression
Western immunoblot analysis was used to detect protein expression of all four NOX subunits (p22phox, p47phox, p67phox, and gp91phox) as well PKC-δ and p-Akt as described previously [23]. Tissue samples from the right ischemic cerebral hemispheres of all experimental and control groups were harvested at 3 or 24 h after rt-PA infusion. Protein extracted from tissue samples were separated by SDS-PAGE and transferred to PVDF membrane. Membranes were incubated with primary antibodies including polyclonal goat anti-p22phox at 1:1000, polyclonal goat anti-p47phox at 1:1000, polyclonal goat anti-p67phox 1:1000, polyclonal goat anti-gp91phox at 1:1000, polyclonal rabbit anti-PKC-δ at 1:5000 (Santa Cruz Biotechnology, Inc.), and polyclonal rabbit anti-phospho-Akt 1:1000 (Cell Signaling Technology, Inc.). Equal protein loading was confirmed and adjusted using β-actin (polyclonal goat anti-β-actin antibody, 1:1000; Santa Cruz Biotechnology, Inc.). Targeted antigens were visualized using standard chemical luminescence methods (Pierce ECL substrate, Thermo Fisher Scientific, Waltham, MA, USA). Quantification of relative target protein expression was performed using the program ImageJ 1.48 (NIH, Bethesda, MD, USA).
Statistical Analysis
All the data were described as mean ± SE. Statistical analysis was performed with SPSS for Windows, version 21.0 (IBM Corporation, Somers, NY, USA). The differences among groups were assessed using one-way analysis of variance or Student’s t test with a significance level of p < 0.05. Post hoc comparison between groups was further detected using the least significant difference (LSD) method.
Results
Physiological Parameters
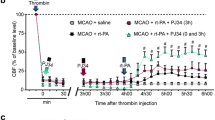
There were no significant differences in blood pH, PaCO2, and MAP among all groups. NBO treatment elevated PaO2 levels to approximately 280 mmHg (p < 0.01) at the conclusion of treatment as compared to groups without NBO treatment (120 mmHg).
Neurological Deficits
At 24 h, a significantly high score (7.7 ± 0.7 out of a possible 12 points) was observed in stroke with sham treatment. While these deficits were significantly reduced by rt-PA administration (5.7 ± 0.3, F [10] = 2.7; p < 0.05; Fig. 1a), TH (3.5 ± 0.6) and EtOH (3.7 ± 0.7) further reduced the neurological deficit (F [5, 30] = 2.7; p < 0.05; Fig. 1b). Furthermore, NBO combined with TH (3.0 ± 0.5) or EtOH (3.2 ± 0.5) exhibited a greater reduction (p < 0.01) in neurological deficits. NOX inhibition with apocynin produced neuroprotection similar to that of EtOH, while their combination significantly enhanced the reduced deficits (F [4, 25] = 3.2; p < 0.01; Fig. 1c). However, when p-Akt was inhibited, the reduced deficit score by EtOH was largely reversed.

Neurological deficit score 24 h after rt-PA administration. a rt-PA treatment significantly reduced the amount of impairment as compared to stroke without treatment (# p < 0.05). b While TH and EtOH monotherapies lead to a reduction in deficit scores compared to rt-PA alone (*p < 0.05), the combination of NBO with either EtOH or TH offered the best neurobehavioral outcome (**p < 0.01). c NOX inhibition with apocynin had a similar efficacy (*p < 0.05) as EtOH monotherapy. However, when combined, apocynin plus EtOH led to the greatest reduction in neurological deficit (**p < 0.01). In contrast, inhibiting Akt phosphorylation diminished neurobehavioral outcome in the EtOH-treated group
Infarct Volume
Ischemic injury produced a substantial infarct volume at 24 h (55.1 ± 3.7 %), which was reduced by rt-PA treatment (37.4 ± 2.8 %, F [10] = 2.8; p < 0.05; Fig. 2a). Compared with rt-PA alone, EtOH (23.9 ± 3.1 %; p < 0.05) and TH (20.8 ± 2.8 %; p < 0.01) monotherapies both further reduced the infarct volume. In contrast, additional NBO (37.5 ± 5.7 %) did not further reduce the volume of the infarct (Fig. 2b) as compared to rt-PA monotherapy (37.4 ± 2.8 %). However, when NBO was used with either TH (9.3 ± 1.3 %) or EtOH (10.4 ± 4.0 %), these combinations resulted in much greater reductions of infarct volume (F [5, 30] = 12.2; p < 0.01; Fig. 2b). While the NOX inhibitor alone was also found to reduce infarct volume (p < 0.05), the combination of EtOH and the NOX inhibitor significantly enhanced neuroprotection (F [4, 25] = 5.6; p < 0.01; Fig. 2c). However, the reduction in infarct volume by EtOH could be reversed (p < 0.05) when p-Akt was inhibited.

Infarct volumes in representative brain sections are shown at 24 h after rt-PA administration. a Compared to no treatment, rt-PA administration effectively (# p < 0.05) reduced infarct volume. b Compared to rt-PA treatment, monotherapies with EtOH and TH yielded significantly smaller infarctions (*p < 0.05 and **p < 0.01), and the co-administration of NBO with either EtOH or TH resulted in the smallest amount of tissue damage (**p < 0.01). c In rt-PA treated rats, NOX inhibition and EtOH alone significantly reduced infarct volumes (*p < 0.05) but not as much as when used in combination (**p < 0.01). However, infarct volume increased when p-Akt was inhibited as compared to EtOH monotherapy ($ p < 0.05)
ROS Levels
Oxidative stress after stroke was significantly increased at the 3-h time point (F [2, 15] = 25.3; p < 0.01; Fig. 3a) and had an even greater elevation at 24 h (F [2, 15] = 43.7; p < 0.01; Fig. 3b). When rt-PA was used, it was only able to reduce (p < 0.01) ROS levels at 24 but not 3 h. NBO only reduced (p < 0.05) the ROS level at 24 h of reperfusion, but EtOH (p < 0.05) and TH (p < 0.01) significantly reduced the ROS at both 3 and 24 h (Fig. 3c, d). Additionally, greater decreases were observed at both time points when NBO was combined with TH or EtOH (F [5, 30] = 4.8; p < 0.01; Fig. 3c) (F [5, 30] = 4.9; p < 0.01; Fig. 3d). While the NOX inhibitor was neuroprotective, superior reduction in ROS levels occurred when NOX inhibitor was used in conjunction with EtOH (F [4, 25] = 5.8; p < 0.01; Fig. 3e). However, the reduced ROS levels by EtOH were significantly (p < 0.05) reversed when PI3K/Akt pathway was inhibited by LY294002.

Untreated stroke resulted in significantly (## p < 0.01) elevated levels of ROS at 3 h (a) and 24 h (b). Thrombolysis with rt-PA reduced oxidative stress only at 24 h (**p < 0.01). Therapeutic adjuvant at 3 h (c) and 24 h (d) showed that EtOH (*p < 0.05) and TH (**p < 0.01) were efficacious at both time points whereas NBO was only efficacious (*p < 0.05) at 24 h. Optimal reduction in ROS production was achieved when either EtOH or TH were combined with NBO. e EtOH treatment paired with NOX inhibition had less oxidative stress (**p < 0.01) than with either treatment alone. However, the reduced ROS levels by EtOH treatment were significantly reversed ($ p < 0.05) when p-Akt was inhibited
NOX Activity and Subunit Protein Expression
Compared to control, there was a significant increase in NOX activity after stroke at 3 and 24 h (F [2, 15] = 9.6; p < 0.01; Fig. 4a) (F [2, 15] = 19.6; p < 0.01; Fig. 4b). Administration of rt-PA only decreased NOX activity slightly but not significantly. Either EtOH or TH but not NBO monotherapies effectively decreased NOX activity at both 3 and 24 h after reperfusion with rt-PA (p < 0.05; Fig. 4c, d). Moreover, when NBO was combined with TH or EtOH, further reduction in NOX activity was resulted at both 3-h (F [5, 30] = 3.6; p < 0.01; Fig. 4c) and 24-h (F [5, 30] = 3.8; p < 0.01; Fig. 4d) time points. At 24 h after thrombolysis, when NOX was inhibited with apocynin, a reduction of NOX activity was confirmed (p < 0.05) and this effect was enhanced by EtOH (p < 0.01) (Fig. 4e). When the PI3K/Akt pathway was inhibited by LY294002 at 24 h of thrombolysis in the EtOH treatment group, however, this reduction in NOX activity was reversed (F [4, 25] = 4.2; p < 0.05; Fig. 4e).

NOX activity was greatly increased (## p < 0.01) after stroke at 3 h (a) and 24 h (b). Thrombolysis with rt-PA did not result in significantly reduced NOX activity. After 3 h (c) and 24 h (d) of thrombolysis, EtOH or TH were both effective (*p < 0.05) in reducing NOX activity. While NBO by itself was not beneficial, if combined with either EtOH or TH, it enhanced NOX attenuation (**p < 0.01). e While apocynin alone inhibited NOX (*p < 0.05), its effect was even greater when paired with EtOH (**p < 0.01). Compared with EtOH monotherapy, however, blunting the PI3K/Akt pathway led to a reverse in NOX activity ($ p < 0.05)
Similar results were obtained when the protein levels of NOX subunits were assessed. Specifically, Figs. 5, 6, 7, and 8a, b show a significant increase in all the protein subunits after stroke, including p22phox at 3 h (F [2, 15] = 11.3; p < 0.01; Fig. 5a) and 24 h (F [2, 15] = 9.2; p < 0.01; Fig. 5b), p47phox at 3 h (F [2, 15] = 12.9; p < 0.01; Fig. 6a) and 24 h (F [2, 15] = 9.3; p < 0.01; Fig. 6b), p67phox at 3 h (F [2, 15] = 36.0; p < 0.01; Fig. 7a) and 24 h (F [2, 15] = 22.6; p < 0.01; Fig. 7b), and gp91phox at 3 h (F [2, 15] = 10.3; p < 0.01; Fig. 8a) and 24 h (F [2, 15] = 16.1; p < 0.01; Fig. 8b). At 3 h after thrombolysis (Figs. 5, 6, 7, and 8a, c), reperfusion or reperfusion plus NBO had no significant impact on the levels of the all four NOX subunits. At 24 h, rt-PA alone did not show effect on p47phox, p67phox, and gp91phox as well, NBO administration provided neuroprotection with decreased levels of p22phox and p47phox (p < 0.01), but had no effect on p67phox and gp91phox (Figs. 5, 6, 7, and 8d). TH treatments decreased the levels of p47phox, p67phox, and gp91phox both at 3 and 24 h after thrombolysis, respectively (F [5, 30] = 10.4; p < 0.01; Fig. 6c) (F [5, 30] = 8.8; p < 0.01; Fig. 6d) (F [5, 30] = 2.8; p < 0.05; Fig. 7c) (F [5, 30] = 4.1; p < 0.05; Fig. 7d) (F [5, 30] = 5.3; p < 0.05; Fig. 8c) (F [5, 30] = 3.4; p < 0.05; Fig. 8d). Similarly, EtOH treatments significantly (p < 0.05) decreased the p22phox, p47phox, p67phox, and gp91phox at the two time points (Figs. 5, 6, 7, and 8c, d). Compared to EtOH monotherapy, NBO in combination with EtOH exhibited an enhanced reduction (p < 0.01) in the four NOX subunits at both 3 and 24 h (Figs. 5, 6, 7, and 8c, d). The combination of NBO with TH also exhibited an enhanced reduction (p < 0.01) in p22phox and gp91phox compared to TH monotherapy at the two time points (Figs. 5, 6, 7, and 8c, d). The NOX inhibition with apocynin at 24 h after thrombolysis resulted in a reduction of NOX subunit protein expression, with a largest reduction being in the expression of gp91phox (p < 0.05; Fig. 8e). EtOH administration in conjunction with apocynin provided a greater (p < 0.01) inhibitory effect on NOX subunit expression of p22phox, p47phox, and gp91phox (Figs. 5, 6, and 8e). In addition, when p-Akt was inhibited at 24 h after thrombolysis, the expressions of the four NOX subunits were markedly elevated compared with those in EtOH treatment group, respectively (F [4, 25] = 6.1; p < 0.05; Fig. 5e) (F [4, 25] = 5.4; p < 0.05; Fig. 6e) (F [4, 25] = 3.8; p < 0.05; Fig. 7e) (F [4, 25] = 4.1; p < 0.05; Fig. 8e).

p22phox subunit expression was significantly (## p < 0.01) elevated after stroke at 3 h (a) and 24 h (b), and these increases were reduced at 24 but not 3 h of reperfusion with rt-PA. c At 3 h of reperfusion, monotherapy with EtOH had a small but significant (*p < 0.05) effect on subunit expression. p22phox levels were significantly reduced when either EtOH or TH were combined with NBO (**p < 0.01). d At 24 h, there were reductions in expression with all treatments and combinations (*p < 0.05, **p < 0.01). e Apocynin by itself was not as effective as when used in combination with EtOH (**p < 0.01). However, inhibition of p-Akt markedly increased p22phox expression when compared with EtOH monotherapy ($ p < 0.05)

p47phox expression was significantly (## p < 0.01) elevated after stroke at 3 h (a) and 24 h (b). c Three hours after reperfusion, TH was the most effective monotherapy at reducing p47phox levels (**p < 0.01). NBO in combination with EtOH was much more effective (**p < 0.01) than either agent alone. d At 24 h, all interventions were effective at reducing p47phox expression. Furthermore, the combination of NBO and TH yielded the greatest p47phox reduction (**p < 0.01). e While apocynin did not have much of an effect by itself, in the presence of EtOH, it significantly reduced p47phox expression (**p < 0.01). However, inhibition of p-Akt could reversed the reduced p47phox expression by EtOH monotherapy ($ p < 0.05)

p67phox expression was drastically (## p < 0.01) elevated after stroke at 3 h (a) and 24 h (b). c At 3 h of rt-PA administration, EtOH or TH but not NBO effectively decreased p67phox (*p < 0.05). However, NBO combined with EtOH resulted in significant reduction of p67phox (**p < 0.01). d At 24 h, EtOH and TH monotherapies effectively decreased p67phox (*p < 0.05) and when NBO was combined with EtOH, p67phox reduction was further enhanced (**p < 0.01). e Apocynin did not have as much of an effect on NOX subunit, as it did when combination with EtOH. Again, the decreased expression of p67phox by EtOH was reversed ($ p < 0.05) when p-Akt was inhibited

gp91phox levels were significantly (## p < 0.01) elevated after stroke at 3 h (a) and 24 h (b). c At 3 h after rt-PA, both EtOH and TH monotherapies effectively decreased gp91phox (*p < 0.05). When NBO was combined with EtOH or TH, further reduction in gp91phox was resulted (**p < 0.01). d Both EtOH and TH monotherapies also effectively decreased gp91phox at 24 h (*p < 0.05), and when NBO was combined with either of them, the reduction in gp91phox was enhanced (**p < 0.01). e Apocynin was effective at reducing gp91phox by itself (*p < 0.05), and this effect was increased by EtOH (**p < 0.01). Inhibition of p-Akt markedly increased gp91phox expression which was reduced by EtOH ($ p < 0.05)
p-Akt Protein
Compared to sham control, there was a decrease (p < 0.01) in p-Akt levels after stroke at both 3 and 24 h. After rt-PA treatment, p-Akt levels were increased at both 3 h (F [2, 15] = 8.2; p < 0.01; Fig. 9a) and 24 h (F [2, 15] = 17.1; p < 0.01; Fig. 9b). EtOH effectively enhanced p-Akt levels at both time points (F [5, 30] = 2.7; p < 0.05; Fig. 9c) (F [5, 30] = 7.7; p < 0.05; Fig. 9d), while TH only at 24 h. This enhanced p-Akt expression could be further increased (p < 0.01) by NBO administration (Fig. 9c, d). As expected, LY294002 administration at 24 h significantly (p < 0.01) decreased p-Akt levels (F [4, 25] = 9.6; p < 0.01; Fig. 9e). Finally, NOX inhibition did not have an impact on p-Akt levels, suggesting that p-Akt is an upstream factor in NOX activation.

p-Akt levels were reduced (## p < 0.01) after stroke at 3 h (a) and 24 h (b), but rt-PA was able to prevent these reductions (**p < 0.01). At both 3 h (c) and 24 h (d), p-Akt expression was increased most effectively when NBO was combined with either EtOH or TH, these increases being greater than EtOH or TH monotherapy. e The PI3K/Akt pathway inhibitor performed as expected and prevented the increase of p-Akt compared with EtOH monotherapy ($$ p < 0.01). Apocynin-induced NOX inhibition did not have an impact on p-Akt
PKC-δ Protein
There was a significant increase in PKC-δ levels at 3 h (F [2, 15] = 8.1; p < 0.01; Fig. 10a) and 24 h (F [2, 15] = 5.5; p < 0.01; Fig. 10b) after stroke. Although after rt-PA administration, all monotherapies using NBO (p < 0.05), EtOH (p < 0.05), or TH (p < 0.01) decreased PKC-δ expression, combination of NBO with TH or EtOH exhibited the greatest reduction at both time points (F [5, 30] = 3.2; p < 0.01; Fig. 10c) (F [5, 30] = 3.9; p < 0.01; Fig. 10d). Treatment with either NOX inhibitor or PI3K inhibitor did not show any effect on the expression of PKC-δ (Fig. 10e), suggesting that PKC-δ may be an upstream factor in the PKC-Akt-NOX pathway.

PKC-δ expression was significantly increased (## p < 0.01) after stroke at both 3 h (a) and 24 h (b), and rt-PA was not sufficient to prevent this upregulation. Monotherapies after rt-PA injection at 3 h (c) and 24 h (d) were all effective at preventing PKC-δ expression with TH (**p < 0.01) being slightly more effective than either NBO or EtOH (*p < 0.05). However, combining NBO with either EtOH or TH led to the largest reduction (**p < 0.01). e Neither p-Akt inhibition nor NOX inhibition led to an increase or reduction in PKC-δ expression
Discussion
Other than rt-PA, adjuvant therapies have had little success in improving neurological outcome of stroke patients. This failure might be attributed to the multifactorial nature of pathologies after stroke. Thus, the need for novel combined therapies for effective outcome after stroke was the main goal of the present investigation. The focal ischemia model with an autologous embolus used here closely mimics events occurring in human thromboembolic stroke. Thrombolysis and reperfusion in this model is initiated by rt-PA administration likely mirrors the prescribed 1996 protocol for stroke intervention as compared to other models [25]. Here, after rt-PA intervention, we confirm the effects of EtOH or TH when used as monotherapies, as previously reported with the monofilament model of focal ischemia [20, 23]. We also, for the first time to our knowledge, demonstrate that low concentration (60 vs. 95 or 100 %) NBO combined with TH or EtOH produces enhanced neuroprotection compared with that resulting from the monotherapies. Another goal of the investigation was to determine whether modulation of NOX via its transduction cascade plays a role in neuroprotection and improved neurological outcome after ischemia/reperfusion. We found that combined therapies that included either NBO + TH or NBO + EtOH resulted in very similar reductions of infarct volume, neurological deficit, level of ROS, NOX activity, and NOX subunit protein expression, as well as PKC-δ and p-Akt expression. These results indicate that PI3K/Akt modulation is involved in EtOH-induced neuroprotection by regulating NOX activity and subunit expression, and that PKC-δ may be involved upstream of Akt in this NOX regulation.
The formation of excessive ROS after stroke is considered the single most significant event in determining the extent of ischemia/reperfusion injury [28]. Because oxidative phosphorylation and energy production are disrupted in the brain through the loss of oxygen and glucose during ischemia, this metabolic dysfunction and concomitant oxidative stress leads to ROS production [28]. In ischemic stroke, once recanalization is established (such as after rt-PA administration), the sudden burst of oxygenation to the anoxic brain further exacerbates the formation of ROS. ROS results in brain damage after ischemia not only through direct damage to cellular proteins, lipids, nucleic acids, and other macromolecular components but also directly as molecular triggers of ischemic cell death [39–41]. NBO has the potential to reduce cerebral edema, inhibit lipid peroxidation, protect the function of blood brain barrier, and promote neuronal survival through increases in the partial pressure of oxygen in the blood and tissues. Conversely, NBO may also increase the production of ROS after reperfusion [42]. Because of these conflicting findings, it would seem that the potential benefit of NBO as an adjuvant after stroke might be counter-indicated, justifying the interruption of its use in clinical trials [9, 12]. However, if administered at levels lower than those previously used, we show here that NBO effects on infarct volume and behavior outcome are negligible. More importantly, when used in combination with other adjuvants, such as EtOH and TH, NBO appeared to confer strong neuroprotection.
A robust neuroprotective role for TH has been demonstrated in various neurological conditions [43–45]. Experimental studies have demonstrated that TH intervention decreases ROS formation, thus providing effective neuroprotection [46–49]. However, TH application in clinical settings may be impractical and therefore its effects remain controversial. We have previously demonstrated the therapeutic benefit of EtOH at a dose of 1.5 g/kg in rats subjected to 2-h MCA occlusion [20]. EtOH’s role as an efficacious neuroprotective strategy has been attributed to its induction of a hibernation-like state that involves a reduction of cerebral energy demand, suppression of metabolic flux, and attenuation of oxidative damage. Our lab further demonstrated that a combination therapy using 95 % NBO plus EtOH after reperfusion more effectively suppressed ROS levels compared to either treatment alone [23]. In this study, although the use of a FiO2 of 60 % did not induce obvious neuroprotection, potential complications such as ROS formation that may arise from higher oxygen concentrations were also not obvious. In addition, FiO2 at 60 % levels can be easily achieved using readily available equipment in the most clinical settings [50]. This concentration is significantly higher than oxygen’s atmospheric level of ~21 % (room air). Comparison of data from the combined therapies further demonstrate that at concentration of 60 %, NBO together with TH or EtOH produce similar and additive effects in reducing ROS formation after reperfusion. In addition, EtOH and TH when combined with NBO conferred better neuroprotection than either agent used alone. Although the mechanism through which NBO in combination with either EtOH or TH might achieve an enhanced effect is presently unclear, our data supports the concept that NBO may augment both EtOH’s and TH’s capability to induce a “hibernation state” and to minimize oxidative damage by partially inhibiting glycolysis and other metabolic pathways [16, 36]. These effects in turn may slow down energy depletion and extend the survival time of cells in the penumbra region [37]. When the post-stroke metabolic dysfunction was inhibited by EtOH or hypothermia, oxygen may have been better able to ameliorate the hypoxic state of cells [38].
Precisely how ROS might be formed after stroke is the subject of intensive current research. In order to cover the shortage of ATP after ischemia, glucose metabolism increases, not only by glycolysis but also by the hexose monophosphate shunt. This pathway generates NADPH, the substrate of NOX, which induces ROS generation after reperfusion in which NOX is the primary source of ROS in reperfusion [27, 51, 52]. NOX is a multi-subunit enzyme, consisting of two membrane-bound elements (p22phox and gp91phox), three cytosolic components (p40phox, p47phox, and p67phox), and a low-molecular-weight G protein (either rac 2 or rac 1) [53, 54]. NOX activation occurs by phosphorylation of its cytoplasmic subunits resulting in the translocation of the subunits from cytosol to the plasma membrane. The subunit gp91phox (also named as NOX2) is thought to be the critical element of NOX in the induction of ischemia/reperfusion injury [53, 55]. In this study, NBO combined with TH or EtOH had an additive effect in reducing both NOX activity and the expression of its subunits. Additive effect of EtOH and NOX inhibitor on NOX activation suggests a key role of NOX inactivation in EtOH-induced neuroprotection.
Protein kinase B (Akt) is a serine/threonine protein kinase and a major downstream molecule of activated phosphoinositide-3 kinase (PI3K). Akt has been implicated in many cellular processes including cell survival, metabolism, and protein synthesis. Activation of Akt occurs via its phosphorylation in the phosphoinositide-3-kinase (PI3K) signaling pathway that is triggered by various stimuli. Akt is thought to play a significant neuroprotective role in ischemic reperfusion [56–59]. Here, we demonstrate decreased p-Akt levels after stroke and show increased p-Akt levels after rt-PA, as well as with other treatments such as EtOH or TH. Specifically, when Akt phosphorylation was inhibited by LY294002 (a PI3K inhibitor) after EtOH treatment, both infarction volume and neurological deficit were exacerbated, suggesting a beneficial role for Akt phosphorylation in cell survival in ischemic stroke. LY294002 also led to an inverse in NOX activity and subunit expression as well as ROS levels reduced by EtOH. In contrast, NOX inhibition had no effect on Akt activation, suggesting that Akt might act in an upstream event of NOX. Taken together, our data strongly supports the concept of Akt activity in promoting cell survival through NOX regulation by EtOH. In support of our findings, a recent study showed that Akt negatively regulated NOX4 activity and decreased TNF-α-induced oxidative stress in endothelial cells of cerebral microvessels from newborn piglets [60]. Furthermore, another study in endoplasmic reticulum-stressed cardiomyocytes showed that Akt activation attenuated superoxide generation, NADPH oxidase p47phox upregulation, apoptosis, and mitochondrial damage [61].
Protein kinase C (PKC), member of a family of serine-threonine kinases that regulates a broad spectrum of cellular functions, has been implicated in several pathologies, including reperfusion injury [62, 63]. Here, PKC-δ was further reduced when NBO was combined with EtOH or TH. This enhanced neuroprotection temporally correlated with the observed decrease in PKC-δ expression after reperfusion. Although it is still unclear how PKC-δ might function detrimentally during reperfusion, studies have shown that inhibition of PKC- δ with δV1-1 leads both to elevated p-Akt expression and increased cell survival [64]. It has been previously shown that PKC-δ regulates monocyte NOX activity through the phosphorylation and translocation of p47phox [65]. In agreement with other work [66], we did not observe significant changes in PKC-δ levels following p-Akt inhibition after ischemia. Moreover, our finding that PKC expression is neither changed by application of a NOX inhibitor nor an Akt inhibitor in EtOH treatment group, suggests that PKC-δ may be an upstream event of Akt in NOX signaling cascade leading to neuroprotection.
The used autologous embolic model requiring thrombolytic intervention (rt-PA) for reperfusion has a strong clinical correlation and provides a high degree of translational applicability of our data. This study demonstrated that at low concentration, NBO (60 %) does not confer significant neuroprotection, even after thrombolysis with rt-PA. However NBO combined with TH or EtOH produces robust neuroprotection in terms of attenuation of infarct volume and neurobehavioral deficits. Therapies that combined NBO with either EtOH or TH resulted in similarly beneficial outcomes which suggested comparable antioxidative injury mechanisms underlying PKC-Akt-NOX pathway, and the possibility that EtOH could be substituted for the more clinically intractable TH in healthcare settings. Since both NBO and EtOH are well-tolerated, widely available, and inexpensive, their combined use is a promising therapy for stroke patients.
References
Cheng YD, Al-Khoury L, Zivin JA (2004) Neuroprotection for ischemic stroke: two decades of success and failure. NeuroRx 1(1):36–45
Minnerup J, Sutherland BA, Buchan AM, Kleinschnitz C (2012) Neuroprotection for stroke: current status and future perspectives. Int J Mol Sci 13(9):11753–11772
Ginsberg MD (2008) Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology 55(3):363–389
Saver JL (2004) Number needed to treat estimates incorporating effects over the entire range of clinical outcomes - Novel derivation method and application to thrombolytic therapy for acute stroke. Arch Neurol 61(7):1066–1070. doi:10.1001/archneur.61.7.1066
Kollmar R, Schwab S (2012) Hypothermia and ischemic stroke. Curr Treat Options Neurol. doi:10.1007/s11940-012-0164-y
Bitterman H (2009) Bench-to-bedside review: oxygen as a drug. Crit Care 13(1):205
Cornet AD, Kooter AJ, Peters MJ, Smulders YM (2013) The potential harm of oxygen therapy in medical emergencies. Crit Care 17(2):313
Bennett MH, Weibel S, Wasiak J, Schnabel A, French C, Kranke P (2015) Hyperbaric oxygen therapy for acute ischemic stroke. Stroke 46(5):e109–e110
Cornet AD, Kooter AJ, Peters MJ, Smulders YM (2012) Supplemental oxygen therapy in medical emergencies: more harm than benefit? Arch Intern Med 172(3):289–290
Pountain SJ, Roffe C (2012) Does routine oxygen supplementation in patients with acute stroke improve outcome? BMJ 345:e6976
Padma M, Bhasin A, Bhatia R, Garg A, Singh M, Tripathi M, Prasad K (2010) Normobaric oxygen therapy in acute ischemic stroke: a pilot study in Indian patients. Ann Indian Acad Neurol 13(4):284
Rønning OM, Guldvog B (1999) Should stroke victims routinely receive supplemental oxygen? A quasi-randomized controlled trial. Stroke 30(10):2033–2037
Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, Smith K (2002) Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med 346(8):557–563
Nikolov NM, Cunningham AJ (2003) Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. Surv Anesthesiol 47(4):219–220
Choi K-E, Hall CL, Sun J-M, Wei L, Mohamad O, Dix TA, Shan PY (2012) A novel stroke therapy of pharmacologically induced hypothermia after focal cerebral ischemia in mice. FASEB J 26(7):2799–2810
Colbourne F, Li H, Buchan AM (1999) Indefatigable CA1 sector neuroprotection with mild hypothermia induced 6 hours after severe forebrain ischemia in rats. J Cereb Blood Flow Metab 19(7):742–749
Karibe H, Chen J, Zarow GJ, Graham SH, Weinstein PR (1994) Delayed induction of mild hypothermia to reduce infarct volume after temporary middle cerebral artery occlusion in rats. J Neurosurg 80(1):112–119
Maier CM, Sun GH, Kunis D, Yenari MA, Steinberg GK (2001) Delayed induction and long-term effects of mild hypothermia in a focal model of transient cerebral ischemia: neurological outcome and infarct size. J Neurosurg 94(1):90–96
Han Z, Liu X, Luo Y, Ji X (2015) Therapeutic hypothermia for stroke: where to go? Exp Neurol 272:67–77
Wang F, Wang Y, Geng X, Asmaro K, Peng C, Sullivan JM, Ding JY, Ji X et al (2012) Neuroprotective effect of acute ethanol administration in a rat with transient cerebral ischemia. Stroke 43(1):205–210. doi:10.1161/STROKEAHA.111.629576
Fu P, Peng C, Ding JY, Asmaro K, Sullivan JM, Guthikonda M, Ding Y (2013) Acute administration of ethanol reduces apoptosis following ischemic stroke in rats. Neurosci Res 76(1-2):93–97. doi:10.1016/j.neures.2013.02.011
Kochanski R, Peng C, Higashida T, Geng X, Hüttemann M, Guthikonda M, Ding Y (2013) Neuroprotection conferred by post‐ischemia ethanol therapy in experimental stroke: an inhibitory effect on hyperglycolysis and NADPH oxidase activation. J Neurochem 126(1):113–121
Geng X, Fu P, Ji X, Peng C, Fredrickson V, Sy C, Meng R, Ling F et al (2013) Synergetic neuroprotection of normobaric oxygenation and ethanol in ischemic stroke through improved oxidative mechanism. Stroke 44(5):1418–1425. doi:10.1161/STROKEAHA.111.000315
Parmar S, Moore-Langston S, Fredrickson V, Kim JM, Rastogi R, Elmadoun O, Ding Y (2015) Neuroprotective mechanisms of oxygen and ethanol: a potential combination therapy in stroke. Curr Med Chem 22(10):1194–1204
Zhang L, Zhang RL, Jiang Q, Ding G, Chopp M, Zhang ZG (2015) Focal embolic cerebral ischemia in the rat. Nat Protoc 10(4):539–547. doi:10.1038/nprot.2015.036
Tang XN, Cairns B, Kim JY, Yenari MA (2012) NADPH oxidase in stroke and cerebrovascular disease. Neurol Res 34(4):338–345
Suh SW, Shin BS, Ma H, Van Hoecke M, Brennan AM, Yenari MA, Swanson RA (2008) Glucose and NADPH oxidase drive neuronal superoxide formation in stroke. Ann Neurol 64(6):654–663
Chen H, Yoshioka H, Kim GS, Jung JE, Okami N, Sakata H, Maier CM, Narasimhan P et al (2011) Oxidative stress in ischemic brain damage: mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid Redox Signal 14(8):1505–1517
Hafeez A, Elmadhoun O, Peng C, Ding JY, Geng X, Guthikonda M, Ding Y (2014) Reduced apoptosis by ethanol and its association with PKC-δ and Akt signaling in ischemic stroke. Aging Dis 5(6):366
Brodie C, Blumberg P (2003) Regulation of cell apoptosis by protein kinase c δ. Apoptosis 8(1):19–27
Aronowski J, Strong R, Shirzadi A, Grotta JC (2003) Ethanol plus caffeine (Caffeinol) for treatment of ischemic stroke preclinical experience. Stroke 34(5):1246–1251
Kollmar R, Henninger N, Bardutzky J, Schellinger PD, Schäbitz W-R, Schwab S (2004) Combination therapy of moderate hypothermia and thrombolysis in experimental thromboembolic stroke—an MRI study. Exp Neurol 190(1):204–212
Qi ZF, Luo YM, Liu XR, Wang RL, Zhao HP, Yan F, Song ZJ, Luo M et al (2012) AKT/GSK3β-Dependent autophagy contributes to the neuroprotection of limb remote ischemic postconditioning in the transient cerebral ischemic rat model. CNS Neurosci Ther 18(12):965–973. doi:10.1111/cns.12016
Gao X, Zhang H, Takahashi T, Hsieh J, Liao J, Steinberg GK, Zhao H (2008) The Akt signaling pathway contributes to postconditioning’s protection against stroke; the protection is associated with the MAPK and PKC pathways. J Neurochem 105(3):943–955
Sharma M, Chuang WW, Sun Z (2002) Phosphatidylinositol 3-kinase/Akt stimulates androgen pathway through GSK3β inhibition and nuclear β-catenin accumulation. J Biol Chem 277(34):30935–30941
Tang XN, Cairns B, Cairns N, Yenari MA (2008) Apocynin improves outcome in experimental stroke with a narrow dose range. Neuroscience 154(2):556–562. doi:10.1016/j.neuroscience.2008.03.090
Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H (1986) Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke 17(3):472–476
Geng X, Elmadhoun O, Peng C, Ji X, Hafeez A, Liu Z, Du H, Rafols JA et al (2015) Ethanol and normobaric oxygen novel approach in modulating pyruvate dehydrogenase complex after severe transient and permanent ischemic stroke. Stroke 46(2):492–499
Taylor JM, Crack PJ (2004) Impact of oxidative stress on neuronal survival. Clin Exp Pharmacol Physiol 31(7):397–406
Ozkul A, Akyol A, Yenisey C, Arpaci E, Kiylioglu N, Tataroglu C (2007) Oxidative stress in acute ischemic stroke. J Clin Neurosci 14(11):1062–1066
Pradeep H, Diya JB, Shashikumar S, Rajanikant GK (2012) Review paper oxidative stress–assassin behind the ischemic stroke. Folia Neuropathol 50(3):219–230
Sjöberg F, Singer M (2013) The medical use of oxygen: a time for critical reappraisal. J Int Med 274(6):505–528
Chen F, Qi Z, Luo Y, Hinchliffe T, Ding G, Xia Y, Ji X (2014) Non-pharmaceutical therapies for stroke: mechanisms and clinical implications. Prog Neurobiol 115:246–269. doi:10.1016/j.pneurobio.2013.12.007
Moore M, Cunningham AJ (2003) Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. Surv Anesthesiol 47(4):220–221
Shankaran S, Laptook AR, Ehrenkranz RA, Tyson JE, McDonald SA, Donovan EF, Fanaroff AA, Poole WK et al (2005) Whole-body hypothermia for neonates with hypoxic–ischemic encephalopathy. N Engl J Med 353(15):1574–1584
Horiguchi T, Shimizu K, Ogino M, Suga S, Inamasu J, Kawase T (2003) Postischemic hypothermia inhibits the generation of hydroxyl radical following transient forebrain ischemia in rats. J Neurotrauma 20(5):511–520
Maier CM, Sun GH, Cheng D, Yenari MA, Chan PH, Steinberg GK (2002) Effects of mild hypothermia on superoxide anion production, superoxide dismutase expression, and activity following transient focal cerebral ischemia. Neurobiol Dis 11(1):28–42
Lei B, Adachi N, Arai T (1997) The effect of hypothermia on H 2 O 2 production during ischemia and reperfusion: a microdialysis study in the gerbil hippocampus. Neurosci Lett 222(2):91–94
Lee SM, Zhao H, Maier CM, Steinberg GK (2009) The protective effect of early hypothermia on PTEN phosphorylation correlates with free radical inhibition in rat stroke. J Cereb Blood Flow Metab 29(9):1589–1600
Slessarev M, Somogyi R, Preiss D, Vesely A, Sasano H, Fisher JA (2006) Efficiency of oxygen administration: sequential gas delivery versus “flow into a cone” methods. Crit Care Med 34(3):829–834
Abramov AY, Scorziello A, Duchen MR (2007) Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci 27(5):1129–1138
Loukogeorgakis SP, van den Berg MJ, Sofat R, Nitsch D, Charakida M, de Groot E, MacAllister RJ, Kuijpers TW et al (2010) Role of NADPH oxidase in endothelial ischemia/reperfusion injury in humans. Circulation 121(21):2310–2316
Babior BM (2004) NADPH oxidase. Curr Opin Immunol 16(1):42–47
Babior BM (1999) NADPH oxidase: an update. Blood 93(5):1464–1476
Brandes RP, Weissmann N, Schröder K (2014) Nox family NADPH oxidases: molecular mechanisms of activation. Free Radic Biol Med 76:208–226
Zhao H, Shimohata T, Wang JQ, Sun G, Schaal DW, Sapolsky RM, Steinberg GK (2005) Akt contributes to neuroprotection by hypothermia against cerebral ischemia in rats. J Neurosci 25(42):9794–9806
Zheng W-H, Kar S, Dore S, Quirion R (2000) Insulin-like growth factor-1 (IGF-1): a neuroprotective trophic factor acting via the Akt kinase pathway. In: Advances in Research on Neurodegeneration. Springer, 261-272.
Xu X, Chua CC, Gao J, Chua K-W, Wang H, Hamdy RC, Chua BH (2008) Neuroprotective effect of humanin on cerebral ischemia/reperfusion injury is mediated by a PI3K/Akt pathway. Brain Res 1227:12–18
Wick A, Wick W, Waltenberger J, Weller M, Dichgans J, Schulz JB (2002) Neuroprotection by hypoxic preconditioning requires sequential activation of vascular endothelial growth factor receptor and Akt. J Neurosci 22(15):6401–6407
Basuroy S, Tcheranova D, Bhattacharya S, Leffler CW, Parfenova H (2011) Nox4 NADPH oxidase-derived reactive oxygen species, via endogenous carbon monoxide, promote survival of brain endothelial cells during TNF-α-induced apoptosis. Am J Physiol Cell Physiol 300(2):C256–C265
Zhang Y, Ren J (2011) Thapsigargin triggers cardiac contractile dysfunction via NADPH oxidase-mediated mitochondrial dysfunction: role of Akt dephosphorylation. Free Radic Biol Med 51(12):2172–2184
Chou W-H, Messing RO (2005) Protein kinase C isozymes in stroke. Trends Cardiovasc Med 15(2):47–51
Zhao M, Xia L, Chen G-Q (2012) Protein kinase cδ in apoptosis: a brief overview. Arch Immunol Ther Exp 60(5):361–372
Bright R, Raval AP, Dembner JM, Pérez-Pinzón MA, Steinberg GK, Yenari MA, Mochly-Rosen D (2004) Protein kinase C δ mediates cerebral reperfusion injury in vivo. J Neurosci 24(31):6880–6888
Bey EA, Xu B, Bhattacharjee A, Oldfield CM, Zhao X, Li Q, Subbulakshmi V, Feldman GM et al (2004) Protein kinase Cδ is required for p47phox phosphorylation and translocation in activated human monocytes. J Immunol 173(9):5730–5738
Wen HC, Huang WC, Ali A, Woodgett JR, Lin WW (2003) Negative regulation of phosphatidylinositol 3-kinase and Akt signalling pathway by PKC. Cell Signal 15(1):37–45
Acknowledgments
This work was partially supported by the Wayne State University Neurosurgery Fund, American Heart Association Grant-in-Aid (14GRNT20460246), and National Nature Science Foundation of China (no. 81501141).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests..
Rights and permissions
About this article

Cite this article
Cai, L., Stevenson, J., Geng, X. et al. Combining Normobaric Oxygen with Ethanol or Hypothermia Prevents Brain Damage from Thromboembolic Stroke via PKC-Akt-NOX Modulation. Mol Neurobiol 54, 1263–1277 (2017). https://doi.org/10.1007/s12035-016-9695-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-9695-7