
Abstract
Obesity is considered one of the greatest risk to human health and is associated with several factors including genetic components, diet, and physical inactivity. Recently, the relationship between obesity and numerous progressive and aging-related neurodegenerative diseases such as Parkinson’s disease (PD) and Alzheimer’s disease (AD) have been observed. Thus, the involvement of the most abundant and heterogeneous group of glial cells in neurodegenerative diseases, the astrocytes, is caused by a combination of the failure on their normal homeostatic functions and the increase of toxic metabolites upon pathological event. Upon brain damage, molecular signals induce astrocyte activation and migration to the site of injury, entering in a highly active state, with the aim to contribute to ameliorating or worsening the pathology. In this regard, the aim of this review is to elucidate the relationship between obesity, Alzheimer’s disease, and Parkinson’s disease and highlight the role of astrocytes in these pathologies.
Similar content being viewed by others

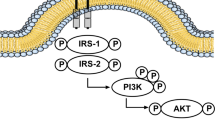
Avoid common mistakes on your manuscript.
Introduction
Obesity is an excessive accumulation of fat stored in adipose and non-adipose tissue as triglycerides that can be broken down into fatty acids, which can negatively affect health by the increased expression of pro-inflammatory markers [1]. Globally, the prevalence of obesity is rising [2–4]. Actually, it is a public health problem, and according to the World Health Organization, it is estimated that more than 1.9 billion adults, 18 years and older, were overweight in 2014 [5]. In fact, globally, there are more than 300 million adults who are obese and 1 billion who are overweight [6]. Obesity, which is considered one of the greatest threats to human health [7], is associated with multitude of risk factors including genetic components, physical inactivity, blood lipid disorders, inflammation, and insulin resistance. All these clusters of common pathologies have been associated to metabolic syndrome, which is a risk factor for neurological diseases [8, 9].
Over the last decade, the population of obese people has also increased in epidemic proportions. For this reason, the probable association between obesity and neurodegenerative diseases has been studied with a main focus on the probability that obesity may lead to neurodegeneration, exacerbate cognitive decline, and increase susceptibility to brain damage [2, 10]. In this regard, a large number of studies have demonstrated that people who suffer from midlife obesity (measured by body mass index or central adiposity) have an augmented risk for developing Alzheimer’s disease (AD) and Parkinson’s disease (PD) [11].
The precise mechanisms of the relationship between fat gain and loss and cognitive functioning remain to be explained. Obesity involves a series of pathological cellular responses due to an increment in basal lipolysis and subsequent release of free fatty acids (FFA) into the bloodstream. Indeed, the increase in FFA produces the activation of non-oxidative metabolic pathways such as ceramides, lysosomal degradation, pattern recognition receptors activation, and endoplasmic reticulum stress. Thus, the activation of these pathways are detrimental to normal cellular homeostasis and cell viability [12], thus resulting in systemic and brain inflammation, particularly in the hypothalamus, leading to cellular dysfunction, lipid droplet formation, and finally cell death [13, 14].
Neurodegenerative diseases (ND) are an important cause of disability, morbidity, and decreased quality of life, constituting the cause of 12 % of total deaths globally [1, 5]. These diseases represent a heterogeneous group of disorders, which are characterized by progressive dysfunction of neurons and astrocytes [15–18]. Recent findings suggest that astrocytes have been strongly involved in the maintenance of brain metabolism, anti-oxidant maintenance, and neuroprotection [17, 19–27]. For this reason, these cells play a critical role in the onset and progression of neurodegenerative diseases [28]. However, the cause of the progressive degeneration of neurons remains unresolved. It has been demonstrated that there is an activation of astrocytes in areas mostly affected by the disease. Both in vitro and in vivo studies suggest that astrocytes can be activated by FFAs [29]. In turn, these compounds can be elevated in subjects with obesity [30, 31]. FFAs can activate Toll-like receptors (TLR) and signaling cascades and generate nuclear translocation of the transcription factor NF-kB (nuclear factor kappa-light chain enhancer of activated B cells) [32]. This factor is implicated in several cellular processes, such as immune and inflammatory responses. After activation and nuclear translocation, NF-kB can promote the production of inflammatory cytokines like interleukin (IL)-6), tumor necrosis factor (TNF), and IL-1 [33]. According to this, in the specific case of interleukin 6, it is important to highlight that it is a cytokine not only implicated in inflammation and infection responses, but also in the adjustment of metabolic, restorative, and neural processes. It has been shown that IL-6 has been classified as a pro-inflammatory cytokine, but in some cases, it might exert restorative or anti-inflammatory activities [34].
Experimental findings identify these pro-inflammatory pathways are important regulators of different neurodegenerative pathologies [35]. Additionally, these cytokines have been recognized as an important mediator in facilitating leukocyte extravasation from the circulation through the blood-brain barrier (BBB) into the CNS parenchyma and that has been associated with neurodegeneration [36, 37]. Taking into account these evidences, the aim of this review is to elucidate the role of obesity and astrocytes in AD and PD development.
Association of Obesity with Neurodegenerative Diseases
Increasing life expectancy is concomitant with increased risk of aging-associated diseases such as neurodegenerative diseases and related health problems such as obesity. Recently, the relationship between obesity and numerous progressive and aging-related neurodegenerative diseases have been observed [38]. This relationship has been assessed in human and animal models with the aim to demonstrate that obesity might be associated to neurodegeneration by stimulating cognitive deterioration and raising the vulnerability to brain injury [39]. Although the exact mechanisms by which obesity negatively affects the brain are poorly understood, previous study has indicated that augmented inflammatory responses are key physiologic features of obesity [40]. In this respect, obesity may lead to brain inflammation and cause protein deposition, oxidative stress, morphological changes in brain cells, and alterations in important metabolic pathways [3, 4, 41].
Obesity and Astrocytes
Two of the main characteristic changes in obese population are insulin resistance and hyperglycemia; while in the first insulin levels are higher in comparison with the levels of glucose, in the second there is an abnormal increase of glucose in the blood [42–46]. The relationship between obesity and astrocytes is primarily generated by hormones. For example, astrocytes are able to synthesize different hormones such as leptin, ghrelin, and insulin [47–49], and as such, these hormones have been intensively studied because of their close relation with obesity and body weight [50, 51]. Indeed, it is shown that obese subjects show higher levels of these hormones than control subjects and this fact is correlated with the accumulation of fat tissue [51].
Different signaling pathways are closely related with the regulation of food intake. For example, one of them is the leptin signaling pathway which plays an important role in the regulation of energy homeostasis and regulation of food intake [52]. There is evidence that leptin signaling in astrocytes can modulate the metabolic response to high-fat diet [53]. According to this, different studies with animal models showed a decrease in food intake and an increase of the hormone ghrelin that is closely related with the stimulation of the hunger after deletion of leptin receptors in the brain [52].
Taking into account that main pathological alteration in obese population is the insulin resistance, it is important to stand out that insulin actions are mediated by the insulin receptor (IR), which is constituted by two alpha sub-units located in the extracellular level [54]. García-Cáceres et al. (2016) used positron emission tomography and glucose monitoring in cerebral spinal fluid in in vivo GFAP-IR KO mice and showed that lack of insulin receptors in hypothalamic astrocytes affects systemic glucose metabolism, thus raising the question of a possible effect in neuroprotection [45]. Moreover, different studies in mice have demonstrated that administration of insulin in the brain reduces body weight, and also showed that mice without insulin receptors become obese [55]. Finally, insulin is also related with dopamine circuits [56]. In this regard, it has been demonstrated that insulin increases the activity of dopamine receptors in mice brain, while there is a decrease in the level of the enzyme tyrosine hydroxylase in obese animals [57].
Obesity and AD
AD is the most common form of dementia and a progressive neurodegenerative disease that is mainly diagnosed by its clinical features [12, 58]. Of the 5.4 million Americans with AD, an estimated 5.2 million people are 65 years of age and older and approximately 200,000 individuals are under 65 years of age. Age is an important risk factor, with one in nine people over 65 years old having AD [59]. It is important to highlight that more women than men have AD and approximately two thirds of Americans with the disease are women [60]. AD is determined clinically by progressive cognitive impairment in two or more domains, such as memory, language, calculations, orientation, and judgment, in which these alterations may be severe enough to induce social or occupational disability [61]. Neuropathologically, the two characteristic findings of the disease are deposits of aggregated amyloid β (Aβ) in neuritic plaques [62] and neurofibrillary tangles (NT), which are produced by hyperphosphorylation of Tau, a microtubule-associated protein [61, 63]. It has been suggested that Aβ could trigger changes in Tau, producing the formation of these neurofibrillary tangles, thus resulting in synaptic loss and neuronal damage [64–66].
The increased prevalence of obesity, as well as reduced age of onset of obesity in the population, may lead to much higher incidence and prevalence of diseases such as AD in younger people, which are normally considered an old-age disease [12]. Also, there is evidence that obesity accelerates memory dysfunction and neuroinflammation in AD [67]. Several studies have exposed that people with obesity have an elevated risk of developing AD and dementia [68, 69]. The overweight in the elderly of 70 to 88 years and older has also been reported as a risk factor for AD [2, 70].
The mechanisms by which overweight increased risk of AD have not been fully understood. There are multifactorial mechanisms that link obesity with AD which includes systemic inflammation, activated astroglia, plaque deposition, and diminished plaque clearance [2, 71–73]. Increased adiposity tissue is associated with insulin resistance, hyperinsulinemia, oxidative stress, and dysregulation of glucose metabolism. Consequently, these metabolic profiles might cause formation and deposition of advanced glycosylation end-products (AGEs) and precursors [72]. AGEs are neurotoxic compounds which increase aggregation by the glycation of amyloid β, as the receptors for AGEs can use amyloid β as ligands. This interaction has been previously shown to play a key role in the pathogenesis of AD [74].
Though the association of obesity and AD has been contemplated here, this should be analyzed in the context that AD may have a reciprocal action in developing hyperinsulinemia, insulin resistance, dyslipidemia, and hypertension. It is important to point that this cluster of risk factors is considered a metabolic syndrome (MS), which has been linked with AD in recent studies [75, 76]. It is important to highlight that obesity is also related with dyslipidemia and disordered fatty acid and cholesterol metabolisms, thus affecting numerous neuronal processes that have been implicated in the development of neurodegenerative disorders [77]. Table 1 shows clinical and animal studies which demonstrate the relationship between obesity and AD. These studies showed that BMI is directly associated with cognitive performance and function including memory [78–83]. Indeed, many studies assessing the brain parenchymal fraction demonstrated that obesity influences brain structure, and volume, thus triggering brain dysfunction, brain atrophy, and cognitive impairment in humans [84–86]. Besides, it has been demonstrated that high-fat diet reduces synaptic plasticity in the hippocampus and cerebral cortex in animal models [87, 88] and increases amyloid and tau aggregates in transgenic mouse models of AD [89, 90], leading to neuroinflammation, reactive gliosis, and predisposition to injury [39, 91]. Finally, studies in brain and adipose tissue in a murine model of high-fat diet-induced obesity showed elevations in amyloid precursor protein (APP), Aβ, and Tau phosphorylation in the hippocampus, demonstrating that high-fat diet-dependent obesity is associated to pro-inflammatory changes in brain and adipose tissue, which is characterized by increased levels of APP [92, 93]. Although more studies on this matter are needed, experimental findings discussed here support the idea that overweight may increase the risk of developing AD through the modulation of cerebral amyloid and tau proteins, and that relationship might be modulated by sex hormones.
Obesity and PD
PD is the second most prevalent neurodegenerative disease after AD and is a chronic and progressive disorder. PD is characterized by the death of dopaminergic neurons in the substantia nigra (SN), as well as intracellular accumulation of aggregates of α-synuclein in neurons of the brainstem, spinal cord, and cortex [101–105]. It is estimated that 10 million people worldwide and about 1 % of the population over 60 years of age are living with PD [61, 106]. It is important to highlight that men are one and a half times more likely to suffer from PD than women [106]. It is believed that at early stages, PD pathology has a focal initiation site that later propagates throughout the brain [107, 108], and initial α-synuclein accumulation occurs without neuronal death or evident symptoms [103]. Although the complete molecular mechanisms of PD progression are not well understood [109], the central focus has always been on the damage of the dopaminergic neurons and reduction of dopamine, but a role for glial cells in mediating pathological or neuroprotective responses in PD is becoming increasingly recognized [61].
Several studies indicated that some factors might be associated with risk of developing PD. These factors include age, genetic factors, environmental toxins, oxidative stress, mitochondrial dysfunction, body shape, low physical activities, and poor diet. Other investigations used a semiquantitative food-frequency questionnaire in 110 PD case patients and 287 control subjects and showed that a high-fat diet, especially that one with increased intake of animal fat, is a risk factor for PD pathogenesis [110]. People with obesity and who are overweight are more physically inactive than normal weight people [111, 112], and lower levels of physical activity may increase the risk of developing PD [113]. On the other hand, other studies have suggested that obesity/overweight might be associated with PD due to disturbances of eating behavior which are associated with abnormal hypothalamic neurotransmission [114, 115].
The relationship between obesity and PD was revealed when overweight subjects demonstrated a depletion in their striatal dopamine receptor availability (D2) [116], and that dopamine played an important role in both obesity and PD, having in common the loss of dopaminergic neurons, and lower dopamine levels in the hypothalamus and striatum [117–119]. The relationship between obesity and dopamine levels can explain in part these findings. The mechanisms associated to obesity and PD are multifactorial. In this context, fat acting as a substrate for lipid nigrostriatal dopaminergic neurotoxins and overweight acting as a systemic metabolic modulator may increase body’s vulnerability to impairment from neurotoxins. Experimental evidences from positron emission tomography (PET) support that obese transgenic animal model exhibits increased striatal dopaminergic neurons susceptibility to methamphetamine and kainic acid [2]. Additionally, dopamine has a key role in the regulation of food intake [114, 115, 120].
Recent epidemiological studies have demonstrated a potential association between obesity/overweight and the risk of PD. In a prospective study with 451 Japanese-American subjects in Hawaii, the authors reported that greater midlife triceps skinfold thickness was correlated with higher risk of PD, which is independent of BMI [121, 122]. Nevertheless, studies correlate BMI with disease severity and cognitive decline and dyskinesia in PD [123], and it was reported that high-fat diet may confer a greater susceptibility to environmental toxins and accelerate the pathogenesis of PD [124]. Finally, Briceño and colleagues carried out a cross-sectional study including 177 healthy controls and 177 PD patients and demonstrated that overweight/obesity was more common among patients with PD, in comparison with non-demented patients [97]. Also, preclinical studies using animal models link obesity and PD. For example, high-fat diet exacerbated the progression of parkinsonism by increasing dopamine depletion in the substantia nigra in mice and also by the reduced capacity of nigral dopaminergic terminals to cope with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced neurotoxicity [124–126]. Although some studies were not able to provide substantial evidences about the association between BMI, weight change, and waist circumference with the risk of PD [127, 128], others showed a clear relationship between obesity and PD and a correlation among BMI increase, inflammation, and PD disease severity [97, 121–126].
Neuroinflammation
Neuroinflammation is a term used to describe a mixture of neurotoxic and neuroprotective responses originated in the CNS by glial cells, as a defense mechanism that is associated with neutralization of an insult, destruction of injured cells, and repair of function and structure of the brain [129, 130]. Neuroinflammation is characterized by an integrated response of the different cells of CNS [131, 132], including neurons, glial cells, and the infiltrating leukocytes [133]. Neuroinflammatory responses may be helpful or harmful, as these mechanisms are associated with normal brain development and function, as well as with neuropathological processes during brain injury and neurodegeneration [134, 135]. There are two types of neuroinflammation. Firstly, acute inflammation, as a defensive response that helps to repair the impaired site, and secondly, chronic neuroinflammation that results from deleterious and more persistent stimuli [136]. Acute neuroinflammation develops quickly with the presence of pain, whereas chronic inflammation develops slowly [130].
Nowadays, it is broadly recognized that not all forms of neuroinflammation are necessarily detrimental for CNS function [133]. An efficient inflammatory response does not only removes pathogens and abnormally aggregated proteins but also results in beneficial, self-limiting, and healing processes [130, 133, 137]. In contrast to acute neuroinflammation, chronic neuroinflammation is a long-lived, persistent response that starts with an initial inflammatory stimulus but becomes self-propagating. Inflammatory factors produced by astrocytes, together with released damage-associated molecular patterns (DAMPs), can further increase inflammation and glial activation, leading to a vicious inflammatory cycle and the damage of local tissues. This long-term inflammation can have disastrous consequences in the CNS, ranging from loss of synapses to impaired cognition and lastly neurodegeneration [138–141]. The concept that the neuroinflammation is prejudicial implies that astroglial activation precedes and causes neuronal degeneration [142].
Obesity and Neuroinflammation
The adipose tissue not only stores fatty acids but also produces and releases a large number of other active compounds such as FFAs, resistin, TNF-α, IL-6, IL-1β, and others. Studies have reported that plasma FFA levels are usually elevated in obesity because the increased adipose tissue mass releases more FFA and its clearance may be reduced, which will further increase the rate of FFA release into the circulation [33].
There is evidence that FFAs can activate inflammatory and innate immune responses and trigger a phenomenon known as lipotoxicity in the brain. Recent studies demonstrated that acute elevation of plasma FFA activated the pro-inflammatory NF-kB signaling resulting in increased astrocytic expression of several inflammatory cytokines such as TNF-α, IL-1β, and IL-6. The brain is very sensitive to inflammatory mediators, and there is substantial data indicating that these inflammatory mediators play a critical role in the inhibition of different signaling pathway and in the induction of endoplasmic reticulum (ER) stress in hypothalamic neurons [143].
Mechanisms Associated to Neuroinflammation in Obesity
Previous studies have shown that hypothalamic dysfunction induced by inflammation causes neural dysregulation and neurodegeneration in obesity [144]. Studies have demonstrated an augmented expression of pro-inflammatory cytokines and activation of IkB kinase-β (IKKβ)/nuclear factor-kβ in the hypothalamus induced by high-fat diet. NF-kB is a critical modulator of immunity and inflammation in the CNS. NF-kB activation is stimulated mainly by IKKβ, in which this phosphorylates and degrades Ikβ proteins, thus liberating NF-kB to enter the nucleus and inducing the transcription of different inflammatory genes. During the immune response and inflammation, IKKβ/NF-κB activation is stimulated by different cell-membrane receptors including Toll-like receptors (TLRs).
It has been demonstrated that long-chain saturated fatty acids, possibly acting through TLR-3 and TLR-4, stimulate the generation of pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-6 [145–147]. Also, it has been reported that TLR-4 is expressed by activated astroglia in diet-induced obese (DIO) mice [148], which also activates NF-κβ signaling [149] thus leading to disrupted leptin and insulin signaling in the hypothalamus [148, 150]. Cytokine receptors such as TNF-α receptors have been shown to be mediating neuroinflammation in overweight. Experimental approaches of loss of function have showed that TNF-α receptor knockout [151, 152] reduced dietary-induced obesity in mice. Also, overnutrition leads to perturbations in the endoplasmic reticulum system. This perturbation activates IKKβ/NF-κβ signaling in the hypothalamus and results in an energy imbalance [153, 154]. ER stress could be a downstream event that ultimately triggers pro-inflammatory processes in the hypothalamus [146]. Recent studies indicated that ER stress and defective autophagy may activate IKKβ/NF-κB signaling pathway to generate hypothalamic inflammation [155, 156]. Furthermore, investigations on inflammatory cytokine expression identified the kinase c-jun N-terminal kinase (JNK) as major intracellular contributors to the induction of inflammation. Compared with lean controls, obese tissues showed increased JNK activity in the liver, muscle, adipose tissue [155, 157], and the hypothalamus [158, 159]. JNK responds to diverse stress signals, including ER stress, pro-inflammatory cytokines, FFAs, and reactive oxygen species (ROS) [157, 160, 161]. Finally, independently of the mechanism involved, the common fact is that the hypothalamus is the main site of inflammation caused by the intake of high-fat diet, thus this process represents an early agent to the development of obesity and insulin resistance [146, 162–164].
Neuroinflammation in AD
Current evidences have shown that many compounds involved in the development of inflammatory processes are present in the CNS of patients with such neurodegenerative diseases [136]. Activated microglia and reactive astrocytes are observed in patients with these diseases, and it seems like that glial activation might have dual effects by degrading Aβ [165–167] and inducing neuroinflammation and neuronal dysfunction. The increase in Aβ concentration in aged transgenic AD mice are associated with increased concentrations of pro-inflammatory cytokines, including TNF-α, IL- 6, IL- 1α, and GM-CSF [168]. These results suggest that pathological accumulation of Aβ is a key factor that drives neuroinflammatory responses in AD.
There is evidence suggesting that the pro-inflammatory environment in the brains AD patients and in mouse models of cerebral amyloidosis induces brain damage. For example, the risk of developing dementia from mild cognitive disability is increased in AD patients with elevated concentrations of TNF-α and reduced concentrations of TGF-β in the CSF [169]. In a clinical trial with elders suffering from metabolic problems, it was shown that elevated levels of inflammation in plasma and serum had a negative effect on cognitive performance [170]. In animal models of AD, neuroinflammation is known as a response to sustained Aβ overproduction and deposition, in which includes involvement of the complement system and production of cytokines [171–173]. Janelsins and cols using quantitative real-time RT-PCR detected early activation of inflammatory processes in the entorhinal cortex and the hippocampus of the triple transgenic model (3xTg) of AD at 3 months of age [174]. The neuroinflammatory process was concurrent with the production and accumulation of intracellular Aβ, but occurred prior to any significant extracellular Aβ plaque deposition, which manifests at about 12 months of age in the 3xTg mice [174]. This neuroinflammation was characterized by a selective trend of increasing expression of TNF-α and monocyte chemoattractant protein-1 (MCP-1), which was not detected for 21 other cytokines tested [174]. Hoozemans and colleagues performed a study by analyzing important characteristics of the neuroinflammatory response in AD patients and control cases in a range between 52 and 97 years old. The authors used immunohistochemistry and antibodies directed against CD68 (KP1), HLA class II (CR3/43), and glial fibrillary acidic protein (GFAP). In this study, the authors found that the association between neuroinflammation and AD is stronger in relatively young AD cases compared with old AD cases, with an age-dependent presence of microglia and astrocytes, which is indicative of a neuroinflammatory response [175]. Finally, inflammation has a significant and important role in metabolic illness being a main risk factor for developing AD. Nevertheless, old individuals with metabolic syndrome did not have an enhanced risk of cognitive disability [176].
Neuroinflammation in PD
The neuroinflammatory processes in PD is characterized by the presence of activated glial cells in the substantia nigra from patients with PD or intoxicated by MPTP [177]. The implication of glucocorticoids (GCs) on this matter has been assessed. For example, an elevation in the systemic level of glucocorticoids (GCs) after the stimulation of the hypothalamic–pituitary–adrenal axis [178] was observed, which is in accordance with another study showing the role of glucocorticoid receptors (GRs) in the death of dopaminergic neurons and in neuroinflammation in models of PD. The results showed a loss of dopaminergic neurons induced by MPTP toxicity [179]. This increased loss of dopaminergic neurons was associated and directly related with an increased microglial and astroglial reaction, an enhanced production and release of pro-inflammatory mediators, and a decreased expression of anti-inflammatory factors [178], suggesting that glucocorticoids may adjust the brain inflammation to balance between a pro- and anti-inflammatory stimuli during neurodegeneration.
Studies performed in transgenic models reported increased TNF-α levels in the substancia nigra of mice overexpressing human α-synuclein, and this increase in TNF-α levels in vivo contrasted with the changes in TNF-α protein levels induced in vitro [180]. Other studies have shown that one of PD genes (DJ-1) is expressed in astrocytes and microglia, and it is known that these expression levels are upregulated in PD patients [181]. Astrocytes from DJ-1 knockout mice produce major levels of cyclooxygenase-2 (COX2) and IL-6 after lipopolysaccharide (LPS) treatment [182]. Mutations in parkin are the most common cause of recessively inherited PD [183]. It is demonstrated that this mutation may lead to the degeneration of dopaminergic neurons induced by LPS in the substancia nigra pars compacta (SNpc) of parkin knockout mice [184]. Aged parkin knockout mice showed enhanced astrogliosis in the striatum and abnormal microglial activation in the midbrain [185]. Altogether, these experimental evidences suggest that parkin plays an outstanding role in the regulation of inflammation in PD [186].
Role of Astrocytes in Neuroinflammation
Astrocytes are the most abundant group of glial cells, representing approximately around 20 to 40 % of the total brain cell population. These glial cells are recognized to modulate extracellular glutamate level, form a “glial scar” after injury and have powerful pro-inflammatory responses as pivotal regulators of CNS inflammatory responses. After a brain injury, molecular signals induce astrocyte activation and migration to the site of the damage [18, 20, 21, 26, 187]. Astrocytes enter a highly active state, which are suggestive to contribute to ameliorating or worsening the pathology [21, 188, 189]. Astrocytes are part of the BBB [24, 190], therefore, these cells influence the entrance of cells into the CNS and also the activity of invading cells [191]. Activated astrocytes produce a number of cytokines and participate in mediating immune responses within the CNS [192]. Table 2 shows different functions of astrocytes.
The involvement of astrocytes in neurodegenerative diseases is associated with the loss of their normal homeostatic functions and the increase of toxic functions induced by damage. Intracellular aggregates in surviving astrocytes in various neurodegenerative diseases have been observed. The presence of these aggregates disturbs astrocytic functions in different ways that could be damaging to neuronal viability [58]. In this respect, astrocyte activation is one of the most important processes of the cellular responses to brain lesion and chronic neurodegeneration. Due to their different shapes, which can reflect their functional phenotypes, astrocytes can adopt from a quiescent state, as seen in the normal uninjured CNS, to reactive-like state, as observed after damage or illness [158, 204–206].
It is important to highlight that inflammation, as stated above, has the ability to influence neuronal homeostasis by modulating intracellular mechanisms such as oxidative stress and ER stress. Oxidative stress is characterized by high levels of superoxide and hydrogen peroxide and has been involved in neuronal damage and cell death related with neurodegenerative diseases [207]. Large amounts of ROS and pro-inflammatory cytokines are produced by activated immune cells. In turn, ROS can activate NF-kB and favor the production of pro-inflammatory cytokines [208]. Inflammation and oxidative stress are highly related and therefore co-exist. There is evidence that high-fat diet is closely related with oxidative stress in diverse brain areas including the hippocampus [209, 210]. Besides, brain oxidative stress is associated with astrocyte activation, production of pro-inflammatory cytokine in the brain, and also with cognitive disability after high-fat diet consumption [91, 100]. Many cytokines, such as interleukins 1 and 6, are involved in the initiation and modulation of reactive astrogliosis, thus contributing to pathological inflammatory responses. Moreover, under different pathological situations, these factors are implicated as mediators of important processes such as neuroprotection and remyelination [211]. In different studies of injury models, interleukins 1β and 6 KO mice showed retarded astrocyte activation and enhanced BBB permeability, suggesting that cytokine-induced astrogliosis is very prominent to restore BBB integrity after trauma [212–216]. Other in vitro studies have demonstrated that cytokines, specifically IL-1, IL-6, and TNF, can favor the production of neuroprotective mediators [217]. There is evidence showing that TLR on cultured astrocytes induces the production of neurotrophic factors [218]. A similar action is displayed by chemokines such as CCL2 and CXCL12 which has an important role in the migration of neural progenitors in brains during development [219]. CCL2 is upregulated in diverse pathological stages in astrocytes and microglia [220, 221]. Other important chemokine in the CNS named CX3CL1 is expressed by astrocytes and neurons and has a role as an inhibitor of microglial-induced toxicity, as evidenced in three different in vivo models of neurotoxicity [222]. Finally, these studies may suggest that due to the production of cytokines and chemokines, reactive astrocytes may induce regeneration in the lesioned CNS (Fig. 1). Moreover, pro-inflammatory cytokines produced by astrocytes such as TNF-α facilitate leukocyte leakage from the bloodstream over the blood-brain barrier into the CNS parenchyma [223–226]. For instance, van Kralingen and colleagues found high levels of inflammatory cytokines in the CNS after injury. Also, in different neurological conditions affecting the integrity of the BBB, elevated levels of specific cytokines like IL-1β and TNF-α have been found [227].

Obesity involves a series of pathological cellular responses due to increment in basal lipolysis and subsequent release of free fatty acids into the bloodstream and the inflammation in the brain, particularly in the hypothalamus. Indeed, the increase in FFA produces endoplasmic reticulum stress, astrocytes activation, and activation of Toll-like receptors, leading to the activation of a signaling cascade and the activation of the transcription factor NF-kB and AP-1 in the nucleus. NF-kB can induce the production of inflammatory cytokines, such as TNF-α, IL-6, IL-1, and CCL2. Obesity also provokes neurodegeneration and a progressive dysfunction of neurons and astrocytes, as common pathological mechanisms in Alzheimer’s disease, which is characterized by deposits of aggregated amyloid β (Aβ) and the presence of Tau protein. On the contrary, in Parkinson’s disease, it is common the intraneuronal aggregates of α-synuclein (called Lewy bodies). These mechanisms lead to neuronal dysfunction and neuronal loss in both diseases
Astrocytes Are Important Regulators of Neuroinflammation
Astrocyte Pro- and Anti-inflammatory Mechanisms
Evidence indicates that there are two types of astrocytes activation such as pro-inflammatory and anti-inflammatory (Fig. 2). Pro-inflammatory (M1), a classical activation mechanism, is characterized by the production of pro-inflammatory proteins like cytokines (Table 3), including interleukins 1β, 6, and 12 and tumor necrosis factor alpha, and other cytotoxic compounds such as nitric oxide (NO), which may promote the increase of pro-inflammatory responses during damage [186]. Also, as widely accepted, upon CNS injury, activated astrocytes can produce and secrete pro-inflammatory molecules that will trigger neuroinflammation, including CCL2 and prostaglandins expressions [228–233].

Inflammatory mediators and cytokine receptors are expressed in the CNS by astrocytes and other glial cells at low levels. Specific factors implicated in reactive astrogliosis comprise large polypeptide growth factors and cytokines such as IL-1, IL-4, IL-6, IL-10, TNF-α, and TGF-β. Glial cells activation is followed by enhanced secretion of these factors, inducing damaging effects in the CNS. As discussed earlier, astrocytes activation produces a different range of downstream mediators, including cytokines, NO, and ROS and loss of fundamental functions, which are important for neuronal homeostasis and synapses maintenance
As described above, there are two types of alternative activation phenotypes. In this regard, the second is the alternative M2 (anti-inflammatory) phenotype, which suppresses an immune response by neutralizing the typical M1 phenotype and promotes the resolution of the damage. The second phenotype (M2) produces different proteins like anti-inflammatory cytokines (Table 4), in which some of them are interleukins and transforming growth factor beta [186]. In addition, fundamental anti-inflammatory roles of astrocytes have now been demonstrated by several experiments in diverse models of CNS injury and disease [206].
Astrocytes and AD
Astrocyte activation is an important component of the cellular responses against brain injuries and chronic neurodegeneration [188]. Also, it is a pathologic response in AD and is related with the accumulation of Aβ and/or the raising number of degenerating synapses and neurons [251]. It has been reported in studies with tissue from patients with AD that activated astrocytes were closely associated and related with amyloid plaques in the molecular layer of the cerebral cortex [252]. Thus, astrocytes might be activated by human Aβ [253], indicating a correlation between this protein and subsequent alterations in astrocyte function. This astrocyte activation process, called astrogliosis, is characterized by increased expression of the astrocyte marker glial fibrillary acidic protein (GFAP), and this process takes place around Aβ deposits in the brain parenchyma and the cerebral microvasculature [251, 254]. Astrocytes can accumulate amyloidogenic material resulting from local neurodegeneration, and then astrocytes might undergo apoptosis and produce the formation of GFAP+ amyloid plaques [253].
There are several studies regarding the capacity of astrocytes to internalize and degrade Aβ deposits under in vitro and ex vivo conditions [165, 173, 255]. For example, some studies have shown the presence of Aβ deposits in post-mortem human AD brain and in animal models of AD [255–257]. Investigations with fluorescently labeled astrocytes transplanted into AD mouse brains [258] showed that glial cells are capable to migrate and internalize deposits of Aβ and that wild-type astrocytes can clear Aβ plaques [259]. Functional experiments also demonstrated the ability of astrocytes to phagocyte and degrade Aβ deposits in vitro [173]. These findings are very significant to note the role of astrocytes in AD. Further, in vitro analyses indicated that treatment of astrocytes with Aβ leads to an increase in calcium-wave signaling between these glial cells [260]. To support these studies, Johnston et al. [261] showed that in cells expressing the familial AD presenilin 1 (PSEN1) mutation, calcium fluctuations in astrocytes occurred at lower ATP and glutamate levels than in wild-type astrocytes. These studies support a model in which calcium signaling between these glial cells is modified by the illness process, which could promote neuronal dysfunction or death under certain conditions [61]. Finally, it is very important to highlight that research about the role of neuroglia in the progression of AD is increasing. Analysis of astrogliosis in old patient’s brains has illustrated a relation between the degree of astrogliosis and cognitive decline [189].
Astrocytes and PD
The role of astrocytes in the progression of PD has not been well characterized, although astrogliosis was detected at the late stages of the disease [262, 263]. It is thought that astrocytes are involved at early stages before neuronal loss is evident and at advanced stages when neurodegeneration occurs [264]. The substantia nigra has low density of astrocytes compared with other brain regions, and it has been speculated that a premature astroglial atrophy may have a role on the development of PD [265].
To date, several studies support the neuroprotective role of astrocytes in PD [24, 27, 61, 266]. As in other neurodegenerative diseases, these studies have revealed an increase in the number of astrocytes expressing GFAP in PD [61] and showing the presence of anti-oxidant pathways that might contribute to their neuroprotective role. The research evidence indicated that in control brains, the levels of glutathione-peroxidase-positive cells were higher in the dopaminergic cell areas, more resistant to PD pathology [61]. The increase in the levels of glutathione-peroxidase-containing cells reflects a protective mechanism against oxidative stress [267]. Also, an accumulation of α-synuclein in the cytoplasm of protoplasmic astrocytes at initiation stage of the disease has been found [268]. This accumulation was more distributed than Lewy bodies and also present areas without Lewy bodies [269], suggesting that astrocytes can capture the altered α-synuclein that has escaped from axon terminals [270]. Also, Lee and colleagues demonstrated in both cell culture and animal models of PD that α-synuclein is transmittable from neuron to neuron [271] and from neuron to astrocytes [270]. In addition, in vivo studies using transgenic mice overexpressing α-synuclein, driven by the neuronal promoter PDGF-β, showed that α-synuclein immunoreactivity was detected in both neurons and astrocytes [270]. In humans, PET studies have shown a pronounced activation of microglia in various regions of the brain in subjects with PD [272, 273]. Importantly, the presence of synuclein-positive astrocytes in PD samples correlated with neuronal cell death in the substantia nigra [274].
Therapeutic Strategies
Astrogliosis as Therapeutic Target
Based on the pathogenic role of astrocytic dysfunction, the strategy to restore or enhance astrocytic functions might be an attractive way to promote neuroprotection in a variety of brain disorders [15]. Various studies using different types of transgenic models have shown that reactive astrogliosis is beneficial and neuroprotective during early stages of neurodegenerative diseases. However, astrocytic function in neuroprotection is greatly compromised during chronic neuroinflammation [275]. New perspectives for therapeutic strategies include the replacement of dysfunctional astrocytes or pharmacological treatments. An explicit example is the replacement of neurotransmitters, such as levodopa for PD or memantine for the treatment of AD [61]. Several experimental studies have revealed that reactive astrocytes can protect CNS cells in different ways such as preventing or reducing oxidative stress by increasing glutathione production [276–280], improving mitochondrial membrane potential and mitochondrial volume [20, 281, 282], inducing neuroprotection via adenosine release [283] and neuroglobin expression [284, 285], and facilitating BBB repair [286]. However, it is important to highlight that reactive astrocytes can play harmful roles during injury or disease, due to the increase of the abnormal effects such as production of reactive oxygen species or some inflammatory cytokines [287–289]. Thereby, reactive astrocytes have a tremendous potential to influence injuries and/or disease effects in a positive or negative way, as determined by specific signaling events and molecular mechanisms [288–290].
Neuroinflammation as Therapeutic Target
Taking into account the important role of neuroinflammation in the initiation and progression of neurodegenerative diseases, it is desirable to develop therapies by targeting the inflammatory pathways mediated by activated glial cells. For example, minocycline, a semi-synthetic tetracycline analog, which acts as a lipophilic molecule, can easily cross the BBB and has anti-inflammatory and neuroprotective properties in multiple inflammation-related neurological diseases [291, 292]. Also, substances such as the glucocorticoids, which are well known for their anti-inflammatory effects, have been widely used in clinical studies for brain inflammation [293].
Other substances used as therapeutic tools are the non-steroidal anti-inflammatory drugs (NSAIDS), such as aspirin, salicylic acid (SA), ibuprofen, and celecoxib [294, 295]. Aspirin has shown to reduce neuroinflammation and neurodegeneration [296], thus promoting the resolution of inflammation [297]. SA is a metabolite product of aspirin, which is widely used for the treatment of myocardial infarction and cardiovascular diseases [295]. Previous clinical studies using NSAIDS did not show encouraging results, but some methodological problems have been proposed to lead to their failure and encourage new investigation targeting neuroinflammation [298]. Lastly, it has been demonstrated that gonadal hormones, such as estradiol and testosterone, selective estrogen receptor modulators (SERMs; raloxifene and tamoxifen), or tibolone, a selective tissue estrogenic activity regulator (STEAR) might regulate the reactive glia upon brain damage [20, 281, 284, 299–302]. Future studies should focus on identifying more specific drug targets with the aim to understand the fundamental processes of inflammation at different stages of the disease progression.
Concluding Remarks
Obesity is considered one of the greatest risk to human health and has a significant role in cognitive dysfunction and aging-associated cognitive disorders including dementia. Thus, the high prevalence of obesity, as well as reduced age of onset in the population, may lead to higher incidence and prevalence of AD and PD. Nowadays, there is evidence that peripheral inflammation induced by obesity and high-fat diet may trigger local inflammation in the hypothalamus altering synaptic plasticity and contributing to the neurodegenerative processes deteriorating cognitive functions. After a brain injury, molecular signals induce astrocyte activation and migration to the site of injury. Astrocytes enter a highly active state that may ameliorate or worsening the pathology at early and late stages, respectively. Recent findings suggest that astrocytes are actively involved in the maintenance of brain metabolism, anti-oxidative maintenance, and neuroprotection during pathology. For this reason, these cells play a critical role in the onset and progression of several neurodegenerative diseases. Thus, the involvement in neurodegenerative diseases of the most abundant and heterogeneous group of glial cell in the brain, the astrocytes, is caused by a combination of the loss of their normal homeostatic functions and the gain of toxic functions in disease that trigger the release and production of pro-inflammatory and anti-inflammatory molecules during pathological diseases of the central nervous system. New studies about the role of astrocytes during early and late stages of neurodegeneration are guaranteed.
References
Chen WW, Zhang X, Huang WJ (2016) Role of neuroinflammation in neurodegenerative diseases (review). Mol Med Rep 13(4):3391–3396. doi:10.3892/mmr.2016.4948
Ashrafian H, Harling L, Darzi A, Athanasiou T (2013) Neurodegenerative disease and obesity: what is the role of weight loss and bariatric interventions? Metab Brain Dis. doi:10.1007/s11011-013-9412-4
Mattson MP, Pedersen WA, Duan W, Culmsee C, Camandola S (1999) Cellular and molecular mechanisms underlying perturbed energy metabolism and neuronal degeneration in Alzheimer’s and Parkinson’s diseases. Ann N Y Acad Sci 893(1):154–175. doi:10.1111/j.1749-6632.1999.tb07824.x
Perl DP, Olanow CW, Calne D (1998) Alzheimer’s disease and Parkinson’s disease: distinct entities or extremes of a spectrum of neurodegeneration? Ann Neurol 44(3 Suppl 1):S19–S31. doi:10.1002/ana.410440705
World Health Organization (WHO) (2011) World health statistics 2011. WHO Press, Geneva, p. 162
World Health Organization (WHO) (2010) World health statistics 2010. WHO Press, Geneva, p. 168
Caveney E, Caveney BJ, Somaratne R, Turner JR, Gourgiotis L (2011) Pharmaceutical interventions for obesity: a public health perspective. doi:10.1111/j.1463-1326.2010.01353.x
Aitlhadj L, Ávila DS, Benedetto A, Aschner M, Stürzenbaum SR (2011) Environmental exposure, obesity, and Parkinson’s disease: lessons from fat and old worms. Environ Health Perspect. doi:10.1289/ehp.1002522
Mardinoglu A, Kampf C, Asplund A, Fagerberg L, Hallström BM, Edlund K, Blüher M, Pontén F et al (2014) Defining the human adipose tissue proteome to reveal metabolic alterations in obesity. J Proteome Res 13(11):5106–5119. doi:10.1021/pr500586e
Wolozin B (2004) Cholesterol and the biology of Alzheimer’s disease. Neuron 41(1):7–10. doi:10.1016/s0896-6273(03)00840-7
Profenno LA, Porsteinsson AP, Faraone SV (2010) Meta-analysis of Alzheimer’s disease risk with obesity, diabetes, and related disorders. Biol Psychiatry 67:505–512. doi:10.1016/j.biopsych.2009.02.013
Naderali EK, Ratcliffe SH, Dale MC (2009) Obesity and Alzheimer’s disease: a link between body weight and cognitive function in old age. Am J Alzheimer’s Dis Other Demen 24(6):445–449. doi:10.1177/1533317509348208
Ford JH (2010) Saturated fatty acid metabolism is key link between cell division, cancer, and senescence in cellular and whole organism aging. Age. doi:10.1007/s11357-009-9128-x
Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz E, Li Z, Hui J et al (2015) Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 160:177–190. doi:10.1016/j.cell.2014.12.019
Barreto G, White RE, Ouyang Y, Xu L, Giffard RG (2011) Astrocytes: targets for neuroprotection in stroke. Cent Nerv Syst Agents Med Chem 11(2):164–173
G. E. Barreto, J. Gonzalez, F. Capani and L. Morales: Role of astrocytes in neurodegenerative diseases. Neurodegenerative Dis ProcessPrevent Protect Monit 257–272 (2011)
Barreto GE, Gonzalez J, Torres Y, Morales L (2011) Astrocytic-neuronal crosstalk: implications for neuroprotection from brain injury. Neurosci Res 71(2):107–113. doi:10.1016/j.neures.2011.06.004
Barreto GE, Sun X, Xu L, Giffard RG (2011) Astrocyte proliferation following stroke in the mouse depends on distance from the infarct. PLoS One 6(11):e27881. doi:10.1371/journal.pone.0027881
Logica T, Riviere S, Holubiec MI, Castilla R, Barreto GE, Capani F (2016) Metabolic changes following perinatal asphyxia: role of astrocytes and their interaction with neurons. Front Aging Neurosci 8:116. doi:10.3389/fnagi.2016.00116
Acaz-Fonseca E, Avila-Rodriguez M, Garcia-Segura LM, Barreto GE (2016) Regulation of astroglia by gonadal steroid hormones under physiological and pathological conditions. Prog Neurobiol. doi:10.1016/j.pneurobio.2016.06.002
Iglesias J, Morales L, Barreto GE (2016) Metabolic and inflammatory adaptation of reactive astrocytes: role of PPARs. Mol Neurobiol. doi:10.1007/s12035-016-9833-2
Garzon D, Cabezas R, Vega N, Avila-Rodriguez M, Gonzalez J, Gomez RM, Echeverria V, Aliev G et al (2016) Novel approaches in astrocyte protection: from experimental methods to computational approaches. J Mol Neurosci 58(4):483–492. doi:10.1007/s12031-016-0719-6
Cabezas R, Avila-Rodriguez M, Vega-Vela NE, Echeverria V, Gonzalez J, Hidalgo OA, Santos AB, Aliev G et al (2016) Growth factors and astrocytes metabolism: possible roles for platelet derived growth factor. Med Chem 12(3):204–210
Cabezas R, Avila M, Gonzalez J, El-Bacha RS, Baez E, Garcia-Segura LM, Jurado Coronel JC, Capani F et al (2014) Astrocytic modulation of blood brain barrier: perspectives on Parkinson’s disease. Front Cell Neurosci 8:211. doi:10.3389/fncel.2014.00211
Romero J, Muniz J, Logica Tornatore T, Holubiec M, Gonzalez J, Barreto GE, Guelman L, Lillig CH et al (2014) Dual role of astrocytes in perinatal asphyxia injury and neuroprotection. Neurosci Lett 565:42–46. doi:10.1016/j.neulet.2013.10.046
Barreto GE, White RE, Xu L, Palm CJ, Giffard RG (2012) Effects of heat shock protein 72 (Hsp72) on evolution of astrocyte activation following stroke in the mouse. Exp Neurol 238(2):284–296. doi:10.1016/j.expneurol.2012.08.015
Cabezas R, El-Bacha RS, Gonzalez J, Barreto GE (2012) Mitochondrial functions in astrocytes: neuroprotective implications from oxidative damage by rotenone. Neurosci Res 74(2):80–90. doi:10.1016/j.neures.2012.07.008
Guillamón-Vivancos T, Gómez-Pinedo U, Matías-Guiu J (2013) Astrocytes in neurodegenerative diseases (I): function and molecular description. Neurologia (Barcelona, Spain) 30:1–11. doi:10.1016/j.nrl.2012.12.007
Pekny M, Wilhelmsson U, Pekna M (2014) The dual role of astrocyte activation and reactive gliosis. Neurosci Lett. doi:10.1016/j.neulet.2013.12.071
Huang S, Rutkowsky JM, Snodgrass RG, Ono-Moore KD, Schneider DA, Newman JW, Adams SH, Hwang DH (2012) Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J Lipid Res 53:2002–2013. doi:10.1194/jlr.D029546
Liu C-C, Liu C-C, Kanekiyo T, Xu H, Bu G (2013) Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 9(2):106–118. doi:10.1038/nrneurol.2012.263
Ajuwon KM, Spurlock ME (2005) Palmitate activates the NF-kappaB transcription factor and induces IL-6 and TNFalpha expression in 3 T3-L1 adipocytes. J Nutr 135:1841–1846
Boden G (2008) Obesity and free fatty acids. Endocrinol Metab Clin N Am. doi:10.1016/j.ecl.2008.06.007
Scheller JR, Chalaris A, Schmidt-Arras D, Rose-John S (2011) The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta-Mol Cell Res 1813:878–888. doi:10.1016/j.bbamcr.2011.01.034
Xanthos DN, Sandkühler J (2014) Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci 15:43–53. doi:10.1038/nrn3617
Ben Haim L, Carrillo-de Sauvage M-A, Ceyzériat K, Escartin C (2015) Elusive roles for reactive astrocytes in neurodegenerative diseases. Front Cell Neurosci 9:278. doi:10.3389/fncel.2015.00278
Buckman LB, Hasty AH, Flaherty DK, Buckman CT, Thompson MM, Matlock BK, Weller K, Ellacott KLJ (2014) Obesity induced by a high-fat diet is associated with increased immune cell entry into the central nervous system. Brain Behav Immun 35:33–42. doi:10.1016/j.bbi.2013.06.007
Whitmer RA (2007) The epidemiology of adiposity and dementia. Curr Alzheimer Res 4:117–122. doi:10.2174/156720507780362065
Bruce-Keller AJ, Keller JN, Morrison CD (2009) Obesity and vulnerability of the CNS. Biochim Biophys Acta Mol basis Dis. doi:10.1016/j.bbadis.2008.10.004
Hotamisligil GS (2006) Inflammation and metabolic disorders. Nature 444:860–867. doi:10.1038/nature05485
Awada R, Parimisetty A, DHellencourt C (2008) Influence of obesity on neurodegenerative diseases. Obésité 3:27–32. doi:10.1007/s11690-008-0100-1
Mathis D (2013) Immunological goings-on in visceral adipose tisuue. Cell Metab 17:851–859. doi:10.1016/j.cmet.2013.05.008
Furuhashi M, Fucho R, Görgün CZ, Tuncman G, Cao H, Hotamisligil GS (2008) Adipocyte/macrophage fatty acid-binding proteins contribute to metabolic deterioration through actions in both macrophages and adipocytes in mice. J Clin Investig 118:2640–2650. doi:10.1172/JC134750
Shanik MH, Xu Y, Skrha J, Dankner R, Zick Y, Roth J (2008) Insulin resistance and hyperinsulinemia: is hyperinsulinemia the cart or the horse? Diabetes Care 31(Suppl 2):S262–S268. doi:10.2337/dc08-s264
García-Cáceres C, Quarta C, Varela L, Gao Y, Gruber T, Legutko B, Jastroch M, Johansson P et al (2016) Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell 166(4):867–880. doi:10.1016/j.cell.2016.07.028
Tomas E, Lin Y-S, Dagher Z, Saha A, Luo Z, Ido Y, Ruderman N (2002) Hyperglycemia and insulin resistance: possible mechanisms. Ann N Y Acad Sci 967:43–51. doi:10.1111/j.1749-6632.2002.tb04262.x
Dixit V, Weeraratna A, Hyunwon Y, Bertak D, Jenkins A, Riggins G, Eberhart C, Taub D (2006) Ghrelin and the growth hormone Secretagogue receptor constitute a novel autocrine pathway in astrocytoma motility. J Biol Chem 281(24):16681–16690. doi:10.1021/jp809000e
Hsuchou H, He Y, Kastin AJ, Tu H, Markadakis EN, Rogers RC, Fossier PB, Pan W (2009) Obesity induces functional astrocytic leptin receptors in hypothalamus. Brain 132:889–902. doi:10.1093/brain/awp029
Heni M, Hennige AM, Peter A, Siegel-Axel D, Ordelheide AM, Krebs N, Machicao F, Fritsche A et al (2011) Insulin promotes glycogen storage and cell proliferation in primary human astrocytes. PLoS One 6:e21594. doi:10.1371/journal.pone.0021594
Brown LM, Gent L, Davis K, Clegg DJ (2010) Metabolic impact of sex hormones on obesity. Brain Res 1350:77–85. doi:10.1016/j.brainres.2010.04.056
Rasmussen MH (2010) Obesity, growth hormone and weight loss. Mol Cell Endocrinol 316:147–153. doi:10.1016/j.mce.2009.08.017
Kim JG, Suyama S, Koch M, Jin S, Argente-arizon P, Gao Y, Garcia-Caceres C et al (2014) Leptin signaling in GFAP-expressing adult glia cells regulates hypothalamic neuronal circuits and feeding. Nat Neurosci 17(7):908–910. doi:10.1038/nn.3725.Leptin
Wang Y, Hsuchou H, He Y, Kastin A, Pan W (2015) Role of astrocytes in leptin signaling. J Mol Neurosci 56:829–839. doi:10.1007/s12031-015-0518-5
Garwood CJ, Ratcliffe LE, Morgan SV, Simpson JE, Owens H, Vazquez-Villaseñor I, Heath PR, Romero IA et al (2015) Insulin and IGF1 signalling pathways in human astrocytes in vitro and in vivo; characterisation, subcellular localisation and modulation of the receptors. Mol Brain 8:51. doi:10.1186/s13041-015-0138-6
Bruning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, Klein R, Krone W et al (2000) Role of brain insulin receptor in control of body weight and reproduction. Science 289:2122–2125
Havrankova J, Roth J, Brownstein M (1978) Insulin receptors are widely distributed in the central nervous system of the rat. Nature. doi:10.1038/272827a0
Figlewicz DP, Szot P, Chavez M, Woods SC, Veith RC (1994) Intraventricular insulin increases dopamine transporter mRNA in rat VTA/substantia nigra. Brain Res 644:331–334. doi:10.1016/0006-8993(94)91698-5
Phatnani H, Maniatis T (2015) Astrocytes in neurodegenerative disease. Cold Spring Harb Perspect Biol 7(6):a020628–a020628. doi:10.1101/cshperspect.a020628
A. S. Association: In Alzheimer´s Association (2016) http://www.alz.org/
Hebert L, Weuve J, Scherr P, Evans D (2013) Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 80(19):1778–1783. doi:10.1212/WNL.0b013e31828726f5
Maragakis NJ, Rothstein JD (2006) Mechanisms of disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol 2(12):679–689. doi:10.1038/ncpneuro0355
Selkoe DJ (2000) The genetics and molecular pathology of Alzheimer’s disease—roles of amyloid and the presenilins. Neurol Clin 18(4):903
Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E (2011) Alzheimer’s disease. Lancet 377:1019–1031. doi:10.1016/S0140-6736(10)61349-9
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science (New York, NY) 297(5580):353–356. doi:10.1126/science.1072994
Small SA, Duff K (2008) Linking aβ and tau in late-onset Alzheimer’s disease: a dual pathway hypothesis. Neuron. doi:10.1016/j.neuron.2008.11.007
Walsh DM, Selkoe DJ (2004) Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron. doi:10.1016/j.neuron.2004.09.010
Takeda S, Sato N, Uchio-Yamada K, Sawada K, Kunieda T, Takeuchi D, Kurinami H, Shinohara M et al (2010) Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci U S A 107(15):7036–7041. doi:10.1073/pnas.1000645107
Gustafson D (2006) Adiposity indices and dementia. Lancet Neurol 5(8):713–720. doi:10.1016/S1474-4422(06)70526-9
Whitmer RA, Gustafson DR, Barrett-Connor E, Haan MN, Gunderson EP, Yaffe K (2008) Central obesity and increased risk of dementia more than three decades later. Neurology 71(14):1057–1064. doi:10.1212/01.wnl.0000306313.89165.ef
Gustafson D, Rothenberg E, Blennow K, Steen B, Skoog I (2003) An 18-year follow-up of overweight and risk of Alzheimer disease. Arch Intern Med 163(13):1524–1528. doi:10.1001/archinte.163.13.1524
Chibber S, Alexiou A, Alama MN, Barreto GE, Aliev G, Ashraf GM (2016) A synopsis on the linkage between age-related dementias and vascular disorders. CNS Neurol Disord Drug Targets 15(2):250–258
Gonzalez-Reyes RE, Aliev G, Avila-Rodrigues M, Barreto GE (2016) Alterations in glucose metabolism on cognition: a possible link between diabetes and dementia. Curr Pharm Des 22(7):812–818
Blach-Olszewska Z, Zaczynska E, Gustaw-Rothenberg K, Avila-Rodrigues M, Barreto GE, Leszek J, Aliev G (2015) The innate immunity in Alzheimer disease- relevance to pathogenesis and therapy. Curr Pharm Des 21(25):3582–3588
Li XH, Lv BL, Xie JZ, Liu J, Zhou XW, Wang JZ (2012) AGEs induce Alzheimer-like tau pathology and memory deficit via RAGE-mediated GSK-3 activation. Neurobiol Aging 33:1400–1410. doi:10.1016/j.neurobiolaging.2011.02.003
Milionis HJ, Florentin M, Giannopoulos S (2008) Metabolic syndrome and Alzheimer’s disease: a link to a vascular hypothesis? CNS Spectrums 13(7):606–613
Razay G, Wilcock GK (1994) Hyperinsulinaemia and alzheimer’s disease. Age Ageing 23(5):396–399. doi:10.1093/ageing/23.5.396
Di Paolo G, Kim T-W (2011) Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat Rev Neurosci 12(5):284–296. doi:10.1038/nrn3012
Cattin L, Bordin P, Fonda M, Adamo C, Barbone F, Bovenzi M, Manto A, Pedone C et al (1997) Factors associated with cognitive impairment among older Italian inpatients. Gruppo Italiano di Farmacovigilanza nell’Anziano (G.I.F.A.). J Am Geriatr Soc 45:1324–1330. doi:10.1111/j.1532-5415.1997.tb02931.x
Cournot M, Marquié JC, Ansiau D, Martinaud C, Fonds H, Ferrières J, Ruidavets JB (2006) Relation between body mass index and cognitive function in healthy middle-aged men and women. Neurology 67:1208–1214. doi:10.1212/01.wnl.0000238082.13860.50
Elias MF, Elias PK, Sullivan LM, Wolf PA, D’Agostino RB (2005) Obesity, diabetes and cognitive deficit: the Framingham heart study. Neurobiol Aging. doi:10.1016/j.neurobiolaging.2005.08.019
Elias MF, Elias PK, Sullivan LM, Wolf PA, D’Agostino RB (2003) Lower cognitive function in the presence of obesity and hypertension: the Framingham heart study. Int J Obes Relat Metab Disord: J Int Assoc Study Obes 27:260–268. doi:10.1038/sj.ijo.802225
Gunstad J, Paul RH, Cohen RA, Tate DF, Spitznagel MB, Gordon E (2007) Elevated body mass index is associated with executive dysfunction in otherwise healthy adults. Compr Psychiatry 48:57–61. doi:10.1016/j.comppsych.2006.05.001
Sabia SV, Kivimaki M, Shipley MJ, Marmot MG, Singh-Manoux A (2009) Body mass index over the adult life course and cognition in late midlife: the Whitehall II Cohort Study. Am J Clin Nutr 89:601–607. doi:10.3945/ajcn.2008.26482
Enzinger C, Fazekas F, Matthews PM, Ropele S, Schmidt H, Smith S, Schmidt R (2005) Risk factors for progression of brain atrophy in aging: six-year follow-up of normal subjects. Neurology 64:1704–1711. doi:10.1212/01.WNL.0000161871.83614.BB
Liang X, Wang Q, Hand T, Wu L, Breyer RM, Montine TJ, Andreasson K (2005) Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer’s disease. J Neurosci: Off J Soc Neurosci 25(44):10180–10187. doi:10.1523/JNEUROSCI.3591-05.2005
Taki Y, Kinomura S, Sato K, Inoue K, Goto R, Okada K, Uchida S, Kawashima R et al (2008) Relationship between body mass index and gray matter volume in 1,428 healthy individuals. Obesity (Silver Spring, MD) 16:119–124. doi:10.1038/oby.2007.4
Lynch CM, Kinzenbaw DA, Chen X, Zhan S, Mezzetti E, Filosa J, Ergul A, Faulkner JL et al (2013) Nox2-derived superoxide contributes to cerebral vascular dysfunction in diet-induced obesity. Stroke 44:3195–3201. doi:10.1161/STROKEAHA.113.001366
Molteni R, Barnard RJ, Ying Z, Roberts CK, Gómez-Pinilla F (2002) A high-fat, refined sugar diet reduces hippocampal brain-derived neurotrophic factor, neuronal plasticity, and learning. Neuroscience 112:803–814. doi:10.1016/S0306-4522(02)00123-9
Julien C, Tremblay C, Phivilay A, Berthiaume L, Emond V, Julien P, Calon FDR (2010) High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiol Aging 31:1516–1531. doi:10.1016/j.neurobiolaging.2008.08.022
Leboucher A, Laurent C, Fernandez F, Burnouf S, Troquier L, Eddarkaoui S, Demeyer D, Caillierez R et al (2013) Detrimental effects of diet-induced obesity on tau pathology are independent of insulin resistance in tau transgenic mice. Diabetes. doi:10.2337/db12-0866
Pistell PJ, Morrison CD, Gupta S, Knight AG, Keller JN, Ingram DK, Bruce-Keller AJ (2010) Cognitive impairment following high fat diet consumption is associated with brain inflammation. J Neuroimmunol 219:25–32. doi:10.1016/j.jneuroim.2009.11.010
Puig KL, Floden AM, Adhikari R, Golovko MY, Combs CK (2012) Amyloid precursor protein and proinflammatory changes are regulated in brain and adipose tissue in a murine model of high fat diet-induced obesity. PLoS One 7:e0030378. doi:10.1371/journal.pone.0030378
Thirumangalakudi L, Prakasam A, Zhang R, Bimonte-Nelson H, Sambamurti K, Kindy MS, Bhat NR (2008) High cholesterol-induced neuroinflammation and amyloid precursor protein processing correlate with loss of working memory in mice. J Neurochem 106:475–485. doi:10.1111/j.1471-4159.2008.05415.x
Mrak RE (2009) Alzheimer-type neuropathological changes in morbidly obese elderly individuals. Clin Neuropathol 28:40–45
Gunstad J, Lhotsky A, Wendell CR, Ferrucci L, Zonderman AB (2010) Longitudinal examination of obesity and cognitive function: results from the Baltimore longitudinal study of aging. Neuroepidemiology 34:222–229. doi:10.1159/000297742
Liang J, Matheson BE, Kaye WH, Boutelle KN (2014) Neurocognitive correlates of obesity and obesity-related behaviors in children and adolescents. Int J Obes 38:494–506. doi:10.1038/ijo.2013.142
Morales-Briceño HA, Cervantes-Arriaga AB, Rodríguez-Violante MAB, Calleja-Castillo JB, Corona TB (2012) Overweight is more prevalent in patients with parkinson’s disease [Excesso de peso é mais frequente em pacientes com doença de Parkinson]. Arq Neuropsiquiatr 70:843–846. doi:10.1590/S0004-282X2012001100004
Rivera P, Pérez-Martín M, Pavón FJ, Serrano A, Crespillo A, Cifuentes M, López-Ávalos MD, Grondona JM et al (2013) Pharmacological Administration of the Isoflavone Daidzein Enhances Cell Proliferation and Reduces High Fat Diet-Induced Apoptosis and gliosis in the rat hippocampus. PLoS One 8:e0064750. doi:10.1371/journal.pone.0064750
Li W, Prakash R, Chawla D, Du W, Didion SP, Filosa JA, Zhang Q, Brann DW et al (2013) Early effects of high-fat diet on neurovascular function and focal ischemic brain injury. Am J Physiol Regul Integr Comp Physiol 304:R1001–R1008. doi:10.1152/ajpregu.00523.2012
Pepping JK, Freeman LR, Gupta S, Keller JN, Bruce-Keller AJ (2013) NOX2 deficiency attenuates markers of adiposopathy and brain injury induced by high-fat diet. Am J Physiol Endocrinol Metab 304:E392–E404. doi:10.1152/ajpendo.00398.2012
Lees AJ, Hardy J, Revesz T (2009) Parkinson’s disease. Lancet. doi:10.1016/S0140-6736(09)60492-X
Jurado-Coronel JC, Avila-Rodriguez M, Echeverria V, Hidalgo OA, Gonzalez J, Aliev G, Barreto GE (2016) Implication of green tea as a possible therapeutic approach for Parkinson disease. CNS Neurol Disord Drug Targets 15(3):292–300
dos Santos AB, Kohlmeier KA, Barreto GE (2015) Are sleep disturbances preclinical markers of Parkinson’s disease? Neurochem Res 40(3):421–427. doi:10.1007/s11064-014-1488-7
Sutachan JJ, Casas Z, Albarracin SL, Stab BR 2nd, Samudio I, Gonzalez J, Morales L, Barreto GE (2012) Cellular and molecular mechanisms of antioxidants in Parkinson’s disease. Nutr Neurosci 15(3):120–126. doi:10.1179/1476830511Y.0000000033
Albarracin SL, Stab B, Casas Z, Sutachan JJ, Samudio I, Gonzalez J, Gonzalo L, Capani F et al (2012) Effects of natural antioxidants in neurodegenerative disease. Nutr Neurosci 15(1):1–9. doi:10.1179/1476830511Y.0000000028
Statistics on Parkinson’s (2016) Parkinson’s Disease Foundation, Inc. http://www.pdf.org/en/parkinson_statistics
Braak H, Del Tredici K, Rüb U, De Vos RAI, Jansen Steur ENH, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24(2):197–211. doi:10.1016/S0197-4580(02)00065-9
Hawkes CH, Del Tredici K, Braak H (2007) Parkinson’s disease: a dual-hit hypothesis. Neuropathol Appl Neurobiol. doi:10.1111/j.1365-2990.2007.00874.x
Nandhagopal R, Kuramoto L, Schulzer M, Mak E, Cragg J, Lee CS, McKenzie J, McCormick S et al (2009) Longitudinal progression of sporadic Parkinson’s disease: a multi-tracer positron emission tomography study. Brain 132(11):2970–2979. doi:10.1093/brain/awp209
Logroscino G, Marder K, Cote L, Tang MX, Shea S, Mayeux R (1996) Dietary lipids and antioxidants in Parkinson’s disease: a population-based, case-control study. Ann Neurol 39:89–94. doi:10.1002/ana.410390113
Hu G, Hu G, Pekkarinen H, Hänninen O, Tian H, Jin R (2002) Comparison of dietary and non-dietary risk factors in overweight and normal-weight Chinese adults. Br J Nutr 88:91–97. doi:10.1079/BJNBJN2002590
Hu G, Pekkarinen H, Hänninen O, Yu Z, Guo Z, Tian H (2002) Commuting, leisure-time physical activity, and cardiovascular risk factors in China. Med Sci Sports Exerc 34(2):234–238. doi:10.1097/00005768-200202000-00009
Chen H, Zhang SM, Schwarzschild MA, Hernán MA, Ascherio A (2005) Physical activity and the risk of Parkinson disease. Neurology 64(4):664–669. doi:10.1212/01.WNL.0000151960.28687.93
Meguid MM, Fetissov SO, Varma M, Sato T, Zhang L, Laviano A, Rossi-Fanelli F (2000) Hypothalamic dopamine and serotonin in the regulation of food intake. Nutrition (Burbank, Los Angeles County, Calif) 16(10):843–857
Schwartz MW, Woods SC, Porte D, Seeley RJ, Baskin DG (2000) Central nervous system control of food intake. Nature 404(6778):661–671. doi:10.1038/35007534
Wang GJ, Volkow ND, Logan J, Pappas NR, Wong CT, Zhu W, Netusll N, Fowler JS (2001) Brain dopamine and obesity. Lancet 357(9253):354–357. doi:10.1016/S0140-6736(00)03643-6
Langston J, Forno L (1978) The hypothalamus in Parkinson’s disease. Int J Neurosci 33(0020–7454):257–259
Shannak K, Rajput A, Rozdilsky B, Kish S, Gilbert J, Hornykiewicz O (1994) Noradrenaline, dopamine and serotonin levels and metabolism in the human hypothalamus: observations in Parkinson’s disease and normal subjects. Brain Res 639(1):33–41
de Weijer BA, van de Giessen E, van Amelsvoort TA, Boot E, Braak B, Janssen IM, van de Laar A, Fliers E et al (2011) Lower striatal dopamine D2/3 receptor availability in obese compared with non-obese subjects. EJNMMI Res 1:37. doi:10.1186/2191-219X-1-37
Martel P, Fantino M (1996) Mesolimbic dopaminergic system activity as a function of food reward: a microdialysis study. Pharmacol Biochem Behav 53(1):221–226. doi:10.1016/0091-3057(95)00187-5
Abbott RD, Ross GW, White LR, Nelson JS, Masaki KH, Tanner CM, Curb JD, Blanchette PL et al (2002) Midlife adiposity and the future risk of Parkinson’s disease. Neurology 59:1051–1057. doi:10.1212/WNL.59.7.1051
Chen H, Zhang S, Schwarzschild M, Hernan M, Willett W, Ascherio A (2004) Obesity and the risk of Parkinson’s disease. Am J Epidemiol 159:547–555. doi:10.1093/aje/kwh059
Bachmann CG, Zapf A, Brunner E, Trenkwalder C (2009) Dopaminergic treatment is associated with decreased body weight in patients with Parkinson’s disease and dyskinesias. Eur J Neurol 16:895–901. doi:10.1111/j.1468-1331.2009.02617.x
Bousquet M, St-Amour I, Vandal M, Julien P, Cicchetti F, Calon F (2012) High-fat diet exacerbates MPTP-induced dopaminergic degeneration in mice. Neurobiol Dis 45:529–538. doi:10.1016/j.nbd.2011.09.009
Choi JY, Jang EH, Park CS, Kang JH (2005) Enhanced susceptibility to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity in high-fat diet-induced obesity. Free Radic Biol Med 38:806–816. doi:10.1016/j.freeradbiomed.2004.12.008
Morris JK, Bomhoff GL, Stanford JA, Geiger PC (2010) Neurodegeneration in an animal model of Parkinson’s disease is exacerbated by a high-fat diet. Am J Physiol Regul Integr Comp Physiol 299:R1082–R1090. doi:10.1152/ajpregu.00449.2010
Driver J, Smith A, Buring J (2008) Prospective cohort study of type 2 diabetes and the risk of Parkinson’s disease. Diabetes 31:8–10. doi:10.2337/dc08-0688
Palacios N, Gao X, McCullough ML, Jacobs EJ, Patel AV, Mayo T, Schwarzschild MA, Ascherio A (2011) Obesity, diabetes, and risk of Parkinson’s disease. Mov Disord: Off J Mov Dis Soc 26:2253–2259. doi:10.1002/mds.23855
Correale J, Villa A (2004) The neuroprotective role of inflammation in nervous system injuries. J Neurol 251:1304–1316. doi:10.1007/s00415-004-0649-z
Farooqui AA, Horrocks LA, Farooqui T (2007) Modulation of inflammation in brain: a matter of fat. J Neurochem 101:577–599. doi:10.1111/j.1471-4159.2006.04371.x
Echeverria V, Grizzell JA, Barreto GE (2016) Neuroinflammation: a therapeutic target of cotinine for the treatment of psychiatric disorders? Curr Pharm Des 22(10):1324–1333
Leszek J, Barreto GE, Gasiorowski K, Koutsouraki E, Avila-Rodrigues M, Aliev G (2016) Inflammatory mechanisms and oxidative stress as key factors responsible for progression of neurodegeneration: role of brain innate immune system. CNS Neurol Disord Drug Targets 15(3):329–336
Carson MJ, Thrash JC, Walter B (2006) Survival. Clin Neurosci 6(5):237–245. doi:10.1016/j.cnr.2006.09.004.The
Ransohoff RM, Schafer D, Vincent A, Blachère NE, Bar-Or A (2015) neuroinflammation: ways in which the immune system affects the brain. Neurotherapeutics 12:896–909. doi:10.1007/s13311-015-0385-3
Adelson JD, Barreto GE, Xu L, Kim T, Brott BK, Ouyang YB, Naserke T, Djurisic M et al (2012) Neuroprotection from stroke in the absence of MHCI or PirB. Neuron 73(6):1100–1107. doi:10.1016/j.neuron.2012.01.020
Streit WJ, Mrak RE, Griffin WST (2004) Microglia and neuroinflammation: a pathological perspective. J Neuroinflam 1:–14. doi:10.1186/1742-2094-1-14
Lo D, Feng L, Li L, Carson MJ, Crowley M, Pauza M, Nguyen A, Reilly CR (1999) Integrating innate and adaptive immunity in the whole animal. Immunol Rev 169:225–239. doi:10.1111/j.1600-065X.1999.tb01318.x
Amor S, Puentes F, Baker D, Van Der Valk P (2010) Inflammation in neurodegenerative diseases. Immunology. doi:10.1111/j.1365-2567.2009.03225.x
Hein AM, O’Banion MK (2009) Neuroinflammation and memory: the role of prostaglandins. Mol Neurobiol. doi:10.1007/s12035-009-8066-z
Kohman RA, Rhodes JS (2013) Neurogenesis, inflammation and behavior. Brain Behav Immun 27:22–32. doi:10.1016/j.bbi.2012.09.003
Rao JS, Kellom M, Kim HW, Rapoport SI, Reese EA (2012) Neuroinflammation and synaptic loss. Neurochem Res. doi:10.1007/s11064-012-0708-2
Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL, Araoz C (1989) Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci U S A 86:7611–7615. doi:10.1073/pnas.86.19.7611
Mayer CM, Belsham DD (2010) Palmitate attenuates insulin signaling and induces endoplasmic reticulum stress and apoptosis in hypothalamic neurons: rescue of resistance and apoptosis through adenosine 5′ monophosphate-activated protein kinase activation. Endocrinology 151:576–585. doi:10.1210/en.2009-1122
Gregor MF, Hotamisligil GS (2011) Inflammatory mechanisms in obesity. Annu Rev Immunol 29:415–445. doi:10.1146/annurev-immunol-031210-101322
Arruda AP, Milanski M, Coope A, Torsoni AS, Ropelle E, Carvalho DP, Carvalheira JB, Velloso LA (2011) Low-grade hypothalamic inflammation leads to defective thermogenesis, insulin resistance, and impaired insulin secretion. Endocrinology 152(4):1314–1326. doi:10.1210/en.2010-0659
Milanski M, Arruda AP, Coope A, Ignacio-Souza LM, Nunez CE, Roman EA, Romanatto T, Pascoal LB et al (2012) Inhibition of hypothalamic inflammation reverses diet-induced insulin resistance in the liver. Diabetes 61(6):1455–1462. doi:10.2337/db11-0390
Sartorius T, Lutz SZ, Hoene M, Waak J, Peter A, Weigert C, Rammensee HG, Kahle PJ et al (2012) Toll-like receptors 2 and 4 impair insulin-mediated brain activity by interleukin-6 and osteopontin and alter sleep architecture. FASEB J 26(5):1799–1809. doi:10.1096/fj.11-191023
Posey KA, Clegg DJ, Printz RL, Byun J, Morton GJ, Vivekanandan-Giri A, Pennathur S, Baskin DG et al (2009) Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab 296:E1003–E1012. doi:10.1152/ajpendo.90377.2008
Hayden MS, Ghosh S (2008) Shared principles in NF-kappaB signaling. Cell 132:344–362. doi:10.1016/j.cell.2008.01.020
Kleinridders A, Schenten D, Könner AC, Belgardt BF, Mauer J, Okamura T, Wunderlich FT, Medzhitov R et al (2009) MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab 10(4):249–259. doi:10.1016/j.cmet.2009.08.013
Pellerin L, Magistretti PJ (2004) Neuroenergetics: calling upon astrocytes to satisfy hungry neurons. Neuroscientist: Rev J Bringing Neurobiol, Neurol Psychiatry 10:53–62. doi:10.1177/1073858403260159
Ridet JL, Alonso G, Chauvet N, Chapron J, Koenig J, Privat A (1996) Immunocytochemical characterization of a new marker of fibrous and reactive astrocytes. Cell Tissue Res 283:39–49. doi:10.1007/s004410050510
Cintra DE, Ropelle ER, Moraes JC, Pauli JR, Morari J, de Souza CT, Grimaldi R, Stahl M et al (2012) Unsaturated fatty acids revert diet-induced hypothalamic inflammation in obesity. PLoS One 7(1):e30571. doi:10.1371/journal.pone.0030571
Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, Myers MG, Ozcan U (2009) Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab 9(1):35–51. doi:10.1016/j.cmet.2008.12.004
Mazzanti M, Sul JY, Haydon PG (2001) Glutamate on demand: astrocytes as a ready source. Neuroscientist: Rev J Bringing Neurobiol, Neurol Psychiatry 7:396–405. doi:10.1177/107385840100700509
Romanatto T, Roman EA, Arruda AP, Denis RG, Solon C, Milanski M, Moraes JC, Bonfleur ML et al (2009) Deletion of tumor necrosis factor-α receptor 1 (TNFR1) protects against diet-induced obesity by means of increased thermogenesis. J Biol Chem 284:36213–36222. doi:10.1074/jbc.M109.030874
Pasti L (1997) A. Volterra, T. Pozzan and G. Carmignoto: intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J Neurosci: Off J Soc Neurosci 17:7817–7830. doi:10.1038/nrn1722
Araque A, Parpura V, Sanzgiri RP, Haydon PG (1999) Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci 22:208–215. doi:10.1016/S0166-2236(98)01349-6
Araque A, Parpura V, Sanzgiri RP, Haydon PG (1998) Glutamate-dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. Eur J Neurosci 10:2129–2142. doi:10.1046/j.1460-9568.1998.00221.x
Bachoo RM, Kim RS, Ligon KL, Maher EA, Brennan C, Billings N, Chan S, Li C et al (2004) Molecular diversity of astrocytes with implications for neurological disorders. Proc Natl Acad Sci U S A 101:8384–8389. doi:10.1073/pnas.0402140101
Milanski M, Degasperi G, Coope A, Morari J, Denis R, Cintra DE, Tsukumo DML, Anhe G et al (2009) Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci: Off J Soc Neurosci 29:359–370. doi:10.1523/JNEUROSCI.2760-08.2009
De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, Saad MJA, Velloso LA (2005) Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology 146(10):4192–4199. doi:10.1210/en.2004-1520
Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, Zhao X, Sarruf DA et al (2012) Obesity is associated with hypothalamic injury in rodents and humans. J Clin Investig 122(1):153–162. doi:10.1172/JCI59660
Velloso LA, Araújo EP, De Souza CT (2008) Diet-induced inflammation of the hypothalamus in obesity. Neuroimmunomodulation 15:189–193. doi:10.1159/000153423
Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, Higgs R, Liu F et al (2004) Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med 10(7):719–726. doi:10.1038/nm1058
Simard AR, Soulet D, Gowing G, Julien JP, Rivest S (2006) Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 49(4):489–502. doi:10.1016/j.neuron.2006.01.022
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P et al (2000) Inflammation and Alzheimer’s disease. Neurobiol Aging. doi:10.1016/S0197-4580(00)00124-X
Patel NS, Paris D, Mathura V, Quadros AN, Crawford FC, Mullan MJ (2005) Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer’s disease. J Neuroinflammation 2:9. doi:10.1186/1742-2094-2-9
Tarkowski E, Andreasen N, Tarkowski A, Blennow K (2003) Intrathecal inflammation precedes development of Alzheimer’s disease. J Neurol Neurosurg Psychiatry 74:1200–1205. doi:10.1136/jnnp.74.9.1200
Yaffe K, Kanaya A, Lindquist K, Simonsick EM, Harris T, Shorr RI, Tylavsky FA, Newman AB (2004) The metabolic syndrome, inflammation, and risk of cognitive decline. JAMA: J Am Med Assoc 292(18):2237–2242. doi:10.1016/j.accreview.2004.12.135
Krstic D, Knuesel I (2013) Deciphering the mechanism underlying late-onset Alzheimer disease. Nat Rev Neurol 9:25–34. doi:10.1038/nrneurol.2012.236
Schwab C, Klegeris A, McGeer PL (2010) Inflammation in transgenic mouse models of neurodegenerative disorders. Biochim Biophys Acta 1802:889–902. doi:10.1016/j.bbadis.2009.10.013
Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J (2003) Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med 9(4):453–457. doi:10.1038/nm838
Janelsins MC, Mastrangelo MA, Oddo S, LaFerla FM, Federoff HJ, Bowers WJ (2005) Early correlation of microglial activation with enhanced tumor necrosis factor-alpha and monocyte chemoattractant protein-1 expression specif- ically within the entorhinal cortex of triple transgenic Alzheimer’s disease mice. J Neuroinflammation 2:23. doi:10.1186/1742-2094-2-23
Hoozemans JJM, Rozemuller AJM, van Haastert ES, Eikelenboom P, van Gool WA (2011) Neuroinflammation in Alzheimer’s disease wanes with age. J Neuroinflammation 8:171. doi:10.1186/1742-2094-8-171
Yaffe K, Weston AL, Blackwell T, Krueger KA (2009) The metabolic syndrome and development of cognitive impairment among older women. Arch Neurol 66(3):324–328. doi:10.1001/archneurol.2008.566
Hirsch EC, Hunot S (2009) Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. doi:10.1016/S1474-4422(09)70062-6
Hirsch EC, Vyas S, Hunot S (2012) Neuroinflammation in Parkinson’s disease. Parkinsonism Relat Disord 18:S210–S212. doi:10.1016/S1353-8020(11)70065-7
Ros-Bernal F, Hunot S, Herrero MT, Parnadeau S, Corvol J-C, Lu L, Alvarez-Fischer D, Carrillo-de Sauvage MA et al (2011) Microglial glucocorticoid receptors play a pivotal role in regulating dopaminergic neurodegeneration in parkinsonism. Proc Natl Acad Sci U S A 108(16):6632–6637. doi:10.1073/pnas.1017820108
Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ (2008) Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol Aging 29:1690–1701. doi:10.1016/j.neurobiolaging.2007.04.006
Bandopadhyay R, Kingsbury AE, Cookson MR, Reid AR, Evans IM, Hope AD, Pittman AM, Lashley T et al (2004) The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson’s disease. Brain 127:420–430. doi:10.1093/brain/awh054
Waak J, Weber SS, Waldenmaier A, Görner K, Alunni-Fabbroni M, Schell H, Vogt-Weisenhorn D, Pham T-T et al (2009) Regulation of astrocyte inflammatory responses by the Parkinson’s disease-associated gene DJ-1. FASEB J: Off Publ Fed Am Soc Exp Biol 23:2478–2489. doi:10.1096/fj.08-125153
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y et al (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392(6676):605–608. doi:10.1038/33416
Frank-Cannon TC, Tran T, Ruhn KA, Martinez TN, Hong J, Marvin M, Hartley M, Treviño I et al (2008) Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J Neurosci 28:10825–10834. doi:10.1523/JNEUROSCI.3001-08.2008
Rodríguez-Navarro JA, Casarejos MJ, Menéndez J, Solano RM, Rodal I, Gómez A, Yébenes JGD, Mena MA (2007) Mortality, oxidative stress and tau accumulation during ageing in parkin null mice. J Neurochem 103:98–114. doi:10.1111/j.1471-4159.2007.04762.x
Wang Q, Liu Y, Zhou J (2015) Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl Neurodegeneration 4:19. doi:10.1186/s40035-015-0042-0
Torrente D, Cabezas R, Avila MF, Garcia-Segura LM, Barreto GE, Guedes RC (2014) Cortical spreading depression in traumatic brain injuries: is there a role for astrocytes? Neurosci Lett 565:2–6. doi:10.1016/j.neulet.2013.12.058
J. Kriz (2013) Neuron-Glia Interaction in Neuroinflammation, 7, 75–89 doi:10.1007/978-1-4614-8313-7
Verkhratsky A, Butt A (2013) Glial physiology and pathophysiology. Glial Physiol Pathophysiol. doi:10.1002/9781118402061
Posada-Duque RA, Barreto GE, Cardona-Gomez GP (2014) Protection after stroke: cellular effectors of neurovascular unit integrity. Front Cell Neurosci 8:231. doi:10.3389/fncel.2014.00231
Mucke L, Eddleston M (1993) Astrocytes in infectious and immune-mediated diseases of the central nervous system. FASEB J: Off Publ Fed Am Soc Exp Biol 7:1226–1232
Aschner M (1998) Immune and inflammatory responses in the CNS: modulation by astrocytes. Toxicol Lett 102–103:283–287. doi:10.1016/S0378-4274(98)00324-5
Kimelberg HK (2007) Supportive or information-processing functions of the mature protoplasmic astrocyte in the mammalian CNS? A critical appraisal. Neuron Glia Biol 3:181–189. doi:10.1017/S1740925X08000094
Sonnewald U, Westergaard N, Schousboe A (1995) Metabolic trafficking between neurons and astrocytes: the glutamate/glutamine cycle revisited. Dev Neurosci 17(4):203–211
Pellerin L, Bouzier-Sore AK, Aubert A, Serres S, Merle M, Costalat R, Magistretti PJ (2007) Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia. doi:10.1002/glia.20528
Boyarsky G, Ransom B, Schlue WR, Davis MB, Boron WF (1993) Intracellular pH regulation in single cultured astrocytes from rat forebrain. Glia 8:241–248. doi:10.1002/glia.440080404
Westergaard N, Schousboe A, Sonnewald U, Petersen SB, Yu AC, Hertz L (1992) Regulatory role of astrocytes for neuronal biosynthesis and homeostasis of glutamate and GABA. Prog Brain Res 94:199–211
Owens T, Babcock AA, Millward JM, Toft-Hansen H (2005) Cytokine and chemokine inter-regulation in the inflamed or injured CNS. Brain Res Rev. doi:10.1016/j.brainresrev.2004.12.007
Frontczak-Baniewicz M, Walski M (2006) Glial scar instability after brain injury. J Physiol Pharmacol 57:97–102
Ransom B, Behar T, Nedergaard M (2003) New roles for astrocytes (stars at last). Trends Neurosci. doi:10.1016/j.tins.2003.08.006
Parpura V, Zorec R (2010) Gliotransmission: Exocytotic release from astrocytes. Brain Res Rev. doi:10.1016/j.brainresrev.2009.11.008
Anderson MF, Blomstrand F, Blomstrand C, Eriksson PS, Nilsson M (2003) Astrocytes and stroke: networking for survival? Neurochem Res 28:293–305. doi:10.1023/A:1022385402197
Dong Y, Benveniste EN (2001) Immune function of astrocytes. Glia 36:180–190. doi:10.1002/glia.1107
Araya R, Kudo M, Kawano M, Ishii K, Hashikawa T, Iwasato T, Itohara S, Terasaki T et al (2008) BMP signaling through BMPRIA in astrocytes is essential for proper cerebral angiogenesis and formation of the blood-brain-barrier. Mol Cell Neurosci 38(3):417–430. doi:10.1016/j.mcn.2008.04.003
Argaw AT, Gurfein BT, Zhang Y, Zameer A, John GR (2009) VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc Natl Acad Sci U S A 106(6):1977–1982. doi:10.1073/pnas.0808698106
Sofroniew MV (2015) Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci 16(5):249–263. doi:10.1038/nrn3898
Barnham KJ, Masters CL, Bush AI (2004) Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov 3:205–214. doi:10.1038/nrd1330
Clark RA, Valente AJ (2004) Nuclear factor kappa B activation by NADPH oxidases. Mech Ageing Dev 125:799–810. doi:10.1016/j.mad.2004.08.009
Freeman LR, Zhang L, Nair A, Dasuri K, Francis J, Fernandez-Kim SO, Bruce-Keller AJ, Keller JN (2013) Obesity increases cerebrocortical reactive oxygen species and impairs brainfunction. Free Radic Biol Med 56:226–233. doi:10.1016/j.freeradbiomed.2012.08.577
Morrison CD, Pistell PJ, Ingram DK, Johnson WD, Liu Y, Fernandez-Kim SO, White CL, Purpera MN et al (2010) High fat diet increases hippocampal oxidative stress and cognitive impairment in aged mice: implications for decreased Nrf2 signaling. J Neurochem 114:1581–1589. doi:10.1111/j.1471-4159.2010.06865.x
Farina C, Aloisi F, Meinl E (2007) Astrocytes are active players in cerebral innate immunity. Trends Immunol. doi:10.1016/j.it.2007.01.005
Herx LM, Yong VW (2001) Interleukin-1 beta is required for the early evolution of reactive astrogliosis following CNS lesion. J Neuropathol Exp Neurol 60:961–971
Herx LM, Rivest S, Yong VW (2000) Central nervous system-initiated inflammation and neurotrophism in trauma: IL-1 beta is required for the production of ciliary neurotrophic factor. J Immunol (Baltimore, MD: 1950) 165:2232–2239. doi:10.4049/jimmunol.165.4.2232
Mason JL, Suzuki K, Chaplin DD, Matsushima GK (2001) Interleukin-1beta promotes repair of the CNS. J Neurosci 21:7046–7052
Penkowa M, Moos T, Carrasco J, Hadberg H, Molinero A, Bluethmann H, Hidalgo J (1999) Strongly compromised inflammatory response to brain injury in interleukin-6-deficient mice. Glia 25:343–357. doi:10.1002/(SICI)1098-1136(19990215)25:4<343::AID-GLIA4>3.0.CO;2-V
Swartz KR, Liu F, Sewell D, Schochet T, Campbell I, Sandor M, Fabry Z (2001) Interleukin-6 promotes post-traumatic healing in the central nervous system. Brain Res 896:86–95. doi:10.1016/S0006-8993(01)02013-3
Liberto CM, Albrecht PJ, Herx LM, Yong VW, Levison SW (2004) Pro-regenerative properties of cytokine-activated astrocytes. J Neurochem. doi:10.1111/j.1471-4159.2004.02420.x
Bsibsi M, Persoon-Deen C, Verwer RWH, Meeuwsen S, Ravid R, Van Noort JM (2006) Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia 53:688–695. doi:10.1002/glia.20328
Tran PB, Miller RJ (2003) Chemokine receptors: signposts to brain development and disease. Nat Rev Neurosci 4:444–455
Ambrosini E, Aloisi F (2004) Chemokines and glial cells: a complex network in the central nervous system. Neurochem Res. doi:10.1023/B:NERE.0000021246.96864.89
Babcock AA, Kuziel WA, Rivest S, Owens T (2003) Chemokine expression by glial cells directs leukocytes to sites of axonal injury in the CNS. J Neurosci 23:7922–7930
Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G et al (2006) Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci 9:917–924. doi:10.1038/nn1715
Gimenez MA, Sim J, Archambault AS, Klein RS, Russell JH (2006) A tumor necrosis factor receptor 1-dependent conversation between central nervous system-specific T cells and the central nervous system is required for inflammatory infiltration of the spinal cord. Am J Pathol 168:1200–1209. doi:10.2353/ajpath.2006.050332
Han HS, Suk K (2005) The function and integrity of the neurovascular unit rests upon the integration of the vascular and inflammatory cell systems. Curr Neurovasc Res. doi:10.2174/156720205774962647
Koehler RC, Gebremedhin D, Harder DR (2006) Role of astrocytes in cerebrovascular regulation. J Appl Physiol (Bethesda, MD: 1985) 100:307–317. doi:10.1152/japplphysiol.00938.2005
Prat A, Biernacki K, Wosik K, Antel JP (2001) Glial cell influence on the human blood-brain barrier. Glia 36:145–155. doi:10.1002/glia.1104
Van Kralingen C, Kho DT, Costa J, Angel CE, Graham ES (2013) Exposure to inflammatory cytokines IL-1β and TNFα induces compromise and death of astrocytes; implications for chronic neuroinflammation. PLoS One 8:e0084269. doi:10.1371/journal.pone.0084269
Journiac N, Jolly S, Jarvis C, Gautheron V, Rogard M, Trembleau A, Blondeau J-P, Mariani J et al (2009) The nuclear receptor ROR(alpha) exerts a bi-directional regulation of IL-6 in resting and reactive astrocytes. Proc Natl Acad Sci U S A 106:21365–21370. doi:10.1073/pnas.0911782106
Le Dréau G, Kular L, Nicot AB, Calmel C, Melik-Parsadaniantz S, Kitabgi P, Laurent M, Martinerie C (2010) NOV/CCN3 upregulates CCL2 and CXCL1 expression in astrocytes through β1 and β5 integrins. Glia 58:1510–1521. doi:10.1002/glia.21025
Medeiros R, LaFerla FM (2013) Astrocytes: conductors of the Alzheimer disease neuroinflammatory symphony. Exp Neurol. doi:10.1016/j.expneurol.2012.10.007
Kim KC, Hyun Joo S, Shin CY (2011) CPEB1 modulates lipopolysaccharide-mediated iNOS induction in rat primary astrocytes. Biochem Biophys Res Commun 409:687–692. doi:10.1016/j.bbrc.2011.05.065
Koyama Y, Mizobata T, Yamamoto N, Hashimoto H, Matsuda T, Baba A (1999) Endothelins stimulate expression of cyclooxygenase 2 in rat cultured astrocytes. J Neurochem 73:1004–1011. doi:10.1046/j.1471-4159.1999.0731004.x
Echeverria V, Clerman A, Dore S (2005) Stimulation of PGE receptors EP2 and EP4 protects cultured neurons against oxidative stress and cell death following beta-amyloid exposure. Eur J Neurosci 22(9):2199–2206. doi:10.1111/j.1460-9568.2005.04427.x
Luheshi NM, Rothwell NJ, Brough D (2009) Dual functionality of interleukin-1 family cytokines: implications for anti-interleukin-1 therapy. Br J Pharmacol 157:1318–1329. doi:10.1111/j.1476-5381.2009.00331.x
Rothhammer V, Quintana FJ (2015) Control of autoimmune CNS inflammation by astrocytes. Semin Immunopathol. doi:10.1007/s00281-015-0515-3
Gadani SP, Cronk J (2013) Interleukin-4: a cytokine to remember. J Immunol 189:4213–4219. doi:10.4049/jimmunol.1202246.Interleukin-4
Hirota H, Kiyama H, Kishimoto T, Taga T (1996) Accelerated nerve regeneration in mice by upregulated expression of interleukin (IL) 6 and IL-6 receptor after trauma. J Exp Med 183(6):2627–2634. doi:10.1084/jem.183.6.2627
Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A (2001) Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol 19:683–765. doi:10.1146/annurev.immunol.19.1.683
de Waal Malefyt R, Figdor CG, Huijbens R, Mohan-Peterson S, Bennett B, Culpepper J, Dang W, Zurawski G et al (1993) Effects of IL-13 on phenotype, cytokine production, and cytotoxic function of human monocytes. Comparison with IL-4 and modulation by IFN-gamma or IL-10. J Immunol (Baltimore, MD: 1950) 151(11):6370–6381
Wynn TA (2003) IL-13 effector functions. Annu Rev Immunol 21:425–456. doi:10.1146/annurev.immunol.21.120601.141142
Jensen CJ, Massie A, De Keyser J (2013) Immune players in the CNS: the astrocyte. J NeuroImmune Pharmacol 8:824–839. doi:10.1007/s11481-013-9480-6
Gorina R, Font-Nieves M, Márquez-Kisinousky L, Santalucia T, Planas AM (2010) Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia 59:242–255. doi:10.1002/glia.21094
Schroder K, Hertzog PJ, Ravasi T, Hume DA (2004) Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 75(2):163–189. doi:10.1189/jlb.0603252
Pfeffer K (2003) Biological functions of tumor necrosis factor cytokines and their receptors. Cytokine Growth Factor Rev 14:185–191. doi:10.1016/S1359-6101(03)00022-4
Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA (2008) Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med 14(6):681–687. doi:10.1038/nm1781
McKimmie CS, Graham GJ (2010) Astrocytes modulate the chemokine network in a pathogen-specific manner. Biochem Biophys Res Commun 394:1006–1011. doi:10.1016/j.bbrc.2010.03.111
Park C, Lee S, Cho IH, Lee HK, Kim D, Choi SY, Oh SB, Park K et al (2006) TLR3-mediated signal induces proinflammatory cytokine and chemokine gene expression in astrocytes: differential signaling mechanisms of TLR3-induced IP-10 and IL-8 gene expression. Glia 53:248–256. doi:10.1002/glia.20278
Calderon TM, Eugenin EA, Lopez L, Kumar SS, Hesselgesser J, Raine CS, Berman JW (2006) A role for CXCL12 (SDF-1alpha) in the pathogenesis of multiple sclerosis: regulation of CXCL12 expression in astrocytes by soluble myelin basic protein. J Neuroimmunol 177:27–39. doi:10.1016/j.jneuroim.2006.05.003
Tohyama M, Sayama K, Komatsuzawa H, Hanakawa Y, Shirakata Y, Dai X, Yang L, Tokumaru S et al (2007) CXCL16 is a novel mediator of the innate immunity of epidermal keratinocytes. Int Immunol 19:1095–1102. doi:10.1093/intimm/dxm083
Smith KJ, Lassmann H (2002) The role of nitric oxide in multiple sclerosis. Lancet Nuerol 1:232–241. doi:10.1016/S1474-4422(02)00102-3
Meda L (2001) Glial activation in Alzheimer’s disease: the role of AÎ2 and its associated proteins. Neurobiol Aging 22(6):885–893. doi:10.1016/S0197-4580(01)00307-4
DeWitt DA, Perry G, Cohen M, Doller C, Silver J (1998) Astrocytes regulate microglial phagocytosis of senile plaque cores of Alzheimer’s disease. Exp Neurol 149(2):329–340. doi:10.1006/exnr.1997.6738
Nagele RG, Wegiel J, Venkataraman V, Imaki H, Wang KC (2004) Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol Aging 25(5):663–674. doi:10.1016/j.neurobiolaging.2004.01.007
Bamberger ME, Landreth GE (2001) Microglial interaction with B-amyloid: implications for the pathogenesis of Alzheimer’s disease. Microsc Res Tech 70:59–70. doi:10.1002/jemt.1121
Nagele RG, D’Andrea MR, Lee H, Venkataraman V, Wang H-Y (2003) Astrocytes accumulate a beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res 971(2):197–209. doi:10.1016/S0006-8993(03)02361-8
Olabarria M, Noristani HN, Verkhratsky A, Rodríguez JJ (2010) Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 58(7):831–838. doi:10.1002/glia.20967
Simpson JE, Ince PG, Lace G, Forster G, Shaw PJ, Matthews F, Savva G, Brayne C et al (2010) Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol Aging 31(4):578–590. doi:10.1016/j.neurobiolaging.2008.05.015
Pihlaja R, Koistinaho J, Malm T, Sikkila H, Vainio S, Koistinaho M (2008) Transplanted astrocytes internalize deposited beta-amyloid peptides in a transgenic mouse model of Alzheimer’s disease. Glia 56(2):154–163. doi:10.1002/glia.20599
Pihlaja R, Koistinaho J, Kauppinen R, Sandholm J, Tanila H, Koistinaho M (2011) Multiple cellular and molecular mechanisms are involved in human Aβ clearance by transplanted adult astrocytes. Glia 59(11):1643–1657. doi:10.1002/glia.21212
Haughey, Mattson (2003) Alzheimer’s amyloid β-peptide enhances ATP/gap junction-mediated calcium-wave propagation in astrocytes. Neruomol Med 3:173–180. doi:10.1385/NMM:3:3:173
Johnston JM, Burnett P, Thomas AP, Tezapsidis N (2006) Calcium oscillations in type-1 astrocytes, the effect of a presenilin 1 (PS1) mutation. Neurosci Lett 395(2):159–164. doi:10.1016/j.neulet.2005.10.088
McGeer PL, McGeer EG (2008) Glial reactions in Parkinson’s disease. Mov Discord. doi:10.1002/mds.21751
Mena MA, García de Yébenes J (2008) Glial cells as players in parkinsonism: the “good,” the “bad,” and the “mysterious” glia. Neuroscientist: Rev J Bringing Neurobiol, Neurol Psychiatry 14(6):544–560. doi:10.1177/1073858408322839
Halliday GM, Stevens CH (2011) Glia: initiators and progressors of pathology in Parkinson’s disease. Mov Disord 26(1):6–17. doi:10.1002/mds.23455
Verkhratsky A, Olabarria M, Noristani HN, Yeh CY, Rodriguez JJ (2010) Astrocytes in Alzheimer’s disease. Neurotherapeutics 7(4):399–412. doi:10.1016/j.nurt.2010.05.017
Barreto GE, Iarkov A, Moran VE (2014) Beneficial effects of nicotine, cotinine and its metabolites as potential agents for Parkinson’s disease. Front Aging Neurosci 6:340. doi:10.3389/fnagi.2014.00340
Damier P, Hirsch EC, Zhang P, Agid Y, Javoy-Agid F (1993) Glutathione peroxidase, glial cells and Parkinson’s disease. Neuroscience 52(1):1–6. doi:10.1016/0306-4522(93)90175-F
Song YJC, Halliday GM, Holton JL, Sullivan SO, McCann H, Path B, Lashley T, Lees AJ et al (2009) Degeneration in different parkinsonian syndromes relates to astrocyte type and astrocyte protein expression. J Neuropathol Exp Neurol 68(10):1073–1083. doi:10.1097/NEN.0b013e3181b66f1b
Braak H, Sastre M, Del Tredici K (2007) Development of α-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol 114(3):231–241. doi:10.1007/s00401-007-0244-3
Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, Hwang D, Masliah E et al (2010) Direct transfer of α-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem 285(12):9262–9272. doi:10.1074/jbc.M109.081125
Desplats P, Lee H-J, Bae E-J, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E et al (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A 106:13010–13015. doi:10.1073/pnas.0903691106
Bartels AL, Willemsen ATM, Doorduin J, de Vries EFJ, Dierckx RA, Leenders KL (2010) [11C]-PK11195 PET: quantification of neuroinflammation and a monitor of anti-inflammatory treatment in Parkinson’s disease? Parkinsonism Relat Disord 16(1):57–59. doi:10.1016/j.parkreldis.2009.05.005
Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, Eggert K, Oertel W et al (2006) In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis 21(2):404–412. doi:10.1016/j.nbd.2005.08.002
Wakabayashi K, Hayashi S, Yoshimoto M, Kudo H, Takahashi H (2000) NACP/alpha-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol 99:14–20. doi:10.1007/PL00007400
Colangelo AM, Alberghina L, Papa M (2014) Astrogliosis as a therapeutic target for neurodegenerative diseases. Neurosci Lett. doi:10.1016/j.neulet.2014.01.014
Chen Y, Vartiainen NE, Ying W, Chan PH, Koistinaho J, Swanson RA (2001) Astrocytes protect neurons from nitric oxide toxicity by a glutathione-dependent mechanism. J Neurochem 77:1601–1610. doi:10.1046/j.1471-4159.2001.00374.x
Sarafian TA, Montes C, Imura T, Qi J, Coppola G, Geschwind DH, Sofroniew MV (2010) Disruption of astrocyte STAT3 signaling decreases mitochondrial function and increases oxidative stress in vitro. PLoS ONE 5:e9532. doi:10.1371/journal.pone.0009532
Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, Johnson JA, Murphy TH (2003) Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci 23:3394–3406
Swanson RA, Ying W, Kauppinen TM (2004) Astrocyte influences on ischemic neuronal death. Curr Mol Med 4:193–205. doi:10.2174/1566524043479185
Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA (2008) Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci: Off J Soc Neurosci 28:13574–13581. doi:10.1523/JNEUROSCI.4099-08.2008
Avila Rodriguez M, Garcia-Segura LM, Cabezas R, Torrente D, Capani F, Gonzalez J, Barreto GE (2014) Tibolone protects T98G cells from glucose deprivation. J Steroid Biochem Mol Biol 144 Pt B:294–303. doi:10.1016/j.jsbmb.2014.07.009
Cabezas R, Avila MF, Gonzalez J, El-Bacha RS, Barreto GE (2015) PDGF-BB protects mitochondria from rotenone in T98G cells. Neurotox Res 27(4):355–367. doi:10.1007/s12640-014-9509-5
Lin JH-C, Lou N, Kang N, Takano T, Hu F, Han X, Xu Q, Lovatt D et al (2008) A central role of connexin 43 in hypoxic preconditioning. J Neurosci Off J Soc Neurosci 28:681–695. doi:10.1523/JNEUROSCI.3827-07.2008
Avila-Rodriguez M, Garcia-Segura LM, Hidalgo-Lanussa O, Baez E, Gonzalez J, Barreto GE (2016) Tibolone protects astrocytic cells from glucose deprivation through a mechanism involving estrogen receptor beta and the upregulation of neuroglobin expression. Mol Cell Endocrinol 433:35–46. doi:10.1016/j.mce.2016.05.024
Toro-Urrego N, Garcia-Segura LM, Echeverria V, Barreto GE (2016) Testosterone protects mitochondrial function and regulates neuroglobin expression in astrocytic cells exposed to glucose deprivation. Front Aging Neurosci 8:152. doi:10.3389/fnagi.2016.00152
Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH et al (1999) Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron 23:297–308. doi:10.1016/S0896-6273(00)80781-3
Chen Y, Swanson RA (2003) Astrocytes and brain injury. J Cereb Blood Flow Metab 23:137–149. doi:10.1097/01.WCB.0000044631.80210.3C
Sofroniew MV, Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol 119:7–35. doi:10.1007/s00401-009-0619-8
Sofroniew MV (2009) Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. doi:10.1016/j.tins.2009.08.002
Sofroniew MV (2005) Reactive astrocytes in neural repair and protection. Neuroscientist: Rev J Bringing Neurobiol, Neurol Psychiatry 11:400–407. doi:10.1177/1073858405278321
Cheng S, Hou J, Zhang C, Xu C, Wang L, Zou X, Yu H, Shi Y et al (2015) Minocycline reduces neuroinflammation but does not ameliorate neuron loss in a mouse model of neurodegeneration. Sci Rep 5:10535. doi:10.1038/srep10535
Noble W, Garwood CJ, Hanger DP (2009) Minocycline as a potential therapeutic agent in neurodegenerative disorders characterised by protein misfolding. Prion 3:78–83
Castaño A, Herrera AJ, Cano J, Machado A (2002) The degenerative effect of a single intranigral injection of LPS on the dopaminergic system is prevented by dexamethasone, and not mimicked by rh-TNF-alpha IL-1beta IFN-gamma. J Neurochem 81:150–157. doi:10.1046/j.1471-4159.2002.00799.x
Di Matteo V, Pierucci M, Di Giovanni G, Di Santo A, Poggi A, Benigno A, Esposito E (2006) Aspirin protects striatal dopaminergic neurons from neurotoxin-induced degeneration: an in vivo microdialysis study. Brain Res 1095:167–177. doi:10.1016/j.brainres.2006.04.013
Mohanakumar KP, Muralikrishnan D, Thomas B (2000) Neuroprotection by sodium salicylate against 1-methyl-4-phenyl-1,2,3, 6-tetrahydropyridine-induced neurotoxicity. Brain Res 864:281–290
Starke RM, Chalouhi N, Ding D, Hasan DM (2015) Potential role of aspirin in the prevention of aneurysmal subarachnoid hemorrhage. Cerebrovasc Dis 39:332–342. doi:10.1159/000381137
Clària J, Serhan CN (1995) Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc Natl Acad Sci U S A 92:9475–9479. doi:10.1073/pnas.92.21.9475
Echeverria V, Yarkov A, Aliev G (2016) Positive modulators of the alpha7 nicotinic receptor against neuroinflammation and cognitive impairment in Alzheimer’s disease. Prog Neurobiol. doi:10.1016/j.pneurobio.2016.01.002
Barreto GE, Santos-Galindo M, Garcia-Segura LM (2014) Selective estrogen receptor modulators regulate reactive microglia after penetrating brain injury. Front Aging Neurosci 6:132. doi:10.3389/fnagi.2014.00132
Barreto G, Santos-Galindo M, Diz-Chaves Y, Pernia O, Carrero P, Azcoitia I, Garcia-Segura LM (2009) Selective estrogen receptor modulators decrease reactive astrogliosis in the injured brain: effects of aging and prolonged depletion of ovarian hormones. Endocrinology 150(11):5010–5015. doi:10.1210/en.2009-0352
Barreto G, Veiga S, Azcoitia I, Garcia-Segura LM, Garcia-Ovejero D (2007) Testosterone decreases reactive astroglia and reactive microglia after brain injury in male rats: role of its metabolites, oestradiol and dihydrotestosterone. Eur J Neurosci 25(10):3039–3046. doi:10.1111/j.1460-9568.2007.05563.x
Lanussa OH, Avila-Rodriguez M, Garcia-Segura LM, Gonzalez J, Echeverria V, Aliev G, Barreto GE (2016) Microglial dependent protective effects of neuroactive steroids. CNS Neurol Disord Drug Targets 15(2):242–249
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Martin-Jiménez, C.A., Gaitán-Vaca, D.M., Echeverria, V. et al. Relationship Between Obesity, Alzheimer’s Disease, and Parkinson’s Disease: an Astrocentric View. Mol Neurobiol 54, 7096–7115 (2017). https://doi.org/10.1007/s12035-016-0193-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-0193-8