
Abstract
Thyroid hormone plays an important role in central nervous system (CNS) development, including the myelination of variable axonal calibers. It is well-established that thyroid hormone is required for the terminal differentiation of oligodendrocyte precursor cells (OPCs) into myelinating oligodendrocytes by inducing rapid cell-cycle arrest and constant transcription of pro-differentiation genes. This is well supported by the hypomyelinating phenotypes exhibited by patients with congenital hypothyroidism, cretinism. During development, myelinating oligodendrocytes only appear after the formation of neural circuits, indicating that the timing of oligodendrocyte differentiation is important. Since fetal and post-natal serum thyroid hormone levels peak at the stage of active myelination, it is suspected that the timing of oligodendrocyte development is finely controlled by thyroid hormone. The essential machinery for thyroid hormone signaling such as deiodinase activity (utilized by cells to auto-regulate the level of thyroid hormone), and nuclear thyroid hormone receptors (for gene transcription) are expressed on oligodendrocytes. In this review, we discuss the known and potential thyroid hormone signaling pathways that may regulate oligodendrocyte development and CNS myelination. Moreover, we evaluate the potential of targeting thyroid hormone signaling for white matter injury or disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Thyroid hormone (TH) regulates neural development, differentiation, and metabolism in mammals [1, 2]. The importance of TH in regulating brain development is manifested in humans under conditions of TH deficiency such as untreated congenital hypothyroidism, or profound maternal hypothyroxinemia due to iodine deficiency, clinically presenting as profound mental retardation and growth restriction [3]. THs including prohormone, thyroxine (T4), and relatively minimal circulating amounts of genomically active tri-iodothyronine (T3) are generated from the thyroid gland [1]. Serum TH-binding proteins (such as albumin, T4-binding globulin, and potentially transthyretin) bound to T4, along with free-T3 can be transported into the central nervous system (CNS) via transporters expressed on epithelial cells of the choroid plexus, endothelial cells of the blood–brain barrier (BBB) and astrocytic foot processes that are in contact with microvessels. This transport mainly occurs through the monocarboxylate transporter (MCT)8 and organic anion-transporting polypeptide 1c1 (OATP1C1) [4]. A low level of T3 in the brain can be compensated by intracellular deiodination of T4 to produce T3 by type 2 deiodinase (Dio2). On the other hand, in a case of cellular hyperthyroidism, type 3 deiodinase (Dio3) deiodinize THs into genomically inactive forms; T4 to reverse T3 and T3 to di-iodothyronine (T2) [1].
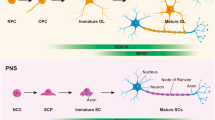
One of the well-studied effects of TH during CNS development is myelination, which is a highly organized process of axon ensheathment by oligodendrocytes to establish fast saltatory conduction via both, facilitation of electrical capacitance (myelin sheath) and molecular clustering of sodium channels at the node of Ranvier and potassium channels at the juxtaparanode (for review, see [5]). It is widely accepted that TH plays a crucial role in oligodendrocyte development and subsequently myelination. Adding T3 in vitro has been widely utilized to potentiate terminal differentiation of both rodent and human oligodendrocytes through rapid cell-cycle arrest in a non-genomic manner and transcription of genes involved in their development upon binding to nuclear TH receptors in a genomic manner [6–9] (Fig. 1). Furthermore, recent data suggest that TH is also involved in regulating early specification of neural precursors to derive oligodendroglial lineage cells, indicating the involvement of TH throughout oligodendrogenesis along with time-dependent specification of oligodendrocyte developmental checkpoints. Importantly, TH is also suspected to be a key player in the context of white matter pathologies, affecting not only oligodendrogenesis in a cell-autonomous manner but also eliciting indirect effects to regulate the metabolism of oligodendrocytes via promoting angiogenesis under hypoxic conditions critical for the maintenance of viable oligodendrocytes [10, 11]. In this review, we discuss how TH regulates oligodendrocyte development and physiology from the extracellular transport to intracellular signaling from both a genomic and potentially non-genomic manner.

Non-genomically and genomically regulated genes by thyroid hormone during oligodendrocyte development. The schematic shows elevation of brain thyroid hormone (TH) levels during oligodendrocyte differentiation. Developmental regulation of oligodendrocyte-associated genes by TH in a non-genomic and genomic expression pattern at different stages of lineage progression. Non-genomic TH signaling is thought to be mediated through putative cytoplasmic thyroid hormone receptor (TR), or αvβ3 integrin dimer, which is tightly regulated to be expressed in pre-myelinating oligodendrocytes. Genomic thyroid hormone signaling is mediated through nucleus TRα1 (expressed throughout oligodendrocyte development) and TRβ1 (increases in expression as oligodendrocytes differentiate)
Thyroid Hormone Signaling in Oligodendrocytes
-
(i)
Extracellular thyroid hormone transport
The cellular transport of THs has long been considered to be mediated by a simple diffusion process across membrane lipid bilayers because of their lipophilic properties. However, the recent discovery of a specific TH transporter, MCT8, suggests that the cellular transport of THs is indeed a physiologically relevant process. So far, MCT8, MCT10, large neutral amino acid transporters (LATs), organic anion-transporting polypeptides (OATPs), and the Na+/taurocholate co-transporting polypeptide (NTCP) protein families were identified to have the capacity to transport THs (for review, see [4]). However, it is MCT8 that has been suggested to be the most specific TH transporter [12], since the others also transport alternate physiological molecules such as amino acids and steroids [4]. In support of this, human mutations of SLC16A2 (encoding MCT8) exhibit severe X-linked inherited psychomotor retardation known as Allan-Herndon-Dudley syndrome (AHDS), which present clinically with increased serum T3 and either normal to reduced T4 levels in the circulation of affected patients [13, 14].
Although THs are trophic during oligodendrocyte development, the import/export of THs in these cells has not been defined. However, indirect evidence implicates MCT8 in oligodendrocyte plasma membrane transport since AHDS patients exhibit delayed myelination measured by longitudinal T2-weighted magnetic resonance imaging (MRI) [15–18]. To study the exact pathophysiological role of MCT8, the slc16a2 knockout mouse was generated [19]. However, despite the matching serum with those of AHDS patients, as well as the altered brain TH profiles (high T3 and low T4 in the circulation/low T3 and low T4 in the brain), no discernable neurological phenotypes were demonstrated in these mutant mice [19]. This species variation in the dependency of the slc16a2 gene suggests that other transporters may indeed compensate for its loss in the knockout mice. Indeed, OATP1C1 enriched in the mouse choroid plexus but not in human [20, 21] was suggested to function in response to MCT8 deficiency facilitating the passage of T4 from the circulation into the brain. This hypothesis was confirmed once prominent neurological phenotypes, including hypomyelination, were detected in the slc16a2/slco1c1 (the latter encodes OATP1C1) double knockout mice [22]. Interestingly, it has been shown that zebrafish lacking slc16a2 exhibit similar neurological deficits that resemble the locomotor developmental abnormalities seen in AHDS patients [23]. Molecular analyses revealed significant reductions in myelin basic protein (mbp) and myelin protein zero (mpz; a critical gene for oligodendrocyte differentiation in zebrafish [24]) genes in this mutant, indicating that in humans and zebrafish, there exists no compensational expression of OATP1C1 in the brain oligodendrocyte development [23]. However, it is unclear whether the neurodevelopmental defects are caused by the dysregulation of MCT8-dependent TH transport at the BBB or at the CNS-specific cells. Therefore, it remains to be confirmed whether the cellular MCT8 of CNS-residing cells, including oligodendrocytes, is responsible for regulating their development. To address this question, a conditional deletion strategy must now be adopted to dissect out the direct role of MCT8 during oligodendrocyte development in vivo.
A recent study has identified that a reduction in the expression of MCT8 in the ventricular zone and sub-ventricular zone (SVZ) cells is observed in post-mortem brains from human fetuses with intra-uterine growth restriction (IUGR) [25]. IUGR is a condition of complicated growth potential most commonly due to the lack of energy supply from uteroplacental failure leading to congenital white matter pathology related to periventricular leukomalacia ultimately causing cerebral palsy (for review, see [26] and hypoxic condition and cytokines have been linked to dysregulation of TH signaling (see ‘Thyroid Hormone as a Treatment for Demyelinating Disease’). This is not surprising as myelination and oligodendrocyte differentiation requires high metabolic support such as oxygen, glucose, and THs from the pre-formed vasculature (for review, see [27]). Furthermore, both guinea pig [28] and rat [29] models of IUGR were shown to exhibit delayed myelination as shown in children with prenatal brain injury [30]. One interpretation of these data could be that the reduction in MCT8 in the germinal zone-derived cells may be limiting the potential to derive appropriate numbers of oligodendroglial lineage cells, thereby stalling or delaying the development of these glial cells and by extension myelinogenesis under IUGR conditions. Moreover, we have recently identified MCT8 expression in oligodendroglial cells derived from human embryonic stem cells that plays an active role in their derivation [31], implicating an important role of MCT8 during oligodendrogenesis (Fig. 2). However, a more sophisticated model of in vivo cell-specific conditional deletion of MCT8 is now required to differentiate the importance of MCT8 during oligodendrogenesis during development and disease. Such models may help identify the how MCT8 downregulation or ablation can modulate the maturation of oligodendrocytes and what effect the BBB plays in this process.

Non-genomic and genomic action of thyroid hormone in oligodendrocytes. Genomic effects of TH: oligodendroglial monocarboxylate transporter 8 (MCT8) may transport T4 and T3 into the cytoplasm where either can be deiodinized into genomically inactive rT3 and T2 or bind to the nuclear TR dimeric complex. Un-liganded TRs can recruit the nucleus co-repressor (Ncor) to repress transcription of genes within the TH response element (TRE). T3 binding of TRs can recruit co-activator (CoA) to regulate TRE-associated genes. Non-genomic effects of TH: THs may signal in a non-genomic manner either via binding to αvβ3 integrin dimeric receptor or cytoplasmic TRβ. Only T3 binds to the S1 site of αvβ3 integrin, and this can phosphorylate and activate phosphatidylinositol 3-kinase (PI3K) and extracellular signal-regulated kinase 1/2 (Erk1/2). Whereas T4 and to a lesser extent T3 binds to the S2 site of αvβ3 integrin and this can phosphorylate and activate Erk1/2 via Src-homology 2 domain containing tyrosine phosphatase (SHP2). Phosphorylated PI3K and Erk1/2 may potentiate the phosphorylation of p85 domain of PI3K and Lyn to dissociate cytoplasmic TRβ, which mediates nucleus trafficking of TRβ for genomic signaling. Activated PI3K and Erk1/2 can also induce p53-dependent cell-cycle arrest
One of the most widely studied genes transcribed under hypoxic conditions is hypoxia-inducible factors (HIFs). Interestingly, a recent report has identified that the persistent stabilization of HIF1/2α in OPCs through Cre-mediated ablation of von Hippel Lindau (VHL; degrades HIF1/2α under normoxia) in Sox10+, Olig1+, or PLP+ oligodendroglia inhibits their differentiation through autocrine activation of Wnt7α/7β [11]. However, the same group also reported that the conditional ablation of HIF1/2α in the abovementioned oligodendroglial cells resulted in reduced angiogenesis, a consequence of reduced paracrine release of Wnt7α/7β (shown to be essential in CNS angiogenesis [32, 33]), leading to axonal injury in the corpus callosum [11].
As HIF1α was also shown to associate with Dio3 to reduce TH signaling, at least in SK-N-AS neuroblastomas under hypoxia [34], it is speculated that under hypoxia-associated developmental injury conditions (such as IUGR-mediated white matter disorders and neonatal brain injuries), reduced MCT8 expression and the active HIF1α in SVZ-derived neural precursors, may reduce TH signaling leading to oligodendrogliopathy and subsequently delayed myelination (Fig. 3). In fact, in humans, post-mortem brain sections of pre-term infants show intra-ventricular hemorrhage (IVH) as sequelae associated with increased Dio3 and reduced Dio2 expression within the germinal zone [35].

Putative cellular hypothyroidism in oligodendrocytes during intra-uterine growth restriction (IUGR) or inflammation-mediated hypoxia. Under normoxic conditions, internalized T3 binds to nuclear TRs to transcribe pro-differentiation genes, thereby mediating oligodendrocyte differentiation. However, under hypoxic conditions such as IUGR and inflammation, persistent HIF1α transcription induces Dio3 and along with a reduced expression of MCT8, results in cellular hypothyroidism. Furthermore, pro-inflammatory cytokines such as TNF-α, IL-1β, and IFN-γ may contribute to dysregulation of thyroid hormone signaling. Un-liganded TRs may repress pro-differentiation genes thereby resulting in stalled oligodendrocyte maturation
Impaired TH signaling is also suspected in adult acquired diseases with demyelination such as multiple sclerosis (MS). Largely, MS can be sub-divided into two forms, acute (relapsing-remitting) and chronic (progressive). Remyelination upon demyelination can occur and is largely responsible for partial recovery of neurological deficits in relapsing-remitting MS whereas this largely fails in progressive MS [36]. In experimental autoimmune encephalomyelitis (EAE)-induced Dark Agouti rats by immunization with recombinant myelin oligodendrocyte glycoprotein, hypoxia was a definitive finding, measured from both in vivo oxygen-sensitive probe and the tissue expression of HIF1α within the inflamed spinal cords, which indeed correlated with neurological deficits [37]. This induction of HIF1α was further ratified through the findings of enlarged microvasculature within the inflamed spinal cords [37]. One plausible hypothesis is that neuroinflammation-driven hypoxia can mediate the induction of HIF1α in oligodendroglial cells, thereby inhibiting their differentiation in autocrine manner via persistent Wnt signaling and local hypothyroidism [11]. Furthermore, increasing evidence suggest for pro-inflammatory cytokine-driven hypothyroidism (see ‘Thyroid Hormone as a Treatment for Demyelinating Disease’), indicating local CNS hypothyroidism driven by multiple factors (Fig. 3).
-
(ii)
Deiodination of thyroid hormone
Intracellular level of THs can be controlled by deiodinases (for review, see [38]). The amount of T3 transported into the CNS is relatively less than T4 during fetal development, and therefore, the majority of T3 in the CNS is generated by the activity of cellular Dio2 during fetal development [39]. In hypothyroidism, the activity of Dio2 increases to compensate reduced T3 availability in the brain [40]. On the other hand, in a hyperthyroidism state, the activity of Dio3 increases to convert excess T3 into T2 and prevent further generation of T3 from T4 [41]. Therefore, deiodination is crucial to maintain the level of T3. However, one should note that this theory is solely based on genomic effects of T3. It is largely unknown whether deiodinases are also involved in regulation of non-genomic effects of TH.
It has been assumed that Dio2 activity in glial cells including astrocytes or tanycytes is a major source of T3 for neurons or oligodendrocytes [42]. This hypothesis is supported by localized messenger RNA (mRNA) expression of dio2 in astroglia and dio3 in neurons [43–45]. Similarly, it is plausible to suggest that oligodendrocytes may receive T3 from astrocytic foot processes as localized expression of Dio3 but not Dio2 has been reported in Olig2-positive oligodendroglia in the human embryonic germinal matrix [35]. However, which transporter regulates the astrocyte-neuron or astrocyte-oligodendrocyte transfer of T3 remains undefined.
-
(iii)
Intracellular thyroid hormone signaling
Role of Co-repressors
The un-liganded TH receptors (TRs) may act as a repressor for T3-responsive genes [46]. In fact, the co-repressors for T3 signaling such as the nuclear receptor co-repressor (Ncor), and silencing mediator of retinoic acid and TH receptors (Smrt), are all essential regulators of embryonic brain development, exerting their effect by repressing genes that are involved in neural lineage specification [47, 48]. As T3 signaling is important in oligodendrocyte development, it is suspected that in hypothyroidism, un-liganded TRs in oligodendrocytes may recruit Ncor to repress genes that are involved in oligodendrocyte differentiation. A recent microarray analysis revealed that in cortical embryonic neural stem cell (NSC) cultures from ncor −/− not smrt −/− mice, significant up-regulation of mbp, plp (encoding proteolipid protein), and nkx2.2 were achieved without altering neuronal genes [49]. As Ncor was shown to enhance deacetylase activity of class I histone deacetylases (HDACs) including HDAC1, HDAC2, HDAC3, and HDAC8 [50], neural stem cells without ncor may have reduced the activity of class I HDAC. Indeed, small interfering RNA (siRNA)-mediated knockdown of hdac2 but not hdac3 in the presence of T3, increased mRNA levels of mbp and plp in rat cortical embryonic NSC cultures, the immortalized oligodendrocyte cell line, CG4, and cultures consisting of embryonic oligodendrocytes and neurons, Oli-Neu [49]. Interestingly, this increase in myelin genes by hdac2 knockdown and T3 treatment was greater than T3 treatment alone and hdac2 knockdown without T3 treatment did not show any up-regulation in these genes [49]. Further studies revealed that HDAC2 was enriched in the U2 enhancer region of an important oligodendroglial gene Sox10, by chromatin immunoprecipitation (ChiP) analysis [49]. This is somewhat in line with previous data which shows histone acetyltransferase activity is required for oligodendrocyte differentiation [51]. Paradoxically, by using Cre-loxP conditional knockout mice of hdac1 and hdac2 in Olig1+ oligodendroglia, the activity of HDAC1/2 was shown to indirectly promote oligodendrocyte differentiation by competing with β-catenin for the transcription of T cell factor (TCF7L2/TCF4; oligodendrocyte-restricted transcription factor) [52]. Although these contradicting results require clear verification with regard to HDAC activity in oligodendrocytes, it seems that in the absence of T3, Ncor negatively regulates early oligodendrocyte differentiation from NSC via HDAC activity.
The co-repressors for T3 signaling not only regulate T3-target gene expression during embryonic development but also exert similar effects in adulthood as experimental induction of hypothyroidism in adult mice resulted in a reduction of mitotic cells and increased the number of cells in interphase G2, in the SVZ [53, 54]. A subsequent mechanistic study revealed that the T3-bound TRα1 expressed on type C DLX2+ transient amplifying cells directly represses Sox2, a gene involved in maintenance of neural stem cells, to promote neurogenesis [53]. However, whether TH signaling plays a similar role during adult OPC development as identified for adult NPCs remains unanswered.
Nuclear Thyroid Hormone Receptors
The genomic effects of THs are mediated by nuclear TRs, which have high affinity for T3 binding. Liganded (T3-bound)-TRs can act as transcription factors by binding to specific TH response elements (TREs) to either activate or repress gene transcriptions. Without T3, un-liganded TRs in the nucleus can repress TH-responsive gene transcription by recruiting co-repressors. Therefore, this balance between co-activators (CoA) and co-repressors associated with TH signaling is dependent on the concentration of T3.
It is well documented that TRα and TRβ are expressed on oligodendrocytes, both in vitro and in vivo [55, 56] and play an important role in their differentiation by gene transcription upon binding to T3 [57, 58]. Early studies revealed that the TRα gene is expressed from early oligodendrocyte development (OPC); whereas, TRβ1 is expressed from later developmental stages of oligodendrocytes (pre-oligodendrocytes), for review, see [59] (Fig. 1). Importantly, during T3-induced oligodendrocyte differentiation, increased expression of TRβ1 not TRα1 in vitro occurs [60, 61]. In support of this, during post-natal rat brain development, the number of oligodendrocytes expressing TRβ1 increases from P0 to P40 when differentiation of oligodendrocytes and myelination occurs [62, 63]. On the other hand, OPCs lacking thra1 (encodes TRα1) failed to differentiate upon TH treatment and thra1 −/− mice showed reduced number of oligodendrocytes in optic nerves at post-natal days 7 and 14 [58]. These results indicate the importance of TRα1 in initial cell-cycle exit upon T3 binding and TRβ1 in driving their terminal differentiation and post-natal myelination.
The TRs are known to function as homodimers [1]. In the optic nerves of 6-month-old mice (a time where OPCs normally disappear from 4 weeks after birth) which lacked all TRβ isoforms showed BrdU+/NG2+ proliferating OPCs whereas these were not found in mice lacking all TRα isoforms [64]. Importantly, mice that were ablated for both TRα and TRβ isoforms exhibited more profound persistence of proliferating OPCs in the optic nerves [64], suggesting that both TRα and TRβ are required for T3-induced normal oligodendrocyte development. However, these knockout mice did not phenocopy the effect of hypothyroidism that can include hypomyelination, quite possibly because of dampened deleterious effects of un-liganded TRs as a physiological result of knocking out TRs. To overcome this, several TRα1 knock-in germline mutant mice were generated, such as TRα1L400R, which targeted the conversion of the nuclear receptor to the transcription repressor. Indeed, the phenotype of these mutants was very similar to mice with hypothyroidism, including severe hypomyelination (for review, see [65]). In an attempt to reveal whether defects in oligodendrocyte lineage cells in these mutants are indirect or cell-autonomous, cnp-Cre (2′,3′-cyclic-nucleotide 3′-phosphodiesterase; thought to target mature oligodendrocytes) and tamoxifen-inducible pdgfra-Cre-ERT2 (thought to target OPCs) lines were crossed with TRα1L400R floxed mice [66]. However, unexpectedly, these conditional mutant models did not target only oligodendrocytes but also other cell types in the cerebellum (cnp-Cre/loxP-TRα1L400R: purkinje cells and oligodendrocytes; pdgfra-Cre/loxP-TRα1L400R: granular neurons, purkinje cells, GABAergic inter-neurons, oligodendrocytes and astrocytes), confounding data interpretation [66]. However, what seems clear from these mutants was that TRα1 indirectly affects cerebellar oligodendrocyte development as these conditional transgenics, along with sycp1 (synaptonemal complex protein 1, targets all cerebellar cells)-Cre/loxP-TRα1L400R and ptf1a (pancreas transcription factor 1a, targets purkinje cells and GABAergic inter-neurons)-Cre/loxP-TRα1L400R mice, exhibited increased numbers of Olig2+ oligodendroglia in the cerebellar white matter at P15 but not in P21, when compared with wild-type controls [66]. However, to reveal whether TRα1 is directly involved in oligodendrocyte development in a cell-autonomous manner, an oligodendrocyte-specific Cre line such as sox10 (expressed throughout the whole oligodendrocyte lineage cells)-Cre may indeed be a more valuable model to uncover the importance of this TR’s influence throughout the entire period of oligodendrogenesis. Furthermore, without conditional deletion of thrb1 in oligodendroglial cells, the direct role of TRβ1 on oligodendrocyte development in vivo is not clear. At least, ChiP sequencing in p15 mouse cerebellum using TRβ antibody revealed oligodendrocyte-specific gene such as mbp and mag [67]. However, it remains to be determined whether these two different isoforms of TRs can regulate common or separate sets of genes during oligodendrocyte development. Although ChiP sequencing of TRα- and TRβ-bound gene promoters in oligodendrocytes may answer these questions, this approach is complicated by the sequence variety of TREs and known non-specificity of commercially available TR antibodies [68]. Strategies such as molecularly tagging TRs with fluorescent reporter such as green fluorescent protein (GFP) [68] or Flag may be useful to identify the TR target genes.
The TRs are known to form heterodimers with other nuclear receptor superfamilies, namely the vitamin D3 receptor [69], the peroxisome proliferator-activated receptor PPARδ [59], and the retinoid X receptor (RXR) [70]. All three of these co-receptors for TRs have been shown to be expressed in oligodendroglial cells [71–73]. Of these co-receptors to TRs, RXR is of significant interest as RXR-TR heterodimerization activity were shown to increase its binding to TRE and induce TR-medicated gene transcription [74]. Furthermore, RXR-TR heterodimer complex can be activated by T3 independently of RXR ligands and even showed synergistic transcriptional activity when both T3 and 9-cis-retinoic acid were applied [75]. RXR has been thought to be particularly important for oligodendrocyte development as RXR signaling was shown to modulate MBP transcript levels in a similar manner to THs [71]. In support of this, Huang et al., recently suggested that RXRγ signaling may play an important role in differentiation of oligodendrocytes and myelination, as siRNA-mediated rxrg (encodes RXRγ) knockdown or RXR-specific antagonists added to rat oligodendrocyte cultures prohibited the formation of the characteristic spider-web-like complex morphology of oligodendrocytes [76]. Moreover, administration of RXR-specific antagonists in co-cultures of dorsal root ganglion neurons with OPCs significantly inhibited myelination. However, the treatment of the RXR agonist, 9-cis-retinoic acids without T3 or T4 showed no changes in oligodendrocyte differentiation or myelination in vitro [76]. In their follow-up study using human embryonic stem cells as a model of oligodendrocyte differentiation, administration of the 9-cis-retinoic acids in the presence of T3 supplied in the differentiation medium significantly increased the yield of MBP+ pre-myelinating oligodendrocytes [77]. These results suggest that retinoic acids probably promote oligodendrocyte differentiation and myelination in concert with THs. It seems that RXR signaling is also important during remyelination (just like TH, see ‘Thyroid Hormone as a Treatment for Demyelinating Disease’) as intraperitoneal administration of 9-cis-retinoic acids upon lysolecithin-induced focal demyelination in the cerebellar peduncle significantly enhanced remyelination [76].
The TRβ agonist, GC-1 or RXR agonist, along with 9-cis-retinoic acids have all been shown to promote both human and rodent oligodendrocyte differentiation in vitro [76–78]. However, their effects in vivo during development are yet to be investigated robustly. Moreover, no in-depth investigation has been performed to clearly delineate the differences and under what circumstances TRβs homo- or hetero-dimerize with RXRs. Simultaneous induction of both TRβ and RXR during differentiation may be warranted to answer this question.
Non-genomic Effects of Thyroid Hormones in Oligodendrocytes
There are rapid metabolic changes (within ∼1 h) upon treatment of oligodendrocytes with THs that cannot be explained by genomic actions per se [6, 7]. These are called non-genomic effects of THs, which regulate cellular proliferation, development and metabolism via protein phosphorylation and kinase activation [1]. For non-genomic effects to occur, THs are required to bind either the membranous integrin αvβ3 dimeric receptor or cytoplasmic TRs [79, 80]. TH signaling through αvβ3 integrin phosphorylates and activates phosphatidylinositol 3-kinase (PI3K)/Akt and Erk1/2 [81]. The αvβ3 integrin has two binding sites for THs; only T3 binds to the S1 site to activate Src and subsequently PI3K, whereas T4 and to a lesser extent T3, binds to the S2 site, thereby activating Erk1/2. Both of these non-genomic actions of THs were shown to translocate TRs to the nucleus from the cytosol [81]. In the absence of THs, TRβ was shown to form a complex within the cytoplasm with the p85 subunit of PI3K and the Src family tyrosine kinase, Lyn [82]. Upon administration of either T3 or T4, dissociated TRβ from this complex and mediated its nuclear translocation through phosphorylation of PI3K or Erk1/2, respectively [82, 83] (Fig. 2). The activation of these two kinases in oligodendrocytes were well-recognized for their role in CNS myelination without altering the number of oligodendrocytes [84, 85]. Although the direct evidence is lacking for oligodendroglial cells, data exists for a potential role of TH in the regulation of non-genomic signaling.
It has been shown that αvβ3 integrin expression is tightly regulated during oligodendrocyte differentiation where its peak expression is at the transition stage (pre-oligodendrocyte) from O2-A progenitors to post-mitotic oligodendrocytes [86], indicating its role in rapid cell-cycle arrest. Furthermore, as genomic effect of THs during oligodendrocyte development is essential for their differentiation (see ‘Regulation of OPC/NPC Differentiation by Thyroid Hormone: Genomic Effect of TH’), it is suspected that TH signaling through αvβ3 integrin in immature oligodendrocytes is an essential step for the accumulation of TRs, especially the β subunit isoform [60] in the nucleus (Fig. 2). Indeed, transfection of human full-length αvβ3 integrin in the CG-4 rat oligodendrocyte cell line showed a reduction of MBP+ mature oligodendrocytes during their differentiation induced by both T4 and T3 [87]. As αvβ3 integrin is shown to physically interact with PDGFRα, a receptor for the major mitogen involved in oligodendrocyte proliferation, i.e., PDGF-AA [88], replacing this mitogen for THs may induce oligodendrocyte differentiation. In fact, this experimental replacement was previously shown to be important for the transition of proliferating OPCs into post-mitotic oligodendrocytes [89, 90] and widely used in culture experiments for both rodents and human oligodendrocyte differentiation (for review, see [91]). Moreover, T3 administration in primary rat OPCs increased phosphorylation of Erk1/2 and Akt within 30 min [92]. Importantly, an intracellular enzyme, Src-homology 2 domain containing tyrosine phosphatase (SHP2) was shown to be involved in this signal, as lentivirus-mediated knockdown of ptpn11 (encodes SHP2) reduced the phosphorylation of Erk1/2 and reduced MBP+ cells under T3-induced differentiation conditions, whereas overexpression of SHP2 increased phosphorylation of Erk1/2 [92]. In addition, Cre-loxP-mediated deletion of SHP2 in Olig2+ oligodendroglial lineage cells in vivo resulted in reduced OPC generation in the embryonic telencephalon and subsequently reduced myelination in the corpus callosum [93]. The phosphorylation of Erk1/2 was reduced in the ventricular zone of these animals. Gain of function mutation of SHP2 in Olig2+ cells increased OPC numbers in the corpus callosum however, resulted in abnormal myelination measured by electron microscopy [93]. Molecular studies revealed an increase of phosphorylation of Erk1/2 in the ventricular zone but not Akt in these animals [93]. These results indicate that T3 binding to the S2 site of αvβ3 integrin and possibly through SHP2, activates the Erk1/2 pathway to translocate TRβ from the cytoplasm to the nucleus for T3-induced gene transcription. However, as major substrates for αvβ3 integrin are extracellular matrix proteins such as vitronectin and fibronectin, it is difficult to interpret the exact role of αvβ3 integrin-dependent TH signaling and more analytical experiments are warranted.
One of the events that are regulated by fast non-genomic actions of THs relates to the role played by TH to regulate the cell cycle. It has been shown that cyclin D kinase inhibitors (CdkI) that are involved in cell-cycle arrest such as p21 and p27 was found to be up-regulated within an hour post-T3 treatment in purified OPCs pre-treated with a protein synthesis inhibitor (to detect the immediate response) derived from P7 rat optic nerves [6]. Further studies revealed that the T3-induced up-regulation of CdkIs are p53-dependent as transduction of OPCs with retrovirus encoding a dominant-negative form of p53 resulted in a significantly reduced appearance of GalC+ mature oligodendrocytes even in the presence of T3 [7]. These results indicate that T3 can induce p53-dependent cell-cycle arrest in OPCs primarily through a non-genomic manner to promote their differentiation. However, it is yet to be determined whether this event is mediated by integrins or cytoplasmic TRs. Pre-treating the OPCs with both a protein synthesis inhibitor such as cycloheximide and integrin αvβ3 antagonists such as tetraiodothyroacetic acid (tetrac) [94], or Arg-Gly-Asp (RGD) peptide [95], may reveal the mechanism of this non-genomic action further.
One of the strong morphogens that may regulate early oligodendrocyte specification is Sonic hedgehog (Shh), a developmentally regulated protein that plays a significant role in initial patterning and specification of both neurons and oligodendrocytes. In the developing forebrain, Shh is required for the maintenance of Nkx2.1+ medial ganglionic eminence progenitors for the production of both inter-neurons and oligodendrocytes. Similarly, Shh is important for the specification of the cells of the ventral neural tube to motor neurons and oligodendrocytes within the spinal cord, (for review, see [96]). TH-induced regulation of Shh-dependent signaling was proposed, since the shh gene expression was observed to be significantly reduced during the development of the rat cerebellum under hypothyroid conditions [97, 98]. At least in neuronal cell culture, acute treatment of T3 induced a rapid increase in histone acetylation within the shh gene promoter [98]. Intriguingly, at least in neuroblastoma and glioblastoma cultures, Shh signaling has been suggested to modulate thyroid hormone signaling by regulating the activity of Dio3 [99]. Therefore, although it requires validation, it seems that in early CNS development, T3 may rapidly induce shh transcription to promote oligodendrogenesis, then to prohibit precocious differentiation of these newly generated oligodendrocytes, via Shh-dependent induction of Dio3 to mediate local hypothyroidism.
Regulation of OPC/NPC Proliferation by Thyroid Hormone
TH can also act on both young and adult NPCs to regulate their proliferation. Experimental models of hypothyroidism in adult rat induced by thyrotoxicant, propyl-thio-uracil (PTU) demonstrated that there is an increase in Ki-67+ and BrdU+ proliferating cells in the SVZ and olfactory bulb [100]. This result was correlated with reductions in mRNA levels of pdgfra and mbp in the optic nerve along with an observed reduction in MBP protein levels [100]. The same group also reported that upon T4 treatment following hypothyroidism, they observed a reduced proliferation of germinal cells within the SVZ and olfactory bulb [100]. Furthermore, ChiP assays upon acute T3 administration in both early embryonic and adult rat, indicated that there is an increase in histone acetylation at the shh promoter [101] in the forebrain of these animals acutely treated with T3 [98]. These data suggest a role for TH in the regulation of OPC proliferation within the germinal matrix.
In addition to the effects of TH regulating OPC proliferation, the TH can promote differentiation of these cells. Magnetic resonance imaging (MRI) and magnetic resonance spectroscopy (MRS) studies showed that delayed myelination is a common feature of children with congenital hypothyroidism [102–104]. This hypothyroidism-related phenotype was shown to be reversed by T4 treatment [104]. This finding was then translated to oligodendrocyte culture where it can be used in vitro for OPCs to exit the cell cycle and differentiate [105]. One plausible explanation for T3 as an intrinsic timer for oligodendrocyte development would be that it may act to reduce mRNA and protein levels of E2F-1, which is a key transcription factor controlling G1 to S phase transition during T3-induced differentiation of OPCs [106]. Furthermore, the binding of T3 to its nuclear receptor can promote the myelination phenotype of oligodendroglial cells through its direct interaction with the promoter region of MBP [8]. Therefore, these results indicate that TH can exert cell-cycle exit in mitotic OPCs to control their differentiation in a timely manner.
Regulation of OPC/NPC Differentiation by Thyroid Hormone: Genomic Effect of TH
As discussed above, TH can directly (cell-autonomous) or indirectly regulate oligodendrocyte differentiation. In the following section, we discuss the cell-autonomous regulation of oligodendrocyte differentiation by oligodendrocyte-specific gene/TRE complexes activated by T3-bound TRs (Figs. 1 and 2).
Myelin Genes
As hypothyroidism leads to hypomyelination in the CNS, it is not surprising that T3 regulates myelin genes. In fact, gene expression studies have revealed that crucial myelin genes such as mbp, plp, myelin-associated glycoprotein (mag), and cnp were reduced in rodent brains upon thyrotoxicant-induced congenital hypothyroidism [107–109]. In early studies using electrophoretic mobility shift assays, in the presence of T3, both TRα and TRβ were found to bind specifically to the promoter region of the mbp gene within the TREs [8, 110]. Of interest, the subsequent study by the same group found that MBP-TRE complex preferentially binds to TRβ when compared with TRα [110]. As oligodendroglial expression of TRβ was found to be increased during their terminal differentiation (see ‘Nuclear Thyroid Hormone Receptors’), TRβ not TRα seems to be involved in transcription of genes that are involved in maturation of oligodendrocytes.
KLF9
Kruppel-like factor 9 (KLF9) has been implicated as an important member of the zinc finger family of transcription factors regulating the CNS development (for review, see [111]. Importantly, KLF9 is shown to be strongly induced upon T3 treatment under both in vitro and in vivo conditions [112, 113]. Furthermore, ChiP assays have revealed the presence of KLF9 within the TREs and the interaction with TRβ1 [114]. Therefore, it seems that T3-bound TRβ1 may potentiate transcription of KLF9 during CNS development. A recent microarray-based whole genome analysis revealed that KLF9 was strongly induced by T3 treatment in purified OPCs derived from P7 rat brains, indicating its role in oligodendrocyte differentiation [9]. In support of this, siRNA-based knockdown of klf9 in OPCs repressed oligodendrocyte differentiation even in the presence of T3 [9]. However, despite its potential role in driving oligodendrocyte differentiation in vitro, developmental myelination was not altered in the corpus callosum, cortical white matter, and optic nerves of klf9 −/− mice. Whereas, delayed remyelination upon cuprizone-mediated demyelination was found in the same mutants [9]. As KLF9 is expressed ubiquitously in the CNS especially in neurons, as well as reported to play a crucial role in neurite outgrowth inhibition and axonal regeneration of both retinal ganglion cells and Purkinje cells [115, 116], oligodendrocyte-specific knockout models are now required to clearly delineate the role of KLF9 in oligodendrocyte differentiation. However, from these data, it is plausible to suggest that during CNS development, T3 signaling may halt the overgrowth of axons upon establishment of neuritic synapses and thereby promote oligodendrocyte differentiation via the transcriptional activities of KLF9 within both cell types.
Enpp2, Autotaxin
Ectonucleotide pyrophosphatase/phosphodiesterase 2 (ENPP2), also known as autotaxin has been defined as one of the T3-induced transcriptional targets [99, 117, 118] in neural cells. During development, autotaxin has been shown to be expressed on cells that reside within the choroid plexus and also within parenchymal oligodendroglial cells. The oligodendroglial expression of autotaxin in particular, has been correlated with the period of active myelination (post-natal day 8 in rat) [119, 120], where serum T3 levels are at their peak [63]. Autotaxin is an extracellular factor, which has been shown to promote the adhesion and complex network formation of pre-myelinating oligodendrocytes [120, 121]. In purified OPCs derived from the P2 rat brain, upon T3 treatment, autotaxin was found to be up-regulated, suggesting that enpp2 may be a T3-responsive gene in oligodendrocytes [122]. However, whether T3 can induce the transcription of enpp2 in oligodendrocytes during active myelination remains unanswered.
Abcd2
The abcd1 gene encodes for the adenosine triphosphate (ATP)-binding cassette transporter superfamily D member 1, also known as adrenoleukodystrophy protein (ALDP) [123]. ALDP is responsible for the import of very long chain fatty acids (VLCFA) into peroxisomes marked for degradation through β-oxidation [123]. The expression of this protein has been previously shown to be restricted to glial cells (oligodendrocytes, astrocytes, and microglia) [124]. The importance of this protein for lipid metabolism is emphasized by X-linked adrenoleukodystrophy (X-ALD) caused by the human loss-of-function mutation of abcd1 [125]. X-ALD is a peroxisomal disorder characterized by inflammatory demyelination in cerebral white matter due to the accumulation of VLCFA [125]. Extracellular VLCFA administration in primary oligodendrocytes has verified its cytotoxic effects through the sustained elevation in intracellular [Ca2+] and potent oxidative stress mechanisms [126], suggesting a primary oligodendrocytopathy in X-ALD patients.
It has been shown that the ALDP-related protein (ALDRP; encoded by abcd2), a protein sharing 63 % sequence homology with ALDP, may be partially redundant leading to the hypothesis that lipid metabolism may be therapeutically normalized in X-ALD patients [127, 128]. Indeed, pharmacological induction of abcd2 was shown to increase the rate of β-oxidation rate of VLCFA in X-ALD patients to homeostatic levels [127, 128] and importantly in oligodendrocytes derived from induced pluripotent stem cells (iPSCs) from X-ALD patients [129]. Interestingly, the abcd2 promoter was found to contain a 4-base pair spacer (DR-4) motif of TRE that binds to the TRβ1 and RXRα heterodimer [130]. Further study revealed that T3 administration induced the expression of abcd2 in the CG4 differentiated oligodendrocyte cell line and increased β-oxidation of VLCFA in X-ALD patient-derived fibroblasts [130]. On the other hand, un-liganded TRβ but not TRα suppressed the transcription of abcd2 [131], implying that the regulation of abcd2 is mediated through TRβ. In support of this, treatment of cells with the TRβ-specific agonists, GC-1 and CGS23425, showed marked increase in abcd2 mRNA [132]. As expression levels of TRβ increase upon oligodendrocyte maturation [59] and β-oxidation of VLCFA is implicated in energy metabolism [133], TRβ-dependent transcription of abcd2 is most likely required for the maintenance of myelinating oligodendrocytes. As T3 may compensate for the loss of abcd1, the severe phenotype exhibited in X-ALD patients suggests dysregulation of TH metabolism in oligodendrocytes. Furthermore, as conditional deletion of the peroxisomal targeting signal type-1 receptor (PEX5) in Cnp-expressing oligodendroglia (cnp-Cre/loxP-pex5) resulted in primary axonopathy preceding inflammatory demyelination [134], TH signaling in oligodendrocytes may be required for the maintenance of axons via axo-glia interactions.
Thyroid Hormone as a Treatment for Demyelinating Disease
The pro-myelinating properties of thyroid hormone has led to its a promise as a physiological molecule capable of treating demyelinating diseases such as MS. Laura Calza’s group has pioneered this research by administering either T4 or T3 subcutaneously into various experimental autoimmune encephalomyelitis (EAE) models [10, 135–137] and from these experiments has partially concluded that clinical EAE scores of neurological impairment are reduced after onset of disease. Molecular analyses of the post-mortem tissue from these experimental animals show increased immunoreactivity against MBP measured by either western immunoblot or immunohistochemistry. Similarly, subcutaneous [138], intraperitoneal [139], or intranasal [140] administration of T3 in a neurotoxicant, cuprizone-mediated demyelination model, all showed enhanced remyelination. In addition, co-administration of HDAC inhibitor, valporic acid, and T4 into dark agouti rat model of EAE increased myelin gene expressions in brain O4+ pre-oligodendrocytes during the remission stage of EAE [141]. Moreover, in other white matter injury model characterized by inflammation and demyelination such as intra-ventricular hemorrhage and hypoxic ischemia, T4 treatment increased myelination during early post-natal developments of rabbit and rat, respectively [35, 142]. Importantly, T4 treatment also showed increased number of O4+/O1+ mature oligodendrocytes in pre-term infants with IVH when compared with IVH alone control [35]. Although these results seem promising, it is difficult to conclude whether the exogenously administered THs are acting directly on the CNS oligodendroglia or indeed through the modulation of other cells that affect the course of these demyelination models, which may specifically include the innate and adaptive immune arms. Regarding this concern, treatment of T3 into MBP-specific T cell lines reduced IL-17+ T cells and adoptive transfer of these cells into naïve small rats but significantly reduced EAE symptoms [141]. Furthermore, even though THs may be beneficial in remyelination in the CNS, an excessive administration of THs may cause local hyperthyroidism in immune organs. Hyperthyroidism may functionally modulate immune cells, reflected in well-documented hyperthyroidism-mediated autoimmune thyrotoxicosis, Grave’s disease. For example, the function of regulatory T cells (Tregs), essential for the regulation of a potential autoimmune response, has been shown to be impaired in patients with Grave’s disease (for review, see [143]). Furthermore, Tregs isolated from the peripheral mononuclear cells of patients with Grave’s disease were found to be apoptotic [144], possibly through a T3-TRβ-dependent induction of precocious cell senescence [145]. Another important aspect to note is that although there is no direct evidence, pro-inflammatory cytokines released from peripheral immune cells that were shown to inhibit oligodendrocyte differentiation [146–149] such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interferon-γ (IFN-γ) can induce local hypothyroidism as these cytokines secreted as a result of lipopolysaccharide-induced inflammation caused dysregulation of Dio2 [150], which may again cause modulation of T3 signaling. Recently, T4 treatment failed to rescue white matter injury including oligodendrocyte differentiation blockade induced by systemic administration of IL-1β [151], which may indicate for the dysregulation of thyroid hormone signaling by IL-1β. Therefore, to reduce side-effects from the peripheral immune arm, local rather than systemic administration of TH may be employed to promote remyelination. Alternatively, innovative nanoparticle-coated THs seem promising for better CNS-targeted drug delivery [152].
Another important point to note is that in a similar manner seen under in vivo hypoxic conditions such as IUGR, which can impair TH transport via a reduction in the expression of MCT8 [25], dysregulation of TH transport is also speculated to be a result of EAE-induced hypoxia [37], thereby further complicating any proposed TH treatment regime during EAE (Fig. 3). In light of this, future studies are required to uncover the implications imposed upon oligodendrocyte development and remyelination, as a consequence of dysregulation of the TH transporters during neuroinflammation. Robust neurobiological data may provide further insights on how treatments targeting the TH signaling cascades can limit inflammatory-mediated oligodendrocyte/myelin damage and further neurodegeneration.
Conclusion
Development of mature, integrating cells within the CNS occurs via a tightly controlled sequence of regionally specific events. Only upon establishment of immature neuronal synaptogenesis, is the emergence of profound oligodendrocyte development. THs are fundamental to the regulation of the timing of oligodendrogenesis proposed through their putative effects upon mitosis and more specifically imposed following the nuclear translocation of TRs (non-genomic) and transcription of genes upon binding to nuclear localized TRs (genomic). Emerging evidence now identifies that dysregulation of TH signaling is a major hallmark of diseases with underlying hypoxia-associated developmental hypomyelination and inflammatory demyelination. Therefore, the design of novel therapeutics to directly enhance TH signaling may indeed hold great potential to enhance developmental myelination or remyelination of denuded axons. However, as hyperthyroidism may lead to pronounced metabolic rates as documented during pathophysiological conditions, the induction of premature cell senescence may indeed be a contraindication of such treatments. Hence, a more cautious and considered design of therapeutic options is required to take advantage of the known TH signaling pathways that may be targeted for clinical use.
References
Cheng SY, Leonard JL, Davis PJ (2010) Molecular aspects of thyroid hormone actions. Endocr Rev 31(2):139–170. doi:10.1210/er.2009-0007
Williams GR (2008) Neurodevelopmental and neurophysiological actions of thyroid hormone. J Neuroendocrinol 20(6):784–794. doi:10.1111/j.1365-2826.2008.01733.x
Moog NK, Entringer S, Heim C, Wadhwa PD, Kathmann N, Buss C (2015) Influence of maternal thyroid hormones during gestation on fetal brain development. Neuroscience. doi:10.1016/j.neuroscience.2015.09.070
Bernal J, Guadano-Ferraz A, Morte B (2015) Thyroid hormone transporters—functions and clinical implications. Nat Rev Endocrinol 11(9):506. doi:10.1038/nrendo.2015.113
Rasband MN, Peles E (2015) The nodes of Ranvier: molecular assembly and maintenance. Cold Spring Harb Perspect Biol. doi:10.1101/cshperspect.a020495
Tokumoto YM, Tang DG, Raff MC (2001) Two molecularly distinct intracellular pathways to oligodendrocyte differentiation: role of a p53 family protein. Embo J 20(18):5261–5268. doi:10.1093/emboj/20.18.5261
Billon N, Terrinoni A, Jolicoeur C, McCarthy A, Richardson WD, Melino G, Raff M (2004) Roles for p53 and p73 during oligodendrocyte development. Development 131(6):1211–1220. doi:10.1242/dev.01035
Farsetti A, Mitsuhashi T, Desvergne B, Robbins J, Nikodem VM (1991) Molecular basis of thyroid hormone regulation of myelin basic protein gene expression in rodent brain. J Biol Chem 266(34):23226–23232
Dugas JC, Ibrahim A, Barres BA (2012) The T3-induced gene KLF9 regulates oligodendrocyte differentiation and myelin regeneration. Mol Cell Neurosci 50(1):45–57. doi:10.1016/j.mcn.2012.03.007
Fernandez M, Giuliani A, Pirondi S, D’Intino G, Giardino L, Aloe L, Levi-Montalcini R, Calza L (2004) Thyroid hormone administration enhances remyelination in chronic demyelinating inflammatory disease. Proc Natl Acad Sci U S A 101(46):16363–16368. doi:10.1073/pnas.0407262101
Yuen TJ, Silbereis JC, Griveau A, Chang SM, Daneman R, Fancy SP, Zahed H, Maltepe E, Rowitch DH (2014) Oligodendrocyte-encoded HIF function couples postnatal myelination and white matter angiogenesis. Cell 158(2):383–396. doi:10.1016/j.cell.2014.04.052
Friesema EC, Ganguly S, Abdalla A, Manning Fox JE, Halestrap AP, Visser TJ (2003) Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J Biol Chem 278(41):40128–40135. doi:10.1074/jbc.M300909200
Friesema EC, Grueters A, Biebermann H, Krude H, von Moers A, Reeser M, Barrett TG, Mancilla EE, Svensson J, Kester MH, Kuiper GG, Balkassmi S, Uitterlinden AG, Koehrle J, Rodien P, Halestrap AP, Visser TJ (2004) Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet 364(9443):1435–1437. doi:10.1016/S0140-6736(04)17226-7
Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S (2004) A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet 74(1):168–175. doi:10.1086/380999
Vaurs-Barriere C, Deville M, Sarret C, Giraud G, Des Portes V, Prats-Vinas JM, De Michele G, Dan B, Brady AF, Boespflug-Tanguy O, Touraine R (2009) Pelizaeus-Merzbacher-Like disease presentation of MCT8 mutated male subjects. Ann Neurol 65(1):114–118. doi:10.1002/ana.21579
Armour CM, Kersseboom S, Yoon G, Visser TJ (2015) Further Insights into the Allan-Herndon-Dudley syndrome: clinical and functional characterization of a novel MCT8 mutation. PLoS One 10(10):e0139343. doi:10.1371/journal.pone.0139343
Tonduti D, Vanderver A, Berardinelli A, Schmidt JL, Collins CD, Novara F, Genni AD, Mita A, Triulzi F, Brunstrom-Hernandez JE, Zuffardi O, Balottin U, Orcesi S (2013) MCT8 deficiency: extrapyramidal symptoms and delayed myelination as prominent features. J Child Neurol 28(6):795–800. doi:10.1177/0883073812450944
La Piana R, Vanasse M, Brais B, Bernard G (2015) Myelination delay and Allan-Herndon-Dudley syndrome caused by a novel mutation in the SLC16A2 gene. J Child Neurol 30(10):1371–1374. doi:10.1177/0883073814555189
Di Cosmo C, Liao XH, Dumitrescu AM, Philp NJ, Weiss RE, Refetoff S (2010) Mice deficient in MCT8 reveal a mechanism regulating thyroid hormone secretion. J Clin Invest 120(9):3377–3388. doi:10.1172/JCI42113
Mayerl S, Visser TJ, Darras VM, Horn S, Heuer H (2012) Impact of Oatp1c1 deficiency on thyroid hormone metabolism and action in the mouse brain. Endocrinology 153(3):1528–1537. doi:10.1210/en.2011-1633
Roberts LM, Woodford K, Zhou M, Black DS, Haggerty JE, Tate EH, Grindstaff KK, Mengesha W, Raman C, Zerangue N (2008) Expression of the thyroid hormone transporters monocarboxylate transporter-8 (SLC16A2) and organic ion transporter-14 (SLCO1C1) at the blood–brain barrier. Endocrinology 149(12):6251–6261. doi:10.1210/en.2008-0378
Mayerl S, Muller J, Bauer R, Richert S, Kassmann CM, Darras VM, Buder K, Boelen A, Visser TJ, Heuer H (2014) Transporters MCT8 and OATP1C1 maintain murine brain thyroid hormone homeostasis. J Clin Invest 124(5):1987–1999. doi:10.1172/JCI70324
Zada D, Tovin A, Lerer-Goldshtein T, Vatine GD, Appelbaum L (2014) Altered behavioral performance and live imaging of circuit-specific neural deficiencies in a zebrafish model for psychomotor retardation. PLoS Genet 10(9):e1004615. doi:10.1371/journal.pgen.1004615
Mobius W, Patzig J, Nave KA, Werner HB (2008) Phylogeny of proteolipid proteins: divergence, constraints, and the evolution of novel functions in myelination and neuroprotection. Neuron Glia Biol 4(2):111–127. doi:10.1017/S1740925X0900009X
Chan SY, Hancox LA, Martin-Santos A, Loubiere LS, Walter MN, Gonzalez AM, Cox PM, Logan A, McCabe CJ, Franklyn JA, Kilby MD (2014) MCT8 expression in human fetal cerebral cortex is reduced in severe intrauterine growth restriction. J Endocrinol 220(2):85–95. doi:10.1530/JOE-13-0400
Jarvis S, Glinianaia SV, Blair E (2006) Cerebral palsy and intrauterine growth. Clin Perinatol 33(2):285–300. doi:10.1016/j.clp.2006.03.009
Morton PD, Ishibashi N, Jonas RA, Gallo V (2015) Congenital cardiac anomalies and white matter injury. Trends Neurosci 38(6):353–363. doi:10.1016/j.tins.2015.04.001
Tolcos M, Bateman E, O’Dowd R, Markwick R, Vrijsen K, Rehn A, Rees S (2011) Intrauterine growth restriction affects the maturation of myelin. Exp Neurol 232(1):53–65. doi:10.1016/j.expneurol.2011.08.002
Reid MV, Murray KA, Marsh ED, Golden JA, Simmons RA, Grinspan JB (2012) Delayed myelination in an intrauterine growth retardation model is mediated by oxidative stress upregulating bone morphogenetic protein 4. J Neuropathol Exp Neurol 71(7):640–653. doi:10.1097/NEN.0b013e31825cfa81
Silbereis JC, Huang EJ, Back SA, Rowitch DH (2010) Towards improved animal models of neonatal white matter injury associated with cerebral palsy. Dis Model Mech 3(11–12):678–688. doi:10.1242/dmm.002915
Lee JY, Kim MJ, Stanley EG, Elefanty AG, Petratos S (2015) Monocarboxylate transporter 8 is expressed on oligodendrocyte progenitors derived from human embryonic stem cells. International Journal of Developmental Neuroscience: the Official Journal of the International Society for Developmental Neuroscience 47 (Pt A):69. doi:10.1016/j.ijdevneu.2015.04.189
Stenman JM, Rajagopal J, Carroll TJ, Ishibashi M, McMahon J, McMahon AP (2008) Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science 322(5905):1247–1250. doi:10.1126/science.1164594
Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA (2009) Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci U S A 106(2):641–646. doi:10.1073/pnas.0805165106
Simonides WS, Mulcahey MA, Redout EM, Muller A, Zuidwijk MJ, Visser TJ, Wassen FW, Crescenzi A, da-Silva WS, Harney J, Engel FB, Obregon MJ, Larsen PR, Bianco AC, Huang SA (2008) Hypoxia-inducible factor induces local thyroid hormone inactivation during hypoxic-ischemic disease in rats. J Clin Invest 118(3):975–983. doi:10.1172/JCI32824
Vose LR, Vinukonda G, Jo S, Miry O, Diamond D, Korumilli R, Arshad A, Zia MT, Hu F, Kayton RJ, La Gamma EF, Bansal R, Bianco AC, Ballabh P (2013) Treatment with thyroxine restores myelination and clinical recovery after intraventricular hemorrhage. J Neurosci 33(44):17232–17246. doi:10.1523/JNEUROSCI.2713-13.2013
Fancy SP, Kotter MR, Harrington EP, Huang JK, Zhao C, Rowitch DH, Franklin RJ (2010) Overcoming remyelination failure in multiple sclerosis and other myelin disorders. Exp Neurol 225(1):18–23. doi:10.1016/j.expneurol.2009.12.020
Davies AL, Desai RA, Bloomfield PS, McIntosh PR, Chapple KJ, Linington C, Fairless R, Diem R, Kasti M, Murphy MP, Smith KJ (2013) Neurological deficits caused by tissue hypoxia in neuroinflammatory disease. Ann Neurol 74(6):815–825. doi:10.1002/ana.24006
Gereben B, Zavacki AM, Ribich S, Kim BW, Huang SA, Simonides WS, Zeold A, Bianco AC (2008) Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr Rev 29(7):898–938. doi:10.1210/er.2008-0019
Bernal J (2007) Thyroid hormone receptors in brain development and function. Nat Clin Pract Endocrinol Metab 3(3):249–259. doi:10.1038/ncpendmet0424
Kaplan MM, McCann UD, Yaskoski KA, Larsen PR, Leonard JL (1981) Anatomical distribution of phenolic and tyrosyl ring iodothyronine deiodinases in the nervous system of normal and hypothyroid rats. Endocrinology 109(2):397–402. doi:10.1210/endo-109-2-397
Kaplan MM, Yaskoski KA (1980) Phenolic and tyrosyl ring deiodination of iodothyronines in rat brain homogenates. J Clin Invest 66(3):551–562. doi:10.1172/JCI109887
Courtin F, Zrouri H, Lamirand A, Li WW, Mercier G, Schumacher M, Goascogne CL, Pierre M (2005) Thyroid hormone deiodinases in the central and peripheral nervous system. Thyroid 15(8):931–942. doi:10.1089/thy.2005.15.931
Guadano-Ferraz A, Obregon MJ, St Germain DL, Bernal J (1997) The type 2 iodothyronine deiodinase is expressed primarily in glial cells in the neonatal rat brain. Proc Natl Acad Sci U S A 94(19):10391–10396
Tu HM, Kim SW, Salvatore D, Bartha T, Legradi G, Larsen PR, Lechan RM (1997) Regional distribution of type 2 thyroxine deiodinase messenger ribonucleic acid in rat hypothalamus and pituitary and its regulation by thyroid hormone. Endocrinology 138(8):3359–3368. doi:10.1210/endo.138.8.5318
Tu HM, Legradi G, Bartha T, Salvatore D, Lechan RM, Larsen PR (1999) Regional expression of the type 3 iodothyronine deiodinase messenger ribonucleic acid in the rat central nervous system and its regulation by thyroid hormone. Endocrinology 140(2):784–790. doi:10.1210/endo.140.2.6486
Mottis A, Mouchiroud L, Auwerx J (2013) Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev 27(8):819–835. doi:10.1101/gad.214023.113
Hermanson O, Jepsen K, Rosenfeld MG (2002) N-CoR controls differentiation of neural stem cells into astrocytes. Nature 419(6910):934–939. doi:10.1038/nature01156
Jepsen K, Solum D, Zhou T, McEvilly RJ, Kim HJ, Glass CK, Hermanson O, Rosenfeld MG (2007) SMRT-mediated repression of an H3K27 demethylase in progression from neural stem cell to neuron. Nature 450(7168):415–419. doi:10.1038/nature06270
Castelo-Branco G, Lilja T, Wallenborg K, Falcao AM, Marques SC, Gracias A, Solum D, Paap R, Walfridsson J, Teixeira AI, Rosenfeld MG, Jepsen K, Hermanson O (2014) Neural stem cell differentiation is dictated by distinct actions of nuclear receptor corepressors and histone deacetylases. Stem Cell Reports 3(3):502–515. doi:10.1016/j.stemcr.2014.07.008
Yang XJ, Seto E (2008) The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol 9(3):206–218. doi:10.1038/nrm2346
Wang J, Weaver IC, Gauthier-Fisher A, Wang H, He L, Yeomans J, Wondisford F, Kaplan DR, Miller FD (2010) CBP histone acetyltransferase activity regulates embryonic neural differentiation in the normal and Rubinstein-Taybi syndrome brain. Dev Cell 18(1):114–125. doi:10.1016/j.devcel.2009.10.023
Ye F, Chen Y, Hoang T, Montgomery RL, Zhao XH, Bu H, Hu T, Taketo MM, van Es JH, Clevers H, Hsieh J, Bassel-Duby R, Olson EN, Lu QR (2009) HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nat Neurosci 12(7):829–838. doi:10.1038/nn.2333
Lopez-Juarez A, Remaud S, Hassani Z, Jolivet P, Pierre Simons J, Sontag T, Yoshikawa K, Price J, Morvan-Dubois G, Demeneix BA (2012) Thyroid hormone signaling acts as a neurogenic switch by repressing Sox2 in the adult neural stem cell niche. Cell Stem Cell 10(5):531–543. doi:10.1016/j.stem.2012.04.008
Lemkine GF, Raj A, Alfama G, Turque N, Hassani Z, Alegria-Prevot O, Samarut J, Levi G, Demeneix BA (2005) Adult neural stem cell cycling in vivo requires thyroid hormone and its alpha receptor. Faseb J 19(7):863–865. doi:10.1096/fj.04-2916fje
Carlson DJ, Strait KA, Schwartz HL, Oppenheimer JH (1994) Immunofluorescent localization of thyroid hormone receptor isoforms in glial cells of rat brain. Endocrinology 135(5):1831–1836. doi:10.1210/endo.135.5.7525253
Baas D, Fressinaud C, Ittel ME, Reeber A, Dalencon D, Puymirat J, Sarlieve LL (1994) Expression of thyroid hormone receptor isoforms in rat oligodendrocyte cultures. Effect of 3,5,3′-triiodo-l-thyronine. Neurosci Lett 176(1):47–51
Billon N, Tokumoto Y, Forrest D, Raff M (2001) Role of thyroid hormone receptors in timing oligodendrocyte differentiation. Dev Biol 235(1):110–120. doi:10.1006/dbio.2001.0293
Billon N, Jolicoeur C, Tokumoto Y, Vennstrom B, Raff M (2002) Normal timing of oligodendrocyte development depends on thyroid hormone receptor alpha 1 (TRalpha1). Embo J 21(23):6452–6460
Sarlieve LL, Rodriguez-Pena A, Langley K (2004) Expression of thyroid hormone receptor isoforms in the oligodendrocyte lineage. Neurochem Res 29(5):903–922
Baas D, Puymirat J, Sarlieve LL (1998) Posttranscriptional regulation of oligodendroglial thyroid hormone (T3) receptor beta 1 by T3. Int J Dev Neurosci 16(6):461–467
Bury F, Carre JL, Vega S, Ghandour MS, Rodriguez-Pena A, Langley K, Sarlieve LL (2002) Coexpression of thyroid hormone receptor isoforms in mouse oligodendrocytes. J Neurosci Res 67(1):106–113
Strait KA, Carlson DJ, Schwartz HL, Oppenheimer JH (1997) Transient stimulation of myelin basic protein gene expression in differentiating cultured oligodendrocytes: a model for 3,5,3′-triiodothyronine-induced brain development. Endocrinology 138(2):635–641. doi:10.1210/endo.138.2.4946
Rodriguez-Pena A (1999) Oligodendrocyte development and thyroid hormone. J Neurobiol 40(4):497–512
Baas D, Legrand C, Samarut J, Flamant F (2002) Persistence of oligodendrocyte precursor cells and altered myelination in optic nerve associated to retina degeneration in mice devoid of all thyroid hormone receptors. Proc Natl Acad Sci U S A 99(5):2907–2911. doi:10.1073/pnas.052482299
Flamant F, Gauthier K (2013) Thyroid hormone receptors: the challenge of elucidating isotype-specific functions and cell-specific response. Biochim Biophys Acta 1830(7):3900–3907. doi:10.1016/j.bbagen.2012.06.003
Picou F, Fauquier T, Chatonnet F, Flamant F (2012) A bimodal influence of thyroid hormone on cerebellum oligodendrocyte differentiation. Mol Endocrinol 26(4):608–618. doi:10.1210/me.2011-1316
Dong H, Yauk CL, Rowan-Carroll A, You SH, Zoeller RT, Lambert I, Wade MG (2009) Identification of thyroid hormone receptor binding sites and target genes using ChIP-on-chip in developing mouse cerebellum. PLoS One 4(2):e4610. doi:10.1371/journal.pone.0004610
Dudazy-Gralla S, Nordstrom K, Hofmann PJ, Meseh DA, Schomburg L, Vennstrom B, Mittag J (2013) Identification of thyroid hormone response elements in vivo using mice expressing a tagged thyroid hormone receptor alpha1. Biosci Rep 33(2):e00027. doi:10.1042/BSR20120124
Bass J, Muirhead S (2000) Radiological case of the month. Pyriform sinus fistula to the left lobe of the thyroid. Arch Pediatr Adolesc Med 154(5):523–524
Brent GA, Williams GR, Harney JW, Forman BM, Samuels HH, Moore DD, Larsen PR (1991) Effects of varying the position of thyroid hormone response elements within the rat growth hormone promoter: implications for positive and negative regulation by 3,5,3′-triiodothyronine. Mol Endocrinol 5(4):542–548. doi:10.1210/mend-5-4-542
Pombo PM, Barettino D, Ibarrola N, Vega S, Rodriguez-Pena A (1999) Stimulation of the myelin basic protein gene expression by 9-cis-retinoic acid and thyroid hormone: activation in the context of its native promoter. Brain Res Mol Brain Res 64(1):92–100
Hall MG, Quignodon L, Desvergne B (2008) Peroxisome proliferator-activated receptor beta/delta in the brain: facts and hypothesis. PPAR Res 2008:780452. doi:10.1155/2008/780452
Baas D, Prufer K, Ittel ME, Kuchler-Bopp S, Labourdette G, Sarlieve LL, Brachet P (2000) Rat oligodendrocytes express the vitamin D(3) receptor and respond to 1,25-dihydroxyvitamin D(3). Glia 31(1):59–68
Hsu JH, Zavacki AM, Harney JW, Brent GA (1995) Retinoid-X receptor (RXR) differentially augments thyroid hormone response in cell lines as a function of the response element and endogenous RXR content. Endocrinology 136(2):421–430. doi:10.1210/endo.136.2.7835272
Kakizawa T, Miyamoto T, Kaneko A, Yajima H, Ichikawa K, Hashizume K (1997) Ligand-dependent heterodimerization of thyroid hormone receptor and retinoid X receptor. J Biol Chem 272(38):23799–23804
Huang JK, Jarjour AA, Nait Oumesmar B, Kerninon C, Williams A, Krezel W, Kagechika H, Bauer J, Zhao C, Evercooren AB, Chambon P, Ffrench-Constant C, Franklin RJ (2011) Retinoid X receptor gamma signaling accelerates CNS remyelination. Nat Neurosci 14(1):45–53. doi:10.1038/nn.2702
Stacpoole SR, Spitzer S, Bilican B, Compston A, Karadottir R, Chandran S, Franklin RJ (2013) High yields of oligodendrocyte lineage cells from human embryonic stem cells at physiological oxygen tensions for evaluation of translational biology. Stem cell reports 1(5):437–450. doi:10.1016/j.stemcr.2013.09.006
Baxi EG, Schott JT, Fairchild AN, Kirby LA, Karani R, Uapinyoying P, Pardo-Villamizar C, Rothstein JR, Bergles DE, Calabresi PA (2014) A selective thyroid hormone beta receptor agonist enhances human and rodent oligodendrocyte differentiation. Glia 62(9):1513–1529. doi:10.1002/glia.22697
Cao X, Kambe F, Moeller LC, Refetoff S, Seo H (2005) Thyroid hormone induces rapid activation of Akt/protein kinase B-mammalian target of rapamycin-p70S6K cascade through phosphatidylinositol 3-kinase in human fibroblasts. Mol Endocrinol 19(1):102–112. doi:10.1210/me.2004-0093
Bergh JJ, Lin HY, Lansing L, Mohamed SN, Davis FB, Mousa S, Davis PJ (2005) Integrin alphaVbeta3 contains a cell surface receptor site for thyroid hormone that is linked to activation of mitogen-activated protein kinase and induction of angiogenesis. Endocrinology 146(7):2864–2871. doi:10.1210/en.2005-0102
Lin HY, Sun M, Tang HY, Lin C, Luidens MK, Mousa SA, Incerpi S, Drusano GL, Davis FB, Davis PJ (2009) l-Thyroxine vs. 3,5,3′-triiodo-l-thyronine and cell proliferation: activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Am J Physiol Cell Physiol 296(5):C980–991. doi:10.1152/ajpcell.00305.2008
Martin NP, Marron Fernandez de Velasco E, Mizuno F, Scappini EL, Gloss B, Erxleben C, Williams JG, Stapleton HM, Gentile S, Armstrong DL (2014) A rapid cytoplasmic mechanism for PI3 kinase regulation by the nuclear thyroid hormone receptor, TRbeta, and genetic evidence for its role in the maturation of mouse hippocampal synapses in vivo. Endocrinology 155(9):3713–3724. doi:10.1210/en.2013-2058
Cao HJ, Lin HY, Luidens MK, Davis FB, Davis PJ (2009) Cytoplasm-to-nucleus shuttling of thyroid hormone receptor-beta1 (Trbeta1) is directed from a plasma membrane integrin receptor by thyroid hormone. Endocr Res 34(1–2):31–42. doi:10.1080/07435800902911810
Flores AI, Narayanan SP, Morse EN, Shick HE, Yin X, Kidd G, Avila RL, Kirschner DA, Macklin WB (2008) Constitutively active Akt induces enhanced myelination in the CNS. J Neurosci 28(28):7174–7183. doi:10.1523/JNEUROSCI.0150-08.2008
Harrington EP, Zhao C, Fancy SP, Kaing S, Franklin RJ, Rowitch DH (2010) Oligodendrocyte PTEN is required for myelin and axonal integrity, not remyelination. Ann Neurol 68(5):703–716. doi:10.1002/ana.22090
Milner R, Ffrench-Constant C (1994) A developmental analysis of oligodendroglial integrins in primary cells: changes in alpha v-associated beta subunits during differentiation. Development 120(12):3497–3506
Blaschuk KL, Frost EE, ffrench-Constant C (2000) The regulation of proliferation and differentiation in oligodendrocyte progenitor cells by alphaV integrins. Development 127(9):1961–1969
Baron W, Shattil SJ, ffrench-Constant C (2002) The oligodendrocyte precursor mitogen PDGF stimulates proliferation by activation of alpha(v)beta3 integrins. Embo J 21(8):1957–1966. doi:10.1093/emboj/21.8.1957
Baas D, Bourbeau D, Sarlieve LL, Ittel ME, Dussault JH, Puymirat J (1997) Oligodendrocyte maturation and progenitor cell proliferation are independently regulated by thyroid hormone. Glia 19(4):324–332
Tokumoto YM, Durand B, Raff MC (1999) An analysis of the early events when oligodendrocyte precursor cells are triggered to differentiate by thyroid hormone, retinoic acid, or PDGF withdrawal. Dev Biol 213(2):327–339. doi:10.1006/dbio.1999.9397
Alsanie WF, Niclis JC, Petratos S (2013) Human embryonic stem cell-derived oligodendrocytes: protocols and perspectives. Stem Cells Dev 22(18):2459–2476. doi:10.1089/scd.2012.0520
Liu X, Li Y, Zhang Y, Lu Y, Guo W, Liu P, Zhou J, Xiang Z, He C (2011) SHP-2 promotes the maturation of oligodendrocyte precursor cells through Akt and ERK1/2 signaling in vitro. PLoS One 6(6):e21058. doi:10.1371/journal.pone.0021058
Ehrman LA, Nardini D, Ehrman S, Rizvi TA, Gulick J, Krenz M, Dasgupta B, Robbins J, Ratner N, Nakafuku M, Waclaw RR (2014) The protein tyrosine phosphatase Shp2 is required for the generation of oligodendrocyte progenitor cells and myelination in the mouse telencephalon. J Neurosci 34(10):3767–3778. doi:10.1523/JNEUROSCI.3515-13.2014
Davis PJ, Davis FB, Blas SD (1982) Studies on the mechanism of thyroid hormone stimulation in vitro of human red cell Ca2+−ATPase activity. Life Sci 30(7–8):675–682
Plow EF, Haas TA, Zhang L, Loftus J, Smith JW (2000) Ligand binding to integrins. J Biol Chem 275(29):21785–21788. doi:10.1074/jbc.R000003200
Rowitch DH, Kriegstein AR (2010) Developmental genetics of vertebrate glial-cell specification. Nature 468(7321):214–222. doi:10.1038/nature09611
Hasebe M, Ohta E, Imagawa T, Uehara M (2008) Expression of sonic hedgehog regulates morphological changes of rat developing cerebellum in hypothyroidism. J Toxicol Sci 33(4):473–477
Desouza LA, Sathanoori M, Kapoor R, Rajadhyaksha N, Gonzalez LE, Kottmann AH, Tole S, Vaidya VA (2011) Thyroid hormone regulates the expression of the sonic hedgehog signaling pathway in the embryonic and adult Mammalian brain. Endocrinology 152(5):1989–2000. doi:10.1210/en.2010-1396
Freitas BC, Gereben B, Castillo M, Kallo I, Zeold A, Egri P, Liposits Z, Zavacki AM, Maciel RM, Jo S, Singru P, Sanchez E, Lechan RM, Bianco AC (2010) Paracrine signaling by glial cell-derived triiodothyronine activates neuronal gene expression in the rodent brain and human cells. J Clin Invest 120(6):2206–2217. doi:10.1172/JCI41977
Fernandez M, Pirondi S, Manservigi M, Giardino L, Calza L (2004) Thyroid hormone participates in the regulation of neural stem cells and oligodendrocyte precursor cells in the central nervous system of adult rat. Eur J Neurosci 20(8):2059–2070. doi:10.1111/j.1460-9568.2004.03664.x
Orentas DM, Hayes JE, Dyer KL, Miller RH (1999) Sonic hedgehog signaling is required during the appearance of spinal cord oligodendrocyte precursors. Development 126(11):2419–2429
Alves C, Eidson M, Engle H, Sheldon J, Cleveland WW (1989) Changes in brain maturation detected by magnetic resonance imaging in congenital hypothyroidism. J Pediatr 115(4):600–603
Gupta RK, Bhatia V, Poptani H, Gujral RB (1995) Brain metabolite changes on in vivo proton magnetic resonance spectroscopy in children with congenital hypothyroidism. J Pediatr 126(3):389–392
Jagannathan NR, Tandon N, Raghunathan P, Kochupillai N (1998) Reversal of abnormalities of myelination by thyroxine therapy in congenital hypothyroidism: localized in vivo proton magnetic resonance spectroscopy (MRS) study. Brain Res Dev Brain Res 109(2):179–186
Barres BA, Lazar MA, Raff MC (1994) A novel role for thyroid hormone, glucocorticoids and retinoic acid in timing oligodendrocyte development. Development 120(5):1097–1108
Nygard M, Wahlstrom GM, Gustafsson MV, Tokumoto YM, Bondesson M (2003) Hormone-dependent repression of the E2F-1 gene by thyroid hormone receptors. Mol Endocrinol 17(1):79–92. doi:10.1210/me.2002-0107
Rodriguez-Pena A, Ibarrola N, Iniguez MA, Munoz A, Bernal J (1993) Neonatal hypothyroidism affects the timely expression of myelin-associated glycoprotein in the rat brain. J Clin Invest 91(3):812–818. doi:10.1172/JCI116301
Ibarrola N, Rodriguez-Pena A (1997) Hypothyroidism coordinately and transiently affects myelin protein gene expression in most rat brain regions during postnatal development. Brain Res 752(1–2):285–293
Barradas PC, Vieira RS, De Freitas MS (2001) Selective effect of hypothyroidism on expression of myelin markers during development. J Neurosci Res 66(2):254–261
Farsetti A, Desvergne B, Hallenbeck P, Robbins J, Nikodem VM (1992) Characterization of myelin basic protein thyroid hormone response element and its function in the context of native and heterologous promoter. J Biol Chem 267(22):15784–15788
Lalonde R, Strazielle C (2007) Brain regions and genes affecting postural control. Prog Neurobiol 81(1):45–60. doi:10.1016/j.pneurobio.2006.11.005
Martel J, Cayrou C, Puymirat J (2002) Identification of new thyroid hormone-regulated genes in rat brain neuronal cultures. Neuroreport 13(15):1849–1851
Cayrou C, Denver RJ, Puymirat J (2002) Suppression of the basic transcription element-binding protein in brain neuronal cultures inhibits thyroid hormone-induced neurite branching. Endocrinology 143(6):2242–2249. doi:10.1210/endo.143.6.8856
Denver RJ, Williamson KE (2009) Identification of a thyroid hormone response element in the mouse Kruppel-like factor 9 gene to explain its postnatal expression in the brain. Endocrinology 150(8):3935–3943. doi:10.1210/en.2009-0050
Moore DL, Blackmore MG, Hu Y, Kaestner KH, Bixby JL, Lemmon VP, Goldberg JL (2009) KLF family members regulate intrinsic axon regeneration ability. Science 326(5950):298–301. doi:10.1126/science.1175737
Avci HX, Lebrun C, Wehrle R, Doulazmi M, Chatonnet F, Morel MP, Ema M, Vodjdani G, Sotelo C, Flamant F, Dusart I (2012) Thyroid hormone triggers the developmental loss of axonal regenerative capacity via thyroid hormone receptor alpha1 and kruppel-like factor 9 in Purkinje cells. Proc Natl Acad Sci U S A 109(35):14206–14211. doi:10.1073/pnas.1119853109
Haas MJ, Mreyoud A, Fishman M, Mooradian AD (2004) Microarray analysis of thyroid hormone-induced changes in mRNA expression in the adult rat brain. Neurosci Lett 365(1):14–18. doi:10.1016/j.neulet.2004.04.028
Jo S, Kallo I, Bardoczi Z, Arrojo e Drigo R, Zeold A, Liposits Z, Oliva A, Lemmon VP, Bixby JL, Gereben B, Bianco AC (2012) Neuronal hypoxia induces Hsp40-mediated nuclear import of type 3 deiodinase as an adaptive mechanism to reduce cellular metabolism. J Neurosci 32(25):8491–8500. doi:10.1523/JNEUROSCI.6514-11.2012
Fuss B, Baba H, Phan T, Tuohy VK, Macklin WB (1997) Phosphodiesterase I, a novel adhesion molecule and/or cytokine involved in oligodendrocyte function. J Neurosci 17(23):9095–9103
Fox MA, Colello RJ, Macklin WB, Fuss B (2003) Phosphodiesterase-Ialpha/autotaxin: a counteradhesive protein expressed by oligodendrocytes during onset of myelination. Mol Cell Neurosci 23(3):507–519
Dennis J, White MA, Forrest AD, Yuelling LM, Nogaroli L, Afshari FS, Fox MA, Fuss B (2008) Phosphodiesterase-Ialpha/autotaxin’s MORFO domain regulates oligodendroglial process network formation and focal adhesion organization. Mol Cell Neurosci 37(2):412–424. doi:10.1016/j.mcn.2007.10.018
Wheeler NA, Lister JA, Fuss B (2015) The autotaxin-lysophosphatidic acid axis modulates histone acetylation and gene expression during oligodendrocyte differentiation. J Neurosci 35(32):11399–11414. doi:10.1523/JNEUROSCI.0345-15.2015
Mosser J, Douar AM, Sarde CO, Kioschis P, Feil R, Moser H, Poustka AM, Mandel JL, Aubourg P (1993) Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature 361(6414):726–730. doi:10.1038/361726a0
Fouquet F, Zhou JM, Ralston E, Murray K, Troalen F, Magal E, Robain O, Dubois-Dalcq M, Aubourg P (1997) Expression of the adrenoleukodystrophy protein in the human and mouse central nervous system. Neurobiol Dis 3(4):271–285. doi:10.1006/nbdi.1997.0127
Moser HW, Mahmood A, Raymond GV (2007) X-linked adrenoleukodystrophy. Nat Clin Pract Neurol 3(3):140–151. doi:10.1038/ncpneuro0421
Hein S, Schonfeld P, Kahlert S, Reiser G (2008) Toxic effects of X-linked adrenoleukodystrophy-associated, very long chain fatty acids on glial cells and neurons from rat hippocampus in culture. Hum Mol Genet 17(12):1750–1761. doi:10.1093/hmg/ddn066
Kemp S, Wei HM, Lu JF, Braiterman LT, McGuinness MC, Moser AB, Watkins PA, Smith KD (1998) Gene redundancy and pharmacological gene therapy: implications for X-linked adrenoleukodystrophy. Nat Med 4(11):1261–1268. doi:10.1038/3242
Pai GS, Khan M, Barbosa E, Key LL, Craver JR, Cure JK, Betros R, Singh I (2000) Lovastatin therapy for X-linked adrenoleukodystrophy: clinical and biochemical observations on 12 patients. Mol Genet Metab 69(4):312–322
Jang J, Kang HC, Kim HS, Kim JY, Huh YJ, Kim DS, Yoo JE, Lee JA, Lim B, Lee J, Yoon TM, Park IH, Hwang DY, Daley GQ, Kim DW (2011) Induced pluripotent stem cell models from X-linked adrenoleukodystrophy patients. Ann Neurol 70(3):402–409. doi:10.1002/ana.22486
Fourcade S, Savary S, Gondcaille C, Berger J, Netik A, Cadepond F, El Etr M, Molzer B, Bugaut M (2003) Thyroid hormone induction of the adrenoleukodystrophy-related gene (ABCD2). Mol Pharmacol 63(6):1296–1303. doi:10.1124/mol.63.6.1296
Weinhofer I, Kunze M, Rampler H, Forss-Petter S, Samarut J, Plateroti M, Berger J (2008) Distinct modulatory roles for thyroid hormone receptors TRalpha and TRbeta in SREBP1-activated ABCD2 expression. Eur J Cell Biol 87(12):933–945. doi:10.1016/j.ejcb.2008.08.002
Genin EC, Gondcaille C, Trompier D, Savary S (2009) Induction of the adrenoleukodystrophy-related gene (ABCD2) by thyromimetics. J Steroid Biochem Mol Biol 116(1–2):37–43. doi:10.1016/j.jsbmb.2009.04.006
Kassmann CM (2014) Myelin peroxisomes—essential organelles for the maintenance of white matter in the nervous system. Biochimie 98:111–118. doi:10.1016/j.biochi.2013.09.020
Kassmann CM, Lappe-Siefke C, Baes M, Brugger B, Mildner A, Werner HB, Natt O, Michaelis T, Prinz M, Frahm J, Nave KA (2007) Axonal loss and neuroinflammation caused by peroxisome-deficient oligodendrocytes. Nat Genet 39(8):969–976. doi:10.1038/ng2070
Calza L, Fernandez M, Giuliani A, Aloe L, Giardino L (2002) Thyroid hormone activates oligodendrocyte precursors and increases a myelin-forming protein and NGF content in the spinal cord during experimental allergic encephalomyelitis. Proc Natl Acad Sci U S A 99(5):3258–3263. doi:10.1073/pnas.052704499
Fernandez M, Paradisi M, Del Vecchio G, Giardino L, Calza L (2009) Thyroid hormone induces glial lineage of primary neurospheres derived from non-pathological and pathological rat brain: implications for remyelination-enhancing therapies. Int J Dev Neurosci 27(8):769–778. doi:10.1016/j.ijdevneu.2009.08.011
D’Intino G, Lorenzini L, Fernandez M, Taglioni A, Perretta G, Del Vecchio G, Villoslada P, Giardino L, Calza L (2011) Triiodothyronine administration ameliorates the demyelination/remyelination ratio in a non-human primate model of multiple sclerosis by correcting tissue hypothyroidism. J Neuroendocrinol 23(9):778–790. doi:10.1111/j.1365-2826.2011.02181.x
Franco PG, Silvestroff L, Soto EF, Pasquini JM (2008) Thyroid hormones promote differentiation of oligodendrocyte progenitor cells and improve remyelination after cuprizone-induced demyelination. Exp Neurol 212(2):458–467. doi:10.1016/j.expneurol.2008.04.039
Harsan LA, Steibel J, Zaremba A, Agin A, Sapin R, Poulet P, Guignard B, Parizel N, Grucker D, Boehm N, Miller RH, Ghandour MS (2008) Recovery from chronic demyelination by thyroid hormone therapy: myelinogenesis induction and assessment by diffusion tensor magnetic resonance imaging. J Neurosci 28(52):14189–14201. doi:10.1523/JNEUROSCI.4453-08.2008
Silvestroff L, Bartucci S, Pasquini J, Franco P (2012) Cuprizone-induced demyelination in the rat cerebral cortex and thyroid hormone effects on cortical remyelination. Exp Neurol 235(1):357–367. doi:10.1016/j.expneurol.2012.02.018
Castelo-Branco G, Stridh P, Guerreiro-Cacais AO, Adzemovic MZ, Falcao AM, Marta M, Berglund R, Gillett A, Hamza KH, Lassmann H, Hermanson O, Jagodic M (2014) Acute treatment with valproic acid and l-thyroxine ameliorates clinical signs of experimental autoimmune encephalomyelitis and prevents brain pathology in DA rats. Neurobiol Dis 71:220–233. doi:10.1016/j.nbd.2014.08.019
Hung PL, Huang CC, Huang HM, Tu DG, Chang YC (2013) Thyroxin treatment protects against white matter injury in the immature brain via brain-derived neurotrophic factor. Stroke 44(8):2275–2283. doi:10.1161/STROKEAHA.113.001552
McLachlan SM, Rapoport B (2014) Breaking tolerance to thyroid antigens: changing concepts in thyroid autoimmunity. Endocr Rev 35(1):59–105. doi:10.1210/er.2013-1055
Nakano A, Watanabe M, Iida T, Kuroda S, Matsuzuka F, Miyauchi A, Iwatani Y (2007) Apoptosis-induced decrease of intrathyroidal CD4(+)CD25(+) regulatory T cells in autoimmune thyroid diseases. Thyroid 17(1):25–31. doi:10.1089/thy.2006.0231
Zambrano A, Garcia-Carpizo V, Gallardo ME, Villamuera R, Gomez-Ferreria MA, Pascual A, Buisine N, Sachs LM, Garesse R, Aranda A (2014) The thyroid hormone receptor beta induces DNA damage and premature senescence. J Cell Biol 204(1):129–146. doi:10.1083/jcb.201305084
Robbins DS, Shirazi Y, Drysdale BE, Lieberman A, Shin HS, Shin ML (1987) Production of cytotoxic factor for oligodendrocytes by stimulated astrocytes. J Immunol 139(8):2593–2597
Selmaj KW, Raine CS (1988) Tumor necrosis factor mediates myelin and oligodendrocyte damage in vitro. Ann Neurol 23(4):339–346. doi:10.1002/ana.410230405
Andrews T, Zhang P, Bhat NR (1998) TNFalpha potentiates IFNgamma-induced cell death in oligodendrocyte progenitors. J Neurosci Res 54(5):574–583
Merrill JE (1991) Effects of interleukin-1 and tumor necrosis factor-alpha on astrocytes, microglia, oligodendrocytes, and glial precursors in vitro. Dev Neurosci 13(3):130–137
Boelen A, Kwakkel J, Platvoet-ter Schiphorst M, Baur A, Kohrle J, Wiersinga WM (2004) Contribution of interleukin-12 to the pathogenesis of non-thyroidal illness. Horm Metab Res 36(2):101–106. doi:10.1055/s-2004-814219
Schang AL, Van Steenwinckel J, Chevenne D, Alkmark M, Hagberg H, Gressens P, Fleiss B (2014) Failure of thyroid hormone treatment to prevent inflammation-induced white matter injury in the immature brain. Brain Behav Immun 37:95–102. doi:10.1016/j.bbi.2013.11.005
Mdzinarishvili A, Sutariya V, Talasila PK, Geldenhuys WJ, Sadana P (2013) Engineering triiodothyronine (T3) nanoparticle for use in ischemic brain stroke. Drug Deliv Transl Res 3(4):309–317. doi:10.1007/s13346-012-0117-8
Acknowledgments
JYL is supported by a Multiple Sclerosis Research Australia Postgraduate Scholarship and the Trish Multiple Sclerosis Research Foundation; SP is supported by a National Multiple Sclerosis Society Project grant (no. RG4398A1/1), an International Progressive Multiple Sclerosis Alliance Challenge award (no. PA0065), and the Trish Multiple Sclerosis Research Foundation administered through the Multiple Sclerosis Research Australia Project grant (no. 15-022).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Lee, J.Y., Petratos, S. Thyroid Hormone Signaling in Oligodendrocytes: from Extracellular Transport to Intracellular Signal. Mol Neurobiol 53, 6568–6583 (2016). https://doi.org/10.1007/s12035-016-0013-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-0013-1