
Abstract
Glucose transporters (GLUTs) at the blood-brain barrier maintain the continuous high glucose and energy demands of the brain. They also act as therapeutic targets and provide routes of entry for drug delivery to the brain and central nervous system for treatment of neurological and neurovascular conditions and brain tumours. This article first describes the distribution, function and regulation of glucose transporters at the blood-brain barrier, the major ones being the sodium-independent facilitative transporters GLUT1 and GLUT3. Other GLUTs and sodium-dependent transporters (SGLTs) have also been identified at lower levels and under various physiological conditions. It then considers the effects on glucose transporter expression and distribution of hypoglycemia and hyperglycemia associated with diabetes and oxygen/glucose deprivation associated with cerebral ischemia. A reduction in glucose transporters at the blood-brain barrier that occurs before the onset of the main pathophysiological changes and symptoms of Alzheimer’s disease is a potential causative effect in the vascular hypothesis of the disease. Mutations in glucose transporters, notably those identified in GLUT1 deficiency syndrome, and some recreational drug compounds also alter the expression and/or activity of glucose transporters at the blood-brain barrier. Approaches for drug delivery across the blood-brain barrier include the pro-drug strategy whereby drug molecules are conjugated to glucose transporter substrates or encapsulated in nano-enabled delivery systems (e.g. liposomes, micelles, nanoparticles) that are functionalised to target glucose transporters. Finally, the continuous development of blood-brain barrier in vitro models is important for studying glucose transporter function, effects of disease conditions and interactions with drugs and xenobiotics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The blood-brain barrier is a structural and chemical barrier between the brain and the systemic circulation that tightly regulates the transport of substances between the blood and brain. This protects the brain from exposure to variations in blood composition and to toxic compounds. Indeed, some molecules that are harmless to peripheral organs and tissues may be toxic to neurons in the brain. At the capillary level, the neurovascular unit at the blood-brain barrier (Fig. 1) contains a network of specialised endothelial cells that are lined by the basal lamina (basement membrane). The overall permeability of the blood-brain barrier is regulated by these endothelial cells and their junctional complexes, which consist of adherens junctions and tight junctions. The end feet of astrocytes encase the large majority of the capillary wall and the astrocytes communicate with the neurons. Pericytes are also in contact with the capillary wall and they communicate with endothelial cells through synapse-like contacts. Resting or ‘ramified’ microglia are also present for sensing neuronal injury and for detecting and fighting infection, whilst maintaining an immunologically silent environment. Together, this unit regulates blood-brain barrier permeability and protects the integrity and function of the brain and central nervous system. The human brain is comprised of about 100 billion neurons and a similar number of glial cells. The neurons, pericytes, astrocytes and microglia account for almost 80 % of the brain volume; the extracellular space occupies 15–30 % and the brain vasculature about 3 % [1]. Under normal physiological conditions, the neurovascular unit prevents bacteria, ions, large molecules and most small molecules crossing from the blood into the brain. Water molecules and small ions (e.g. Na+, K+, Cl−) cross the blood-brain barrier via channels. Small gaseous molecules (e.g. oxygen, carbon dioxide) and small lipophilic molecules (less than 500 Da, logP oct 2–4, less than 5 hydrogen bond donors) [2, 3] and that are not substrates of active efflux transporters (e.g. anaesthetics, ethanol, nicotine, caffeine) can cross the endothelial cells by passive diffusion. Other molecules can only cross into the brain if they are carried by transport proteins or by receptor- or adsorptive-mediated transport processes. Similarly, most waste products have to leave the brain by the way of active efflux transporters. Under conditions where the integrity of the blood-brain barrier is compromised (e.g. inflammation, traumatic brain injury, ischemic stroke), then larger and hydrophilic substances may be able to pass. A number of comprehensive reviews/descriptions of the structure and functions of the blood-brain barrier and neurovascular unit are available [4–11].

Major components of the neurovascular unit at the blood-brain barrier. a This figure was reproduced from Wong et al. (2013) [9], which was originally published in Frontiers in Neuroengineering. Copyright by Wong, Ye, Levy, Rothstein, Bergles and Searson 2013. b This figure was reproduced with permission from Abbott et al. (2010) [7], which was originally published in Neurobiology of Disease. Copyright by Elsevier Inc. 2009
It has been estimated that 10–15 % of all proteins in the neurovascular unit are transporters [12]. Transport proteins on the endothelial cells are found in both the luminal (blood-facing) and abluminal (brain-facing) membranes to allow the passage of nutrients, peptides and ions and the efflux of drugs and waste or harmful compounds, examples of which are shown in Fig. 2. Some of the proteins function through a mechanism of facilitated diffusion that allows bi-directional transport, whilst others are sodium-dependent active transporters that achieve unidirectional concentrative transport. Some of the examples shown in Fig. 2 have a mechanism of receptor-mediated transcytosis. The majority of the proteins are expressed at both the luminal and abluminal membranes, whilst some are found only at one side of the endothelial cell. The full complement of transport mechanisms at the blood-brain barrier in humans is yet to be clarified, but those that transport glucose are especially important. The human brain is almost entirely dependent upon glucose as an energy source, taking in around 100–150 g of glucose per day [21]. Indeed, the normal adult brain constitutes around 2 % of the body weight yet consumes around 20 % of glucose in the body [22], and it has been estimated that endothelial cells at the blood-brain barrier transport around ten times their weight of glucose per minute to support the glucose requirements of the brain [23]. Glucose transport to the brain involves numerous interactions of solutes, transporters, enzymes and cell signalling processes within the complex architecture of the neurovascular unit at the blood-brain barrier. The transport of the highly hydrophilic glucose molecule across the blood-brain barrier is principally achieved by the sodium-independent facilitative transporter GLUT1, which is shown at the top of Fig. 2 in both the luminal and abluminal membranes. The brain rapidly catabolises glucose and this creates a downhill gradient for transport of glucose by GLUT1 from the blood towards the brain interstitial fluid. Given the highly important role of GLUT1 in feeding energy to the brain, it is clear that GLUT1 is essential for maintaining normal neurological functions and anything affecting the normal expression or functioning of GLUT1 can have severe consequences on human health. Other glucose transporter (GLUT) and sodium-dependent transporters (SGLTs) are also involved in glucose transport at the neurovascular unit of the blood-brain barrier. Transport proteins for glucose at the neurovascular unit of the blood-brain barrier will be the main focus of this article.

Transport proteins on endothelial cells of the blood-brain barrier. Examples of major transport proteins found in the luminal and abluminal membranes of endothelial cells are illustrated by ovals and grouped based on the nature of their substrates as follows. Transporters of nutrients (green): facilitative bi-directional transporters GLUT1 (glucose), MCT1 (lactate), L1 (large essential neutral amino acids) and Y+ (cationic essential amino acids) and sodium-dependent concentrative transporters A (small non-essential neutral amino acids), ASC (non-essential amino acids) and EAATs 1–3 (glutamate). Transporters of peptides (yellow): receptor-mediated transcytosis of insulin and transferrin by the insulin receptor (IR) and the transferring receptor (TFR), respectively, unidirectional transport of amyloid-β by LRP1 and facilitative bi-directional transport of encephalins by PTS1. ABC transporters (pink): active drug efflux proteins ABCB1 (P-glycoprotein) and ABCG2 and unidirectional thyroid transporters MCT8 and OATP1C1. Transporters of ions (blue): Na+,K+-ATPase. The arrows indicate the direction(s) of transport. This diagram was constructed based on information given in Kido et al. (2000), Hawkins et al. (2006), Deane et al. (2009), Zlokovic (2011), Pardridge (2012), Banks et al. (2012), Bien-Ly et al. (2014), Strazielle and Ghersi-Egea (2015), Wittmann et al. (2015) [4, 13–20]
Whilst mutations and the binding of chemical inhibitors can affect the structure, function and activity of glucose transporters, conditions such as hypoglycemia, hyperglycemia, diabetes, neurodegenerative disorders (e.g. Alzheimer’s disease), traumatic brain injury and cerebral ischemia can alter their expression, distribution and activity at the blood-brain barrier. Some of these are of course interlinked, and there are potential benefits from intervening with therapeutic drugs that use glucose transporters at the blood-brain barrier as their molecular target. Along with other solute carriers, glucose transporters at the blood-brain barrier are also potential routes of entry for delivering drugs and pro-drugs to the brain for preventing or treating neurological disorders and central nervous system diseases including mental disorders, migraine, epilepsy, neurodegenerative diseases such as Alzheimer’s and Parkinson’s, cerebrovascular diseases such as cerebral ischemia and stroke, cancer, inflammatory diseases such as multiple sclerosis, brain trauma and infections such as meningitis. It is generally not feasible to perform in vivo studies on human glucose transporters at the blood-brain barrier, so in vitro models have to be used, which is an ongoing theme of development. These topics will be covered in this article.
Glucose Transporters at the Blood-Brain Barrier
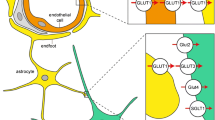
Two different types of glucose transporter are found in the neurovascular unit at the blood-brain barrier. By far the most prevalent are sodium-independent bi-directional facilitative transporters from the solute carrier 2 (SLC2) family of which 14 isoforms (GLUTs 1–14) have been identified and reviewed extensively [24–33]. The GLUT family proteins are members of the sugar porter sub-family of the large and widespread major facilitator superfamily (MFS) of secondary transport proteins [34–36]. They share a high-sequence similarity (19–65 % identity, 39–81 % homology) [37] and a number of structural features including 12 putative transmembrane-spanning α-helices and a single-site of N-linked glycosylation. The others are sodium-dependent unidirectional concentrative transporters from the solute carrier 5 family (SLC5) family of which 12 isoforms (SGLTs 1–12) have been identified [38, 39]. The SGLT family sugar transporters have 14 putative transmembrane-spanning α-helices and a single-site of N-linked glycosylation. Under normal circumstances, glucose crosses the endothelial cells of the blood-brain barrier by way of GLUT1, as already described. Once in the brain, extracellular space glucose is rapidly taken up by the different types of brain cells including astrocytes, microglia and neurons. The distribution and expression levels of different isoforms of both the GLUT and SGLT transporters is cell type-specific and also dependent on developmental and physiological conditions. GLUTs 1–8 and SGLTs 1 and 2 in the cells of the neurovascular unit at the blood-brain barrier have been identified at various locations and levels of expression (Table 1), where GLUT1 and GLUT3 are the major glucose transporters (Fig. 3).

Movement of glucose molecules across the neurovascular unit at the blood-brain barrier. Under normal physiological conditions, glucose molecules (black dots) move from a higher concentration in the blood flow (4–6 mM) across endothelial cells and the basal membrane to a lower concentration (1–2 mM, [80–85]) in the brain extracellular (interstitial) fluid and then into astrocytes, neurons and microglia. Glucose is constantly being ‘pulled’ in this direction by the high-energy demands of brain cells and its metabolism by the action of hexokinase. Facilitative glucose transporters (GLUTs, ovals) and sodium-dependent glucose transporters (SGLTs, oblongs) are responsible for transporting glucose across membranes as shown. The most abundant glucose transporter is GLUT1 (red), which is present in red blood cells and endothelial cells in heavily glycosylated form (55 kDa, thick border) and in astrocytes, neurons and microglia in low glycosylated form (45 kDa, thin border). In neurons, GLUT3 (pink) is the major glucose transporter. In microglia, GLUT5 (green) is the major GLUT transporter, which transports fructose and only has a low affinity for glucose. Other glucose transporters have been detected on cells at the neurovascular unit of the blood-brain barrier (Table 1) as indicated: GLUT2 (purple), GLUT4 (blue), GLUT6 (gold), GLUT7 (pale green), GLUT8 (cyan), SGLT1 (yellow), SGLT2 (orange)
Facilitative Glucose Transporter GLUT1
Given the widespread distribution of GLUT1 and its crucial roles in the human body, not least in the blood-brain barrier, GLUT1 has become one of the most extensively studied of all membrane transport proteins. GLUT1 was the first equilibrative glucose transporter to be identified, purified and cloned [40–43]; and a recent X-ray crystal structure of human GLUT1 at 3.2 Å resolution has been determined with the protein in an inward-open conformation (PDB 4PYP) [44]. The structure constitutes an overall MFS and predicted GLUT protein fold with 12 transmembrane-spanning α-helices arranged in two distinct N- and C-terminal domains of six helices, cytoplasmic N- and C-terminal ends, a large intracellular loop between helices 6 and 7 and a single-site of N-linked glycosylation on one of the extracellular loops (Fig. 4a). It also has an intracellular helical bundle comprised of four short α-helices that connects the N- and C-terminal domains, which was also seen in structures of the homologous proton-coupled active bacterial sugar porter proteins XylE [45] and GlcP [37]. The structure of GLUT1 has allowed an accurate mapping of disease-associated mutations and provided further insight into the alternating access mechanism of transport in GLUT proteins and its relation to the transport mechanism in homologous active sugar porters [44]. The preferred substrates of GLUT1 are hexose and pentose sugars that adopt a pyranose conformation including d-glucose in both its α- and β-pyranose forms [46] (Fig. 4a), which are recognised with equal affinity [47]. GLUT1 transports d-glucose with apparent affinities (K m app values) of around 1.5 mM, 1–2 and 3 mM when examined in erythrocytes [48], reconstituted in liposomes [49, 50] and when expressed in Xenopus laevis oocytes [51–54], respectively. In addition to d-glucose, GLUT1 transports glucose analogues including 2-deoxy-d-glucose and 3-O-methyl-d-glucose (Fig. 4b), other hexoses including galactose, mannose and glucosamine and it also transports the oxidised form of vitamin C, dehydroascorbic acid (Fig. 4b), in order to confer mitochondrial protection against oxidative injury [55–57]. Interestingly, GLUT1 has been identified as a facilitator for the uptake of trivalent arsenicals such as arsenite [As(OH)3] and methylarsenite [CH3As(OH)2], which have a different translocation pathway in GLUT1 compared with that of glucose transport [58]. Transport of the radiolabelled glucose analogue 2-deoxy-2-[18F]fluoro-d-glucose (Fig. 4b) by GLUT1 provides the basis for measuring glycolytic activity in cells and tissues by the nuclear medicine technique of positron emission tomography (PET), which allows the diagnosis and monitoring of a wide range of human diseases [59]. The transport activity of GLUT1 is inhibited by a number of different compounds including cytochalasin B, forskolin, phloretin and other flavonoids, maltose and mercuric chloride, which all have low micromolar affinities [60–64], and these have been used in a range of experimental studies of GLUT1 sugar transport and function.

The human facilitative glucose transport protein GLUT1. a Crystal structure of GLUT1 illustrated in a cell membrane catalysing the bi-directional transport of d-glucose. The transported glucose can be metabolised by the glycolytic pathway, the first step being conversion to glucose-6-phosphate catalysed by hexokinase. The structure of GLUT1 is coloured with the N-terminus in blue and the C-terminus in red, which was drawn using PDB file 4PYP and PDB Protein Workshop 3.9. b Structures of transported glucose analogues (i) 2-deoxy-d-glucose, (ii) 3-O-methyl-d-glucose and (iii) 2-deoxy-2-fluoro-d-glucose and the structure of dehydroascorbic acid (iv), which is also transported by GLUT1
At the blood-brain barrier, a high density of GLUT1 is found in both luminal and abluminal membranes of endothelial cells in heavily glycosylated, high molecular weight form (55 kDa). This isoform of GLUT1 is also found in human erythrocytes (Fig. 3). Quantitative measurements suggest an asymmetric distribution of GLUT1 at the luminal and abluminal membranes and up to 40 % of the GLUT1 protein may be sequestered within the cell cytoplasm at any given time [65, 66] (Fig. 3). A number of other studies have quantified the relative amounts of GLUT1 in luminal and abluminal membranes and cytoplasm from humans and from other mammals with variable results [67–74]. A change in these ratios for the distribution of GLUT1 is a probable mechanism for achieving a change in the rate of glucose transport across the blood-brain barrier in response to changes in energy demand and other physiological conditions. An increased luminal to abluminal ratio is likely to favour an increase in glucose transport to the brain. Some studies have suggested that the conformations of GLUT1 are different in the luminal and abluminal membranes of brain endothelial cells. One of these studies demonstrated that the different conformations originate from differential phosphorylation of GLUT1 and not from alternative splicing or altered glycosylation [75].
GLUT1 provides steady transport for the high demand of glucose required to supply the high rate of aerobic metabolism in the brain and for maintaining neuronal homeostasis. There are a number of saturable and unsaturable components that regulate the transport of glucose from the blood to the brain. The facilitated transport of glucose by GLUT1 is saturable, whilst there are also unsaturable intra- and inter-cellular unsaturable diffusion processes that have to occur (Fig. 3). The half-saturation constant of glucose uptake into the brain (K t ) is around 8 mM [76]. The diffusion coefficient of d-glucose in the cytosol of a single astrocyte has been measured with an apparent value (D app) of 2.38 ± 0.41 × 10−10 m2 s−1 at 22–24 °C compared with a D app value for d-glucose in water of 6.7 × 10−10 m2 s−1 at 24 °C [77]. Although diffusion of d-glucose in the cytoplasm is hindered by around threefold compared with that in aqueous solution, cytosolic diffusion is likely to contribute to the movement of glucose across endothelial cells and from endothelial cells to astrocytes and neurons at the blood-brain barrier. Hexokinase I (brain hexokinase), the enzyme involved in the first step of glycolysis, which catalyses the conversion of glucose to glucose-6-phosphate (Fig. 4a), has a K m for glucose of around 40–50 μM [78, 79]. In the extracellular (interstitial) fluid of the healthy human brain, glucose levels are around 1–2 mM [80–85], so the capacity of hexokinase for glucose is significantly greater than the transport by GLUT1 [86]. Under normal circumstances, it is generally considered that brain glycolysis is not limited by glucose transport, but by phosphorylation of glucose to glucose-6-phosphate. Some studies using in vitro models of the blood-brain barrier have suggested that the location and concentration of hexokinase can alter the ratio of GLUT1 at the luminal and abluminal membranes [71]. The concentration and expression of GLUT1 in endothelial cells, which appears to be under both transcriptional and post-transcriptional control [87, 88], is regulated by the circulating concentrations of glucose, variations in energy demands during different stages of brain development and changes in physiological conditions [89–91]. For example, GLUT1 plays crucial roles in the development of the blood-brain barrier and in other early stages of brain development, during which there are increased levels of GLUT1 expression associated with increases in cerebral glucose utilisation and energy demands [91–95]. In contrast, a decrease in the efficiency of GLUT1 upregulation has been associated with hot flashes during menopause. A decline in oestrogen levels at menopause causes the upregulation of GLUT1 to be less efficient, and it is proposed that there is a consequent overcompensation in neurobarrier coupling with an excess neurovascular response, or a hot flash [96].
Elsewhere in the neurovascular unit at the blood-brain barrier, GLUT1 is also the major glucose transporter in the membranes of astrocytes, but here it is present in its less glycosylated, lower molecular weight form (45 kDa) [70, 97, 98] (Table 1 and Fig. 3). Whilst some studies suggest that the different forms of GLUT1 do not appear to differ in their protein structure or kinetic characteristics [99], others suggest that the different levels of glycosylation have implications for transport activity [100], intracellular targeting and protein stability [101], substrate affinity [102] and GLUT1 trafficking [103]. The 45 kDa form of GLUT1 has also been detected at low levels in microglia [104, 105] and in neurons, especially in the foetal brain [93, 106].
Other GLUT-Facilitative Glucose Transporters
GLUT3 is the major glucose transporter in neurons (Fig. 3). In the brain GLUT3 is localised almost exclusively to neurons; hence, it was originally referred to as the ‘neuronal glucose transporter’, and GLUT3 localisation shows good correlation with neuron function along both dendrites and axons [107–110]. The concentrations of total GLUT1 (55 and 45 kDa) and GLUT3 in whole brain samples have been calculated to be approximately equal [111, 112]. Compared with GLUT1, GLUT3 is considered to be a higher affinity and higher capacity glucose transporter to supply the high-energy demands of neurons. A combination of a lower K m value and higher capacity of GLUT3 provides neurons with preferential access to the available glucose [113]. Indeed, brain tumour-initiating cells adapt to restricted nutrition through preferential glucose uptake via GLUT3 so that they can outcompete for the available glucose. Hence, GLUT3, but not GLUT1, correlates with poor survival in brain tumours and other cancers [114]. Like GLUT1, GLUT3 also transports the glucose analogues 2-deoxy-d-glucose and 3-O-methyl-d-glucose, other hexoses including galactose and mannose, and transport activity is inhibited by cytochalasin B, forskolin and phloretin with low micromolar affinities [109]. GLUT3 also has important glucose transport roles in other types of cells including sperm, preimplantation embryos and circulating white blood cells [113]. Crystal structures of human GLUT3 have recently been determined in complex with d-glucose at 1.5 Å resolution in an outward-occluded conformation (PDB 4ZW9) and in complex with the exofacial inhibitor maltose at 2.6 Å in an outward-open conformation (PDB 4ZWC) and at 2.4 Å in an outward-occluded conformation (PDB 4ZWB) [115].
GLUT5 is the major GLUT transporter found in microglia [116, 117] and the only other place that it has been detected in the neurovascular unit at the blood-brain barrier is at low levels in endothelial cells [118] (Table 1 and Fig. 3). Interestingly, GLUT5 principally functions as a fructose transporter with an apparent K m value of around 6 mM for fructose and it has very low capacity to transport glucose [119]. GLUT5 is also insensitive to cytochalasin B and phloretin [27, 120]. The main functions of GLUT5 in the body include direct absorption of fructose in the small intestine and recovery of fructose from glomerular filtration in the kidney [121, 122], whilst its roles and mechanisms of regulation in the brain are not yet understood. Crystal structures of rat and bovine GLUT5 have recently been determined at 3.3 Å resolution in an open outward conformation (PDB 4YBQ) and at 3.2 Å resolution in an open inward conformation (PDB 4YB9), respectively [123].
The remaining GLUT transporters identified in the neurovascular unit at the blood-brain barrier are found at much lower levels than those of GLUTs 1, 3 and 5 (Table 1 and Fig. 3). GLUT2 is abundantly expressed in the pancreas and liver, where it serves as glucose sensor, and it is also found in the brain at low levels in astrocytes [124, 125] and in neurons [126–128] suggesting that it has glucose sensing roles in these regions of the blood-brain barrier. GLUT2 is a low-affinity transporter for glucose (K m ~ 17 mM) and also for fructose, mannose and galactose, whilst it is a high-affinity transporter for glucosamine (K m ~ 0.8 mM) [32]. Recent results have demonstrated the physiological importance of GLUT2 in glucose uptake and availability during brain development and confirm the involvement of GLUT2 in brain glucose sensing [129]. The insulin-responsive glucose transporters GLUT4 and GLUT8 have both been detected at low levels in the neurovascular unit of the blood-brain barrier, GLUT4 in astrocytes [130], endothelial cells [131, 132] and neurons [133–135] and GLUT8 in endothelial cells and neurons [136–138] (Table 1 and Fig. 3). Both GLUT4 and GLUT8 have shown increased expression in the developing mammalian brain [137]. It also appears that insulin translocates GLUT4 from the cytosol to the plasma membrane to transport glucose into cells and GLUT8 from the cytosol to rough endoplasmic reticulum to recover redundant glucose to the cytosol after protein glycosylation [128]. Indeed, GLUT8 appears to have a preference for catalysing transport of sugars through intracellular membranes [139]. GLUT6 (formerly called GLUT9) has been detected in the brain, specifically in neurons [140, 141], and GLUT7 has been detected in astrocytes [111, 112] (Table 1 and Fig. 3). The physiological functions of GLUTs 4, 6, 7 and 8 in the brain are not yet understood.
Sodium-Dependent Glucose Transporters
In the neurovascular unit at the blood-brain barrier, the sodium-dependent glucose transporter SGLT1 has been detected in endothelial cells [142–145] and in neurons [146–148] and SGLT2 has also been detected in endothelial cells [12] (Table 1 and Fig. 3). In most cases, this appears to be under conditions of stress such as oxygen/glucose deprivation or ischemia. SGLT1 and SGLT2 use the sodium electrochemical gradient to drive the transport of glucose uphill against its concentration gradient across membranes [149]. Both the sodium ions and glucose molecules pass through the protein in the same direction and it is the Na+, K+-ATPase pump that provides the sodium electrochemical gradient. SGLT1 has a coupling ratio of 2 Na+ ions/1 glucose molecule, has a high affinity but a low capacity for transporting glucose and is also capable of transporting galactose [150, 151]. One study identified SGLT1 at just the abluminal side of endothelial cells where it may be positioned to transport glucose from the brain extracellular fluid into the endothelial cells [66, 152]. SGLT2 has a coupling ratio of 1 Na+ ion/1 glucose molecule, a lower affinity for transporting glucose than SGLT1 and does not transport galactose [153, 154]. Given the energy demands of the SGLT glucose transporters, a question remains as to why they are needed at the blood-brain barrier along with the more abundant facilitative glucose transporters. Indeed, a number of studies have detected no expression or transport activity of SGLTs in the endothelial cells of the blood-brain barrier under normal physiological conditions. As mentioned above, one of the most sensitive methods for measuring the uptake of glucose into the brain is to monitor the uptake of the radiolabelled glucose analogue 2-deoxy-2-[18F]fluoro-d-glucose ([18F]FDG) using PET [59]. Because FDG is only transported by GLUTs and not SGLTs, its accumulation reflects only facilitated glucose transport. Two new glucose radiotracers were developed that allowed SGLT transport activity to be measured. α-Methyl-4-[18F]fluoro-4-deoxy-d-glucose ([18F]Me-4FDG) is highly specific for SGLTs and not transported by GLUTs, whilst 4-[18F]fluoro-4-deoxy-d-glucose ([18F]4-FDG) is transported by both SGLTs and GLUTs (Fig. 5a) [155]. The use of these tracers to measure SGLT activity in specific regions of rat brain showed that uptake was consistent with the distribution of SGLT proteins detected by immunohistochemical assays (Fig. 5b). These measurements proved the presence of functional SGLTs in a number of brain structures under normal physiological conditions, which included neurons but not endothelial cells at the blood-brain barrier [155]. The physiological roles and regulation of the sodium-dependent glucose transporters in the brain are not yet understood, not least at the blood-brain barrier. The apparent presence of SGLTs under stress conditions (e.g. oxygen/glucose deprivation or ischemia) at the blood-brain barrier may serve as a mechanism to protect neurons.

Glucose analogue PET radiotracers transported by SGLTs. a Structures of 4-[18F]fluoro-4-deoxy-d-glucose ([18F]4-FDG) and α-methyl-4-[18F]fluoro-4-deoxy-d-glucose ([18F]Me-4-FDG). b MicroPET image of SGLT activity in the midbrain of rat shown by the accumulation of [18F]4-FDG 1 h after intravenous injection (left) compared with a cryosection (right) to define the brain substructure: cortex (CX), hippocampus (HP), amygdala (AP), hypothalamus (HTH) and thalamus (TH). The picture in (b) was reproduced with permission from Yu et al. (2010) [155], which was originally published in American Journal of Physiology-Cell Physiology. Copyright by the American Physiological Society 2010
Effects of Conditions, Diseases and Mutations on Glucose Transporters at the Blood-Brain Barrier
Conditions such as hypoglycemia and hyperglycemia, especially associated with diabetes, and oxygen/glucose deprivation associated with cerebral ischemia have effects on the regulation, expression and distribution of glucose transporters at the blood-brain barrier. In contrast, a reduction in glucose transporters at the blood-brain barrier is one of the pathophysiological changes that occurs before the onset of the other events and symptoms in Alzheimer’s disease and is also a potential causative effect in the vascular hypothesis of the disease. Mutations in glucose transporters can affect their transport activity at the blood-brain barrier, notably those identified in GLUT1 deficiency syndrome. Some recreational drug compounds affect the expression and/or activity of glucose transporters at the blood-brain barrier. These topics will be considered in this section of the article, some of which are interlinked.
Effects of Hypoglycemia
Hypoglycemia, or low blood glucose, can be caused by a number of factors and conditions such as starvation, kidney failure, liver disease, some tumours, severe infections, inborn errors of metabolism and certain drugs including alcohol. The most common incidences of hypoglycemia occur in diabetics, which is often due to the medications used to treat diabetes such as insulin, sulfonylureas and biguanides along with eating less than usual, exercising more than usual or drinking alcohol. As already discussed, the brain requires a constant supply of glucose moving from the bloodstream into the interstitial fluid and neurons in order to maintain essential neurological functions and metabolic processes. When glucose levels fall below normal (typically at 65 mg/dl or 3.6 mM), initial effects are usually subtle reductions in mental efficiency followed by impairments of action and judgement. Other initial symptoms include hunger, sweating, shakiness and weakness. If there is no intervention and glucose levels continue to fall, then effects become more severe including cognitive impairments, seizures, coma (typically at 10 mg/dl or 0.55 mM) and death. All of these responses are defensive or adaptive mechanisms that aim to raise the blood sugar by way of glycogenolysis and gluconeogenesis or the provision of alternative energy sources. An expected pathophysiological result of hypoglycemia would be an upregulation and increased expression and activity of glucose transporters at the blood-brain barrier in order to maintain the supply of glucose for neurological functions. Indeed, this is the prevailing observation resulting from a number of experimental studies.
Using rat in vivo models of insulin-induced chronic hypoglycemia, an increase in expression of GLUT1 messenger RNA (mRNA) and protein (around 50 % in both cases) in endothelial cells at the blood-brain barrier has been demonstrated [156]. Experiments using isolated rat brain microvessels demonstrated a 23 % increase in total GLUT1 protein and a 52 % increase in luminal GLUT1 protein in insulin-induced hypoglycemic animals [157]. Hence, these results suggested both an increase in GLUT1 synthesis at the blood-brain barrier and a redistribution of GLUT1 to the luminal membrane for achieving enhanced glucose uptake into the brain under hypoglycemic conditions. The same study showed no effects of hypoglycemia on GLUT3 mRNA or protein expression [157]. A recent in vitro study using the human brain microvascular endothelial cell line hCMEC/D3 demonstrated that cultures exposed to hypoglycemic conditions for 3 h had a significant decrease in expression of GLUT1 and this was returned to normal or marginally increased levels after 24 h of exposure [158]. By contrast, the expression level of SGLT1 was unchanged at 3 h and significantly increased after 24 h of exposure to hypoglycemia. The expression level of GLUT4 remained unchanged. A previous study had demonstrated that low cytosolic glucose levels enhance the activity of an SGLT-like transporter in bovine brain endothelial cells [143], and a later study using confluent brain endothelial cells co-cultured with astrocytes demonstrated a combined role for both GLUT1 and SGLT1 transporters at the blood-brain barrier during oxygen/glucose deprivation [145]. A number of other studies have demonstrated an upregulation or increased expression of GLUT1 and/or GLUT3 in brain under hypoglycemic conditions [159–162], therefore confirming their roles in correcting the glucose levels required for maintaining neurological functions.
Effects of Hyperglycemia
Hyperglycemia, or high blood glucose, is most commonly caused by untreated diabetic conditions due to an insufficient production of insulin or response to insulin. Other conditions that can cause hyperglycemia include pancreatitis and pancreatic cancer, certain hormone-secreting tumours, Cushing’s syndrome, acute events such as stroke or myocardial infarction and also certain drugs. Whilst normal blood glucose levels are typically 80 to 110 mg/dl or 4 to 6 mM, hyperglycemia is defined as levels of greater than 126 mg/dl or 7.0 mM when fasting and greater than 200 mg/dl or 11.0 mM 2 h after meals [163]. The initial main symptoms of hyperglycemia are increased thirst, increased frequency of urination and increased hunger. Chronic and long-term periods of hyperglycemia can lead to a number of serious complications including damage to the kidneys, nervous and circulatory systems, retina, feet and legs. An expected pathophysiological result of hyperglycemia would be a downregulation and decreased expression and activity of glucose transporters at the blood-brain barrier in order to avoid neuronal damage. There have been conflicting results obtained from experimental studies on the effects of hyperglycemia on glucose transporters, however.
Some studies have reported a downregulation of glucose transporter expression and/or activity at the blood-brain barrier under conditions of hyperglycemia. A study examining both glucose transporter activity and microvessel glucose transporter concentration at the blood-brain barrier demonstrated a 44 % decrease in transporter activity in parallel with a 44 % decrease in cerebral blood flow and a 77 % decrease in transporter concentration under conditions of experimental diabetes. It was suggested that the primary mechanism underlying the downregulation is a post-transcriptional inhibition of glucose transporter mRNA translation [164]. A further study demonstrated that under conditions of chronic hyperglycemia in rat brain, the average density of GLUT1 was decreased by 7.5 % and local densities of GLUT1 were decreased in 12 out of 28 brain structures. Positive correlations were found between levels of local cerebral glucose utilisation and local GLUT1 densities during control conditions and during chronic hyperglycemia. Densities of GLUT3 were unchanged [69]. A more recent study investigated the influence of blood glucose levels on the mRNA and protein levels of GLUT1 and GLUT3 in the brain of diabetic rats. Compared with normal controls, those with chronic hyperglycemia showed reductions of 46 and 75 % in GLUT1 and GLUT3 mRNA, respectively, and the abundance of GLUT1 and GLUT3 proteins had a negative correlation with the blood glucose level. Furthermore, the density of microvessels in the brain of diabetic rats was not changed significantly compared with normal controls [165].
In contrast, a number of studies have reported no change in the expression and/or activity of glucose transporters at the blood-brain barrier under conditions of hyperglycemia. The study using isolated rat brain microvessels described above that showed an increase and redistribution of GLUT1 during hypoglycemia also showed no significant changes in regional brain glucose uptake and no changes in total microvessel GLUT1 or luminal GLUT1 concentrations under conditions of hyperglycemia. Levels of GLUT3 mRNA or protein expression were also unchanged [157]. A study of blood-brain barrier glucose permeability and regional brain glucose metabolism (CMR(glc)) under conditions of acute hyperglycemia in normal human subjects was performed using [18F]FDG PET, and the Kety-Schmidt technique was used for measurement of cerebral blood flow (CBF). The results demonstrated no major adaptational changes in the maximal transport velocity or affinity to blood-brain barrier glucose transporters under hyperglycemia. Furthermore, hyperglycemia did not change the global CBF or CMR(glc) [166]. Brain extracellular fluid glucose levels in diabetic and control awake/freely moving rats were measured under conditions of normal glycaemia and acute hyperglycemia using a microdialysis technique. The extracellular fluid/plasma glucose ratio (0.27 to 0.34) was remarkably similar in all groups, resulting in proportional elevations of brain extracellular fluid glucose in the hyperglycaemic groups. It was concluded that there is no significant protective adaptation of the blood-brain barrier to the transfer of glucose during chronic hyperglycemia [167]. Hence, the brain tissue may be chronically exposed to elevated levels of glucose in poorly controlled diabetes and therefore may be subject to the same long-term adverse effects of hyperglycemia seen in peripheral tissues. The same study also showed that brain extracellular fluid levels of lactate and β-hydroxybutyrate were increased in diabetic rats as compared with controls [167]. In a study that used proton magnetic resonance spectroscopy to measure glucose concentration in the occipital cortex of patients with poorly controlled diabetes and healthy volunteers at the same levels of plasma glucose, brain glucose concentrations of patients with poorly controlled diabetes were lower but not statistically different from those of healthy subjects. The authors concluded that chronic hyperglycemia in diabetes does not alter brain glucose concentrations in human subjects [168], hence suggesting no changes in glucose transport properties of the blood-brain barrier. The study described above using the human brain microvascular endothelial cell line hCMEC/D3 that showed an increase in SGLT1 during hypoglycemia also showed no changes in GLUT1 or SGLT1 expression in cultures exposed to hyperglycaemic conditions. The expression level of GLUT4 was upregulated by hyperglycemia, however [158].
The conflicting results obtained from experimental studies on the effects of hyperglycemia on glucose permeability of the blood-brain barrier and on the expression levels of glucose transporters may be due to the different experimental techniques, animal and in vitro models and methods of analysis that have been used. It is desirable that a consensus is reached about the effects of hyperglycemia (and hypoglycemia) on glucose permeability of the human blood-brain barrier and on the regulation and expression of its constituent glucose transporters. This may require the use of common experimental methods and models, for example, development of robust in vitro models using human cell lines since in vivo measurements on human subjects are generally not feasible.
Effects on Alzheimer’s Disease
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder that causes deterioration of memory and other cognitive functions. It is the most common cause of dementia in the elderly, accounting for an estimated 60–80 % of dementia cases (www.alz.org/downloads/ facts_Fig.s_2012.pdf) [169], and is a major cause of morbidity and mortality in modern westernised societies. The main neuropathological characteristics associated with the disease are extracellular neuritic plaques containing β-amyloid peptide (Aβ) derived from β-amyloid precursor protein (β-APP) and intracellular neurofibrillary tangles (containing hyperphosphorylated tau protein). There are a number of complex interactions between vascular and neuronal factors that come together to produce the pathology of AD, but a detailed description of these is beyond the scope of this review. In addition to the link between stroke and dementia, there are clear links between cardiovascular risk factors and heart disease with the onset and development of AD. These include atherosclerosis, hypertension, diabetes and obesity [170–176]. In the classical amyloid hypothesis of AD, accumulation of Aβ in the brain is the primary driving force for AD pathogenesis [177]. It is now becoming apparent that amyloid deposition is the downstream result and not the cause of AD and that therapeutic developments for AD should be also be targeting other factors [178–182]. An alternative vascular hypothesis of AD, also known as the two-hit vascular hypothesis (Fig. 6), has emerged in which the pathological accumulation of Aβ in the brain is secondary to primary vascular damage [15, 183–188]. In this hypothesis, there is an initial disturbance in the microcirculation of the brain including reduced CBF, hypoxia and blood-brain barrier dysfunction. This activates a cascade of events leading to neuronal damage, neurodegeneration, cognitive decline and dementia. Of particular relevance to this current review is that pathophysiological changes associated with AD include reduced brain glucose metabolism and reduced expression of glucose transporters at the blood-brain barrier [188]. Indeed, reduction in glucose transporters at the blood-brain barrier has been detected before the onset of other AD pathophysiological events and symptoms. Glucose transporters are therefore molecular markers for the onset of AD, and restoration or increase in GLUT1 is a possible approach for preventing, delaying or treating AD.

The ‘two-hit’ vascular hypothesis of Alzheimer’s disease. This figure was reproduced with permission from Zlokovic (2011) [15], which was originally published in Nature Reviews Neuroscience. Copyright by Nature Publishing Group 2011
A decrease in brain glucose metabolism [189–195] and a decrease in brain glucose transport activity [196–199] associated with AD have both been recognised for some time. As a consequence, the nuclear medicine tool of [18F]FDG PET, which can measure glucose uptake and glycolytic activity in the brain [59], has become routine for the early detection and monitoring of AD before the onset of AD pathophysiological changes and progression [200–205]. A number of studies have demonstrated that the reduction in glucose transport activity at the blood-brain barrier and in the brain associated with AD is due to a decrease in the levels of GLUT1 and GLUT3 proteins [5, 206–211]. In the case of GLUT1, it appears that a significant decrease in protein expression levels is accompanied by no significant changes in GLUT1 mRNA levels, which suggests a post-transcriptional mechanism for the decline [209]. A number of studies have reported a direct correlation between a decrease in brain glucose metabolism and in expression levels of GLUT1 and GLUT3 with a decrease in O-GlcNAcylation of nucleocytoplasmic proteins and with hyperphosphorylation of tau in AD pathogenesis and progression [211–218]. The question remains as to whether the reduced glucose metabolism and reduced glucose transporter expression are causative factors in the progression of AD or a response to reduced neuronal activity in AD. Evidence so far suggests that the former is more likely to be the case. In a further demonstration of how GLUT1 reductions influence the pathogenesis of AD, a recent study has shown that a GLUT1 deficiency in mice overexpressing β-APP leads to early cerebral microvascular degeneration, reduced blood flow and blood-brain barrier breakdown along with accelerated Aβ pathology and reduced Aβ clearance leading to diminished neuronal activity and cognitive behaviour, neuronal loss and neurodegeneration [219]. This confirms GLUT1 as a potential therapeutic target for preventing, delaying or treating AD.
There is an emerging consensus in the evidence that links type 2 diabetes with dementia and neurodegenerative disorders such as AD and that type 2 diabetes contributes significantly to the onset or progression of AD [220–227]. Indeed, the links between AD and diabetes are so strong that AD has been referred to as ‘type 3 diabetes’, which has been justified by the fact that AD appears to represent a form of diabetes that selectively affects the brain and involves molecular and biochemical features that are common with both type 1 and type 2 diabetes [228–231]. Biological mechanisms that are associated with both type 2 diabetes and AD include insulin resistance, impaired glucose uptake and metabolism, amyloidosis, oxidative stress, brain atrophy and the formation of advanced glycation end products and tau phosphorylation. As we have already seen, a reduction in glucose transporter expression at the blood-brain barrier plays an important role in the cascade of events for the onset and progression of both diabetes and AD [232].
Effects of Cerebral Ischemia
Cerebral ischemia occurs when blood flow to the brain is insufficient to meet metabolic demand. This can result from cerebral artery occlusion that interrupts blood flow and results in oxygen/glucose deprivation to the brain (cerebral hypoxia/hypoglycemia) and ultimately to the death of brain tissue or cerebral infarction/ischemic stroke. The ischemia can be confined to a specific region of the brain (focal ischemia) or be more widespread in the brain (global ischemia). Cerebral ischemia initiates a cascade of molecular events in the neurons and in cerebrovascular endothelial cells including energy depletion, dissipation of ion gradients, calcium overload, excitotoxicity, oxidative stress, inflammation and accumulation of ions and fluid [233, 234]. These events are associated with a disruption of the blood-brain barrier, which can lead to vasogenic cerebral edema [235], a primary cause of stroke-associated mortality. Glucose transporters at the blood-brain barrier are involved in the cascade of events resulting from cerebral ischemia and are potential therapeutic targets for post-ischemia treatment.
A number of studies have investigated the effects of cerebral ischemia on the expression levels and regulation of brain glucose transporters. In rat brain, GLUT1 overexpression occurs rapidly and widely in microvessels and parenchyma following global cerebral ischemia, which may be associated with an immediate early-gene form of response to cellular stress [236]. Cerebral hypoxia-ischemia leads to overexpression of GLUT1 in cerebral microvessels of both damaged and undamaged hemispheres during both early and late stages in the recovery period, whilst expression of GLUT3 is enhanced in penumbral regions, such as piriform cortex and amygdala [237, 238]. Hypoxic conditions associated with cerebral ischemia promote the upregulation of GLUT1 in brain endothelial cells and this is triggered by the production of vascular endothelial growth factor (VEGF) mediated by the phosphoinositide-3 kinase/Akt pathway [239]. In mouse brain, activation of the metabolic stress pathway results in rapid stimulation of blood-brain barrier endothelial cell sugar transport by acute upregulation of GLUT1 levels, possibly involving AMP-activated kinase activity [240]. Diabetic conditions combined with cerebral ischemia have produced even higher overexpression of GLUT1 and of GLUT3 mRNA and protein, although expression tended to decrease with increased blood glucose levels. Hence, it was considered that in the treatment of diabetic patients with cerebral ischemia, blood glucose control should not be too strict; otherwise, the upregulation of GLUT1 and GLUT3 induced by ischemia may not meet the energy requirements of the cells [241]. A study examining the effects of hypoxic ischemia and hypoxia on substrate transporter concentrations and function in postnatal murine brain detected a transient increase in neuronal GLUT3 in response to hypoxic ischemia after 4 h of reoxygenation [242]. This increase was associated with no changes in GLUT1, SGLT1 or SGLT2. At 24 h of reoxygenation, the increase in GLUT3 disappeared. Hypoxia alone in the absence of ischemia was associated with a transient but modest increase in GLUT3. It was concluded that hypoxic cerebral ischemia is associated with a transient compensatory increase in GLUT3 that protects glucose delivery for maintaining neuronal energy metabolism [242]. In a study using cultured astrocytes, GLUT1 was expressed primarily and GLUT3 was detected only at extremely low levels under normal physiological conditions. Interestingly, exposure of the astrocytes to ischemic stress increased the expression levels of both GLUT1 and GLUT3. It was also observed that astrocytic GLUT3 was responsible for the enhanced storage of intracellular glucose during reperfusion, resulting in a protection to lethal ischemic stress [243]. Based on current understanding, the upregulation of cerebral GLUT1 and GLUT3 is considered as a potential preventative neuroprotive therapy for ischemia [244]. Because hyperglycemia is an indicator of severe stroke and this promotes further ischemia in the brain, cerebral GLUTs are also considered as therapeutic targets for post-ischemic stroke treatments [245]. Related to this, a recent study has demonstrated that nicotine pre-exposure reduces ischemia reperfusion-enhanced GLUT1 transporter function and expression at the blood-brain barrier in a focal brain ischemia mouse model. This suggests that nicotine exposure prior to stroke could create an enhanced glucose-deprived state at the neurovascular unit, thus providing an additional vulnerability to enhanced stroke injury [246].
As described earlier, a number of studies have demonstrated the detection and/or upregulation of SGLT1 in blood-brain barrier endothelial cells and neurons. In the majority of cases, this was under stress conditions, especially oxygen/glucose deprivation and ischemia [143–146, 148]. Along with increased expression of the facilitative glucose transporters (GLUT1 and GLUT3), upregulation of SGLT1 is therefore a further inborn mechanism that is switched on to increase the supply of glucose for maintaining the energy demands of neurons during cerebral ischemia. Upregulation of SGLT1 is therefore a potential therapeutic strategy for post-ischemia treatment, whilst inhibition of SGLT1 during stroke has the potential to improve stroke outcome [145]. This is because cerebral ischemia can be exacerbated by post-ischemic hyperglycemia, which may involve SGLTs [247]. SGLT3 does not transport glucose, instead it depolarises the plasma membrane when glucose is bound, suggesting that SGLT3 is a glucose sensor. A study using a mouse model of focal cerebral ischemia (middle cerebral artery occlusion) suggested that cerebral SGLT3 suppresses neuronal damage by activation of cholinergic neurons, which are neuroprotective. In contrast, other cerebral SGLTs may be involved in the development of ischemia [248]. A further study has indicated that SGLTs are not involved in neuronal cell death under non-hyperglycaemic conditions and that post-ischemic hyperglycaemic conditions may be necessary for the SGLT-mediated exacerbation of cerebral ischemic neuronal damage [249].
Effects of Mutations in Glucose Transporters
Mutations in GLUT1, and in other glucose transporters, that have deleterious effects on proper expression, folding, structure and function of the protein have potential to disrupt glucose transport across the blood-brain barrier and have severe effects on neuronal function. The best-known example is the relatively recently recognised GLUT1 deficiency syndrome (G1DS) [250], which results from mutations in the gene that expresses GLUT1. An impaired function of the GLUT1 protein at the blood-brain barrier reduces the amount of glucose available to brain cells affecting brain development and function. This was originally identified in two children with persistent hypoglycorrhachia (low concentrations of glucose in cerebrospinal fluid), seizures and delayed development that responded dramatically to treatment with a ketogenic diet [250]. Further studies have demonstrated that the condition is inherited in an autosomal dominant or autosomal recessive manner [251–253] and neurological problems present in young children, which include difficulties in movement and speech and delay in development and intellectual disability [254–262]. GLUT1 defects are also increasingly being recognised as the cause of some genetic generalised epilepsies and other neurological disorders including early-onset absence epilepsy [263–265], familial idiopathic generalised epilepsy [266, 267], paroxysmal exercise-induced dyskinesia and paroxysmal choreoathetosis/spasticity [268–271]. Around 36 residue positions in the GLUT1 protein have been identified with mutations (substitutions or deletions) in G1DS of which the majority are charged or polar residues (Fig. 7). The mutated residues are generally found in three distinct clusters at different locations within the crystal structure of GLUT1: residues responsible for substrate binding, residues located at the interface between the transmembrane domain and the intracellular helical bundle and residues lining the transport path [44]. Mutations at these positions may interfere with the proper recognition of glucose or disrupt the molecular mechanism of glucose transport and therefore compromise the normal functioning of GLUT1.

Mutations in GLUT1 deficiency syndrome. Amino acid sequence of human GLUT1 (P11166, GTR1_HUMAN) taken from the UniProt KnowledgeBase (http://www.uniprot.org/) and coloured to indicate helical regions based on the crystal structure of GLUT1 [44]: transmembrane helix (grey), extramembrane helix (green), intramembrane helix (cyan). Residues putatively involved in direct interactions with glucose based on the crystal structure of the Escherichia coli xylose transporter XylE with bound glucose [45] are highlighted in red. Residues in GLUT1 that have shown mutations in GLUT1-deficiency syndrome [44] are indicated with a cross below the sequence
The experimental diagnosis of G1DS is based on low to normal lactate levels and low glucose levels (hypoglycorrhachia) in the cerebrospinal fluid confirmed by molecular analysis of the GLUT1 (SLC2A1) gene and by glucose uptake studies and immunoreactivity in human erythrocytes [272, 273]. GLUT1 function in erythrocytes is assayed by measuring the uptake of 14C-labelled 3-O-methyl-d-glucose (0.5 mM, 1 μCi/ml, 4 °C, pH 7.4) with an uptake cut-off point of 60 % (recently increased to 74 %) relative to controls for defining abnormal glucose transport [274, 275]. Results between 35 and 74 % of controls are typically diagnostic [276]. [18F]FDG PET scans of the brain in individuals with G1DS reveal some abnormalities including a global reduction of glucose uptake with more severe hypometabolism in the medial temporal lobes and the thalami, where the thalamic hypometabolism is accentuated by the relative uptake of glucose in the basal ganglia [277]. The primary treatment for G1DS is a ketogenic diet, whereby ketone bodies use a different transporter to cross the blood-brain barrier and provide the brain with an alternative source of energy [254, 255, 257, 278–282]. G1DS has also been indicated as a cause of permanent ketosis in which there is upregulation of monocarboxylic acid transporters (MCT1) at the blood-brain barrier provoked by neuroglycopenia allowing ketone body utilisation by the brain [283].
Effects of Recreational Drugs
Some recreational drug compounds, including alcohol, nicotine and methamphetamine, have been shown to affect the expression and/or activity of GLUT1 and GLUT3 at the blood-brain barrier. In cultured rat astrocytes, exposure to ethanol inhibited glucose uptake and reduced the number of glucose transporters, as indicated by binding studies with cytochalasin B [284]. A decrease in GLUT1 protein levels was confirmed by western blotting analysis. In contrast, GLUT1 mRNA levels were increased by exposure to ethanol, so it was concluded that ethanol acts at the post-transcriptional level in reducing the expression and activity of GLUT1 in astrocytes [284]. Similarly in cultured neurons, exposure to ethanol reduced the expression levels and glucose transport activities of both GLUT1 and GLUT3 [285]. Acute ethanol administration in rat brain resulted in decreased levels of both GLUT1 and GLUT3 expression but no change in affinity [286]. More recently, it has been demonstrated that the inhibitory effects of alcohol on glucose transport across the blood-brain barrier lead to neurodegeneration and that there is a preventive role of acetyl-l-carnitine [286]. Using cultures of human brain endothelial cells and neurons, ethanol exposure resulted in a decrease in glucose uptake along with a decrease in GLUT1 expression. In animal experiments, a chronic alcohol intake suppressed the transport of glucose into the frontal and occipital regions of the brain, which was validated by a significant decrease in GLUT1 protein expression in brain microvessels, whilst other measurements showed a breakdown in the integrity of the blood-brain barrier. Administration of the neuroprotective agent acetyl-l-carnitine prevented the adverse effects of alcohol on glucose uptake, blood-brain barrier damage and neuronal degeneration [287]. A chronic administration of nicotine in rat brain causes a general stimulation of brain metabolism along with distinct increases in protein densities for GLUT1 and GLUT3 and in increase in local cerebral glucose utilisation, whilst capillary densities remain unchanged [288, 289]. It has already been mentioned how nicotine exposure prior to stroke could create an enhanced glucose-deprived state at the neurovascular unit, thus providing an additional vulnerability to enhanced stroke injury [246]. Studies have shown how the psycho-stimulant drug methamphetamine impairs endothelial GLUT1 transport and causes blood-brain barrier dysfunction. A low concentration of methamphetamine (20 μM) increased the expression of GLUT1 in human brain endothelial cells without affecting the uptake of glucose, whilst a higher concentration (200 μM) decreased both the glucose uptake and GLUT1 protein levels [290]. The methamphetamine-induced decrease in GLUT1 protein was correlated with decreases in levels of blood-brain barrier tight junction proteins, and both of these effects were suppressed by addition of the neuroprotective agent acetyl-l-carnitine [290]. A decrease in neuronal glucose uptake by methamphetamine was associated with a decrease in levels of GLUT3. In astrocytes, a low concentration of methamphetamine (20 μM) increased glucose uptake whilst a higher concentration (200 μM) inhibited glucose uptake. These dual effects of methamphetamine on glucose uptake were correlated with changes in expression levels of astrocytic GLUT1 [291]. The difference in sensitivity between neurons and astrocytes to methamphetamine exposure appears to be in the adaptability of the cells to fatty acid oxidation as an alternative source of energy during glucose limitation. The effect of acetyl-l-carnitine for enhanced production of ATP from fatty oxidation in glucose-free culture conditions validated the adaptive nature of the cells, and the results suggest that deprivation of glucose-derived energy may contribute to neurotoxicity in users of methamphetamine [291].
Glucose Transporters at the Blood-Brain Barrier as Therapeutic Targets and Carriers for Drug Delivery
Solute carrier proteins are increasingly being recognised as important therapeutic drug targets [292] and this includes glucose transporters at the blood-brain barrier. As already discussed in the previous sections, the upregulation of glucose transporters is a potential strategy for the treatment of hypoglycemic conditions and the downregulation or inhibition of glucose transporters is a potential strategy for treatment under hyperglycaemic conditions. The restoration or increase in the expression levels of GLUT1 is a possible approach for preventing, delaying or treating AD. Because glucose transporters at the blood-brain barrier are involved in the cascade of events resulting from cerebral ischemia, they are potential therapeutic targets for post-ischemia treatment. Hyperglycemia is an indicator of severe stroke and this promotes further ischemia in the brain, so cerebral GLUTs are considered as therapeutic targets for post-ischemic stroke treatments. The upregulation of SGLT1 is a potential therapeutic strategy for post-ischemia treatment, whilst inhibition of SGLT1 during stroke has the potential to improve stroke outcome. GLUT1 is highly overexpressed in most types of cancer cells, including brain tumours, in order to satisfy their greatly enhanced uptake of glucose and rates of glycolysis [59]. A reduction in the expression and/or activity of GLUT1 in tumour cells is therefore a potential therapeutic strategy in cancer therapy. In addition to being direct therapeutic targets, glucose transporters at the blood-brain barrier are potential routes of entry for the delivery of drugs to the brain and central nervous system.
Drug Delivery via Glucose Transporters
Whilst the barrier function of the blood-brain barrier is critical for regulating transport of metabolites to the brain and for protecting the brain and central nervous system from harmful substances, it also acts as a significant roadblock for delivering drugs to the brain. There are a significant number of neurological disorders and central nervous system diseases including mental disorders, migraine, epilepsy, neurodegenerative diseases such as Alzheimer’s and Parkinson’s, cerebrovascular diseases such as cerebral ischemia and stroke, cancer, inflammatory diseases such as multiple sclerosis, brain trauma and infections such as meningitis. Only a relatively few of these are currently treatable with small molecule drug therapy, which is largely due to the restrictions provided by the blood-brain barrier. Hence, there are significant ongoing efforts to develop strategies for enabling therapeutic drugs to cross the blood-brain barrier and to reach their target sites in the brain and central nervous system. A major approach, known as carrier-mediated transport, is to target and make use of endogenous transport proteins for the delivery of drugs across the blood-brain barrier [10, 16, 293–299]. Recent concepts and strategies include nanoscale drug delivery systems [300], improving brain penetration of anticancer drugs by minimising drug efflux at the blood-brain barrier [301], transporter-conscious drug design [302] and dual-track screening of small molecule libraries for drug compounds that have both an affinity for a neural cell drug receptor target and an affinity for a blood-brain barrier transporter target [303]. Transport proteins are generally highly stereospecific for their substrates, so neuroactive drugs themselves are often not recognised or transported by endogenous transporters at the blood-brain barrier. Hence, the pro-drug approach has been used to overcome this [304–310], which has two main strategies: (i) modification of a drug to give a ‘pseudonutrient’ structure that is able to be recognised and transported by an endogenous transporter and (ii) conjugation of a drug with a nutrient that is able to be transported by an endogenous transporter (Fig. 8). Following transport, the drugs are released by enzymatic cleavage from their pro-drugs after being targeted into the brain or central nervous system [309]. Because the capacity of glucose transporters at the blood-brain barrier is around 15 and 50 times higher than those of monocarboxylic acid and neutral amino acid transporters, respectively [311], they are prime targets for the delivery of pro-drugs to the brain, especially GLUT1 [312].

Pro-drug strategy for delivering drugs across blood-brain barrier membranes via endogenous transport proteins
Conjugation of Drugs with GLUT1 Substrates
A number of neuroactive drugs have been conjugated with glucose in order to target GLUT1 for transport across the blood-brain barrier. Four derivatives of the chemotherapy drug chlorambucil were conjugated with d-glucose and all of the resultant compounds inhibited 14C-glucose uptake into erythrocytes by GLUT1 in a concentration-dependent manner [313]. One of these compounds, 6-O-4-[bis(2-chloroethyl)amino]benzenebutanoyl-d-glucopyranose (Fig. 9a), achieved a 160-fold higher inhibition of 14C-glucose uptake by GLUT1 than did glucose itself and also inhibited 3H-cytochalasin B binding to erythrocytes with 1000-fold higher efficiency than glucose. Whilst inhibition of glucose uptake by this compound was reversible, uptake measurements using the 14C-labelled compound led to the conclusion that this compound is a non-transported inhibitor of GLUT1 [313] and therefore not suitable for drug delivery across the blood-brain barrier. In an aim to achieve hydrophilic analogues of the anticancer compound busulphan (1,4-butanediol dimethanesulphonate) with enhanced selectivity and brain penetration, a number of O-methylsulphonyl derivatives of d-glucose were synthesised as potential alkylating agents for targeted drug delivery to the brain (Fig. 9b) [314]. Compounds with mesylation at the 4-OH and 6-OH positions of d-glucose had slightly diminished affinity for GLUT1, whilst mesylation at the 3-OH position resulted in complete loss of activity [314].

Drugs conjugated with GLUT1 substrates: d-glucose conjugates. a Chlorambucil conjugate 6-O-4-[bis(2-chloroethyl)amino]benzenebutanoyl-d-glucopyranose [313]. b O-Methyl-sulphonyl derivatives with mesylation at the (i) 3-OH, (ii) 4-OH and (iii) 6-OH positions [314]. c 7-Chlorokynurenic acid conjugates [318, 319]: (i) d-glucopyranos-3′-yl ester, (ii) d-glucopyranos-6′-yl ester, (iii) galactopyranos-6′-yl ester. d Dopamine conjugate at the 3-OH position [321]. e 6-OH position conjugates with (i) ketoprofen, (ii) indomethacin and (iii) ibuprofen [322, 323]
Conjugation of d-glucose to neuroactive enkephalin peptides, which are opioid agonists used to treat pain, was successful in decreasing their lipophilicity and in increasing their penetration of the rat blood-brain barrier [315]. Similar glycopeptides administered to mice produced more potent analgesic properties than the unglycosylated peptides [316, 317]. It is presumed that the increased penetration of the blood-brain barrier and improved analgesic properties originates from transport of the glycopeptides by GLUT1. The antidepressant drug 7-chlorokynurenic acid is itself not able to cross the blood-brain barrier so it was coupled with d-glucose and d-galactose, and the resultant ester conjugates (Fig. 9c) were tested for protective effects in mice against seizures induced by N-methyl-d-aspartate (NMDA). It was found that the d-glucopyranos-6′-yl ester was more potent than the d-glucopyranos-3′-yl ester whilst the galactopyranos-6′-yl ester was not protective at all. Based on these results, a role of GLUT1 was suggested for the brain uptake of the active glucosyl conjugates considering that GLUT1 has a lower affinity for d-galactose than for d-glucose [318, 319]. In a similar manner, glycosyl conjugates of dopamine and l-DOPA were obtained using a succinyl linker as esters at the C-3 position of glucose and at the C-6 position of galactose as potential anti-Parkinson pro-drugs. The dopamine derivatives were more active in reversing reserpine-induced hypolocomotion in rats than l-DOPA or its conjugates, whilst all compounds reduced morphine-induced locomotion [320]. Based on these results, it was suggested that glycosyl conjugation of dopamine allowed this drug to cross the blood-brain barrier via GLUT1 and it was later demonstrated that the C-3 glucose conjugate of dopamine (Fig. 9d) is a transportable substrate of GLUT1 [321].
Conjugation of the nonsteroidal anti-inflammatory drugs (NSAIDs) ketoprofen and indomethacin with glucose at the 6-OH position (Fig. 9e) produced pro-drugs that were able to significantly inhibit the GLUT1-mediated uptake of glucose. These conjugates were also able to cross the blood-brain barrier in a temperature-dependent manner, suggesting that their brain uptake is carrier-mediated, most likely via GLUT1 [322]. In order to improve the delivery of the NSAID ibuprofen across the blood-brain barrier, a number of conjugates to d-glucose were prepared via ester bonds at positions C-2, C-3, C-4 and C-6 [323]. All four pro-drug compounds were moderately stable in pH 7.43 buffer solution, rat plasma and brain tissue extracts. The feasibility of the compounds to undergo enzymatic cleavage by esterase in biological samples to regenerate the original drug was also assessed. In vivo experiments showed that the levels of ibuprofen in plasma after the injection of the pro-drugs was several times higher than after the injection of ibuprofen and that the maximal concentration of ibuprofen in brain after administration of the C-6 conjugate (Fig. 9e) was threefold higher than that of the control group. Futhermore, the concentration of ibuprofen was kept stable in brain for around 4 h for all four conjugates and this was suggested to be beneficial for treatment of AD [323]. It is presumed that delivery of these pro-drugs to the brain is via GLUT1. In a different study, a number of l-ascorbic acid derivatives of ibuprofen (Fig. 10a) were synthesised as pro-drugs to improve delivery of ibuprofen to the brain via GLUT1 and the sodium-dependent vitamin C transporter SVCT2 [324]. In vivo distribution measurements following intravenous administration of the pro-drugs and naked ibuprofen showed that the pro-drugs exhibited excellent transportability across the blood-brain barrier and significantly increased the level of ibuprofen in the brain. Biodistribution and pharmacokinetic parameters suggested that an l-ascorbic acid thiamine disulphide delivery system (Fig. 10b) is a promising carrier for enhancing drug delivery to the brain [324].

Drugs conjugated with GLUT1 substrates: l-ascorbic acid conjugates. a Conjugate with ibuprofen. b l-Ascorbic acid thiamine disulphide system for delivering ibuprofen [324]
Nano-enabled Delivery Systems
Nano-enabled delivery systems are a promising approach for improving the uptake and targeted delivery of drugs to the brain. The various nanocarriers that can be used to encapsulate drugs, either alone or in combination with targeting ligands, include polymer-drug conjugates, liposomes, micelles, bolaamphiphiles, microspheres, dendrimers, nanogels, bionanocapsules and nanoparticles [297, 325–329]. Glucose derivatives of cholesterol have been synthesised as a material for preparing liposomes to encapsulate drugs and improve their delivery to the brain by targeting GLUT1 at the blood-brain barrier. Here, ethylene glycols of different chain lengths were used as linkers between the glucose and cholesterol moieties (Fig. 11a) [330, 331]. In one study, the fluorescent model drug coumarin 6 was loaded into liposomes composed of phospholipids and glucose-derived cholesterols with different linker lengths (GLU200-LIP, GLU400-LIP, GLU1000-LIP, GLU2000-LIP) prepared by a thin-film dispersion-ultrasound method. An in vitro blood-brain barrier model was developed to evaluate the transendothelial ability of the different liposomes crossing the blood-brain barrier. The biodistribution of the liposomes in mouse brain was also measured by in vivo methods. The liposomes GLU400-LIP, GLU1000-LIP and GLU2000-LIP all showed potential for brain targeting and the one with intermediate chain length, GLU1000-LIP, exhibited the strongest brain delivery capacity (Fig. 11b) [332]. Some multivalent glucosides with high affinity as ligands for targeting liposomes to the brain via GLUT1 have recently been synthesised. These liposomes loaded with the chemotherapy drug docetaxel significantly increased the level of docetaxel in mouse brain compared to naked docetaxel and empty liposomes as a control [333]. Liposomes modified with p-aminophenyl-α-d-mannopyranoside have been investigated as carriers for delivering encapsulated drugs across the blood-brain barrier via GLUT1 and have also shown potential for targeting various functional regions of the brain. Such liposomes loaded with a fluorescent dye showed efficient penetration through the blood-brain barrier and accumulation in mouse brain with a distinct spatiotemporal pattern [334]. A further investigation of the relationship between the brain distribution of these mannose-derivatised liposomes and glucose transporters showed that cellular uptake was significantly improved by GLUT1 and GLUT3 overexpression. The results indicated that transcytosis by GLUT1 and GLUT3 is the likely pathway for transport of the liposomes into brain and the specific brain distribution of the liposomes was closely related to the non-homogeneous distribution of GLUT1 and GLUT3 [335]. A recent study using mannose-derivatised liposomes encapsulating the antidepressant drug sertraline demonstrated how optimisation of ultrasound parameters can maximise mannosylation capacity, sertraline entrapment and vesicle size. Also, transendothelial ability was increased by 2.5-fold by mannosylation through binding with GLUT1 [336].

Liposomes from glucose derivatives of cholesterol for drug delivery. a Structure of a glucose derivative of cholesterol with ethylene glycols of different chain lengths used as the linker between the glucose and cholesterol moieties [328, 329]. b In vivo imaging of mice anaesthetised at 2, 6 and 12 h after intravenous injection of liposomes with different linker lengths (GLU200-LIP, GLU400-LIP, GLU1000-LIP, GLU2000-LIP) loaded with the fluorescent dye 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine (DIR). Ex vivo imaging in mouse brain 1 h after the intravenous injection of the different DIR-loaded liposomes is also shown at the bottom. The colour bar indicates the intensity of the near-infrared fluorescence signal. The pictures in (b) were adapted from Xie et al. (2012) [332], which was originally published in International Journal of Nanomedicine. Copyright by Xie et al. 2012
Nanoscale micelles functionalised with dehydroascorbic acid have been developed for targeting GLUT1 to deliver drugs across the blood-brain barrier [337]. The micelles were also designed with a disulphide linkage that forms a bio-responsive inner barrier. This maintains the stability of the micelles in the blood circulation and prevents leakage of encapsulated drug molecules before reaching target cells in the brain. Once within the cells, drug release is triggered by the high intracellular level of glutathione. Such micelles combine the dual characteristics of GLUT1-targeting and selective control of drug deposition in the brain. Dehydroascorbic acid-derivatised micelles containing the antifungal drug itraconazole were highly effective against intracranial infection [337]. Similar dehydroascorbic acid-derivatised micelles have been developed for treating the highly aggressive cancer malignant glioma [338] and have shown a ‘one-way’ continuous accumulation within tumour cells [339] and therefore potential for delivering drugs to cancer sites in the brain and central nervous system via GLUT1. Pluronic P105 polymeric micelles, derivatised for targeting GLUT1 and the folic acid receptor, and encapsulating the chemotherapy drug doxorubicin have been prepared for enhancing blood-brain barrier transport and accumulation of the drug in glioma cells. Intravenous injection of these micelles produced a high suppression ratio of tumour growth, and the results showed promise for treating brain tumours [340].
Glucose-coated gold nanoparticles have been investigated as potential carriers of drugs across the blood-brain barrier endothelium and subsequently into astrocytes [341]. These nanoparticles were 4 nm in size with a 2-nm gold core and the coating with glucose was for targeting GLUT1. It was found that the transport rate of these nanoparticles across the human brain endothelium was at least three times faster than across non-brain endothelia. The nanoparticles moved across the apical and basal plasma membranes and through the cytosol with relatively little vesicular or paracellular migration, and antibiotics that interfere with vesicular transport did not block their migration. In a culture system that included human brain endothelial cells and primary human astrocytes, the glucose-coated nanoparticles traversed the endothelium then moved through the extracellular matrix and localised in the astrocytes [341]. Because movement of the nanoparticles was not blocked by antibiotics that interfere with endocytosis or by cytochalasin B, it was concluded that transcytosis and GLUT1 are not responsible for transfer of the nanoparticles across the membranes and that this transfer must be dependent on some other biophysical property of the nanoparticles [341]. Nanoparticles of poly(ethylene glycol)-co-poly(trimethylene carbonate) functionalised with 2-deoxy-d-glucose were dual-targeted to GLUT1 for drug delivery in glioma treatment. Derivatisation with 2-deoxy-d-glucose was aimed at enhancing penetration of the blood-brain barrier via GLUT-mediated transcytosis and improving drug accumulation in the glioma via GLUT-mediated endocytosis [342]. These nanoparticles, which had a size of 71 nm, were encapsulated with the anticancer drug paclitaxel. Compared with non-glucosylated nanoparticles, a significantly higher amount of the glucosylated nanoparticles were internalised by glioma cells through caveolae-mediated and clathrin-mediated endocytosis. Transport ratios across an in vitro blood-brain barrier model and the cytotoxicity of glioma cells after crossing the blood-brain barrier were significantly greater for the glucosylated nanoparticles than for unglycosylated nanoparticles. In this case, a role for GLUT1 in the enhanced transport of the glycosylated nanoparticles across the blood-brain barrier was confirmed. Furthermore, in vivo fluorescent imaging indicated that glucosylated nanoparticles had high specificity and efficiency in intracranial tumour accumulation, and the anti-glioblastoma efficacy of the paclitaxel-loaded glucosylated nanoparticles was significantly greater compared with that of Taxol and paclitaxel-loaded unglycosylated nanoparticles [342].
In Vitro Models of the Human Blood-Brain Barrier for Investigating Glucose Transporters
Other than using relatively non-invasive imaging techniques such as PET, it is generally not feasible to perform direct in vivo studies on the expression, distribution and function of glucose transporters and their interactions with drugs at the blood-brain barrier in humans. In vivo measurements on the blood-brain barriers from mice, rats, pigs, cows and primates can be performed but these suffer from high costs and have ethical implications. Hence, a large number of in vitro models of the blood-brain barrier have been developed with varying levels of complexity. The model should ideally mimic the in vivo blood-brain barrier as closely as possible, but since this is so complex, the in vitro models are generally much more simplified. The in vitro models are therefore not able to completely replicate the in vivo conditions of the blood-brain barrier, so it is important to recognise their limitations when designing experiments and interpreting the data. Functional features that should be included in an ideal and complete in vitro model of the blood-brain barrier include an ability to express tight junctions between endothelial cells, negligible paracellular diffusion between endothelial cells, selective and asymmetric permeability to physiologically important ions (Na+, K+, Cl−), functional expression of endogenous transport and receptor proteins, responsiveness to stimuli and an ability to reproduce the effects of a range of physiological and pathophysiological conditions that affect the blood-brain barrier in vivo [343]. The most basic in vitro models are cell culture-based static systems, which can comprise just a monolayer of endothelial cells or this can be co-cultured with astrocytes, pericytes, microglia or neurons (Fig. 12a). The co-cultures provide an environment that more closely mimics the in vivo neurovascular unit by introducing interactions between the different cell types. These systems can be adapted for measuring the transendothelial electrical resistance (TEER) of the endothelial cell layers and for measuring the flux of tracer compounds or drugs across the endothelial cells (Fig. 12b, c). Whilst these systems are relatively simple, their contents can be carefully controlled. Some in vitro models use primary cultures from human, cow, pig, mouse and rat, but in addition to brain endothelial cells, these can contain contaminating cells including brain pericytes, fibroblasts, smooth muscle cells and leptomeningeal cells [344, 345]. The contaminating cells disturb the development of tight monolayers and overgrow the brain endothelial cells during long‐term cultivation. The preparation of primary cultures is also expensive, time-consuming and requires special expertise [346]. Cell line-based models using immortalised brain endothelial cell lines from human, cow, mouse or rat have more recently been developed, and these overcome many of the problems of primary cultures. The first stable and well‐characterised human brain endothelial cell line was hCMEC/D3, which shows many characteristics of the in vivo blood-brain barrier [347]. In all of these systems, the matrix can be modified to include chemical and other components to replicate in vivo conditions at the blood-brain barrier, stress conditions such as hypoglycemia, hyperglycemia and hypoxia, disease states or to test the effects of drugs. A further factor to consider in the development of an in vitro model is the shear stress generated by blood flow under physiological conditions, which affects transporter and tight junction protein expression as well as endothelial barrier function [348]. Dynamic blood-brain barrier models, which include shear stress, have therefore been developed of which there are three main types: cone-plate apparatus [349], dynamic in vitro models [350–354] and microfluidic in vitro models [355–359]. A detailed description of in vitro models of the blood-brain barrier is beyond the scope of this work, but numerous reviews on this theme are available [343, 346, 360–373]. These demonstrate that this is still a highly developmental area, and further progress is needed to provide more sophisticated and robust in vitro models of the blood-brain barrier.

Basic cell culture in vitro models of the blood-brain barrier. a In vitro reconstituted static blood-brain barrier models using culture inserts: mono-culture of brain endothelial cell layer, co-culture of brain endothelial cell layer with a second cell type (e.g. astrocytes) on the other side of the porous filter or at the bottom of the culture well, triple culture with brain endothelial cell layers in the upper side of the inserts with the second cell type at the other side of filter and the third type in the culture well. b Measurement of transendothelial electrical resistance (TEER) of brain endothelial cell monolayers grown in cell culture inserts with a pair of electrodes. c Flux of tracer compounds or drugs from the upper (luminal) compartment to the lower (abluminal) compartment through a brain endothelial monolayer can be measured during given time intervals and endothelial permeability coefficients can be calculated. This figure was adapted from Deli (2007) [362], which was originally published in Handbook of Neurochemistry and Molecular Neurobiology. Copyright by Springer-Verlag 2007
Human Brain Endothelial Cell Line hCMEC/D3
The human brain endothelial cell line hCMEC/D3 [347] has become one of the most abundantly used cell lines for in vitro models of the blood-brain barrier [374], including for the study of glucose transporters. Its popularity arises from its stability and ease of growth and the fact that it retains the expression of most metabolising enzymes, tight junction proteins, transporters and receptors expressed in vivo at the blood-brain barrier. Furthermore, it can be adapted for drug uptake and active transport studies and for studying the brain endothelium response to human pathogens and inflammatory stimuli [374–377]. In order to validate the hCMEC/D3 cell line as a blood-brain barrier model, transcriptomic profiles of hCMEC/D3 cells and human brain microvascular endothelial cells (BEC) have been compared with published transcriptional data from freshly isolated mouse BECs [378]. The analysis revealed that some important types of proteins, including tight junction proteins, transporters and receptors are expressed at very low levels in the hCMEC/D3 and human BEC cells compared with the fresh mouse BECs. For example, GLUT1 was highly expressed in mouse BECs but present at very low levels in both hCMEC/D3 and human BEC cells. This trend was seen for a number of SLC and ABC transporters that have crucial functions and are expressed at high levels at the blood-brain barrier (Fig. 13). It was concluded that the hCMEC/D3 and human BEC cells lose their unique protein expression profile when outside of their native environment at the neurovascular unit and display a more generic endothelial cell phenotype [379]. A subsequent quantitative proteomic analysis of transporters, receptors and junction proteins in the hCMEC/D3 cell line has been performed [379]. From 91 target molecules, 12 transporters, 2 receptors, 1 junction protein and 1 membrane marker were present at quantifiable levels in plasma membranes of hCMEC/D3 cells. The transport proteins included GLUT1. After normalisation based on Na+/K+-ATPase expression, the differences in protein expression levels between the hCMEC/D3 cells and human brain microvessels were within fourfold for the large majority of the proteins. GLUT1 expression was 15-fold higher in the hCMEC/D3 cells than in human umbilical vein endothelial cells (HUVECs) used as reference non-brain endothelial cells [379]. The results of this proteomic analysis suggest that the expression levels of some important transporters in the hCMEC/D3 cell line, including GLUT1, may be closer to the levels in the native blood-brain barrier than suggested by the transcriptomic analysis.

Expression levels of blood-brain barrier genes in human hCMEC/D3 cells. The x-axis shows the ratio between expression levels in mouse BECs and hCMEC/D3 cells, and the y-axis shows the ratio between mouse BECs and hCMEC/D3 cells multiplied by the expression levels in mouse BECs. Expression levels were first normalised against the expression level of the ribosomal protein L4 (RBL4) in each cell type. Genes in the upper right hand corner are therefore highly expressed in mouse BECs and much more in comparison to the hCMEC/D3 cells. All genes for mouse BECs versus hCMEC/D3 are displayed as grey dots. Tight junction genes (red square), SLC members (blue dot), ABC members (yellow triangle) and receptors (green diamond) are highlighted. All crucial blood-brain barrier genes (enlarged symbols) are expressed at much lower levels in the hCMEC/D3 cells than in the mouse BECs. This figure was reproduced from Urich et al. (2012) [378], which was originally published in PLoS One. Copyright by Urich et al. 2012
Characterisation and modulation of glucose uptake in a human blood-brain barrier model using the hCMEC/D3 cell line have been performed [380]. [3H]-2-deoxy-d-glucose uptake was sodium- and energy-independent and regulated by Ca2+ ions and calmodulin but not by MAPK kinase pathways. This suggests that [3H]-2-deoxy-d-glucose uptake is via facilitative GLUT proteins and this was confirmed by a decrease in uptake by the known GLUT1 competitive inhibitor quercetin and the related compound myricetin. Progesterone and estrone decreased [3H]-2-deoxy-d-glucose uptake, and protein kinases A and C and protein tyrosine kinase also seemed to be involved in modulating the uptake [380]. A study using the hCMEC/D3 cell line to investigate the effects of altered glycaemia on the blood-brain barrier endothelium [160] has already been mentioned in previous sections. Parallel monolayers of hCMEC/D3 cells were exposed to normal, hypo- or hyperglycaemic conditions (5.5, 2.2 or 35 mM d-glucose, respectively); and the expression levels and distribution of a number of proteins, including glucose transporters, were followed during exposure over 3–24 h. Cultures exposed to hypoglycemic conditions for 3 h had a significant decrease in expression of GLUT1, which was returned to normal or marginally increased levels after 24 h [160]. In contrast, SGLT1 levels were unchanged at 3 h and significantly increased after 24 h. Hyperglycemic conditions produced no changes in the expression levels of GLUT1 or SGLT1, but they did produce an upregulation of GLUT4 [160].
Brain Endothelial Cells from Human Stem Cells
Human blood-brain barrier endothelial cells prepared from human embryonic stem cells (hESCs) or induced human pluripotent stem cells (iPSCs) have recently been described that show similar characteristics compared with the in vivo blood-brain barrier and therefore a good promise as the basis for in vitro models [381]. The hESCs or iPSCs are first incubated with medium-favouring neural differentiation and then with medium-favouring endothelial differentiation to acquire the blood-brain barrier properties. The resultant pure endothelial cells have well-organised tight junctions, appropriate expression of nutrient transporters and polarised efflux transporter activity, respond to astrocytes and have a molecular permeability that correlates well with an in vivo rodent blood-brain barrier [381]. Possible limitations of this model may come from a low reproducibility of paracellular permeability and transendothelial electrical resistance across replicates, which may be affected by the type and history of the stem cell lines; and the stability of the model for periods of up to only 7 days might preclude its general use for drug screening and toxicology studies. As an alternative, a human blood-brain barrier model using human cord blood-derived haematopoietic stem cells has been developed [382]. These stem cells were initially differentiated into endothelial cells followed by the induction of blood-brain barrier properties by co-culture with pericytes. The resultant endothelial cells express tight junction proteins and transporters typically observed in brain endothelium and maintain the expression of most in vivo blood-brain barrier properties for at least 20 days. This model also showed good reproducibility with similar paracellular permeability for cells derived from three different donors and in three different laboratories. Furthermore, results showed a good correlation between the in vitro predicted ratio of concentrations of unbound drug in brain and plasma obtained with this model and the in vivo ratio of concentrations of unbound drugs in cerebrospinal fluid and plasma reported in humans [382].
Conclusions
Whilst much is known about the distribution, function and regulation of glucose transporters at the blood-brain barrier, a further understanding of their complex relationships with foremost disorders such as diabetes, Alzheimer’s disease and cerebral ischemia is still necessary. This will allow the roles of glucose transporters as potential therapeutic targets and as routes of entry for drug delivery to the brain and central nervous system to be exploited to their full potential in the prevention and/or treatment of neurological and neurovascular disorders and brain tumours. In vitro models of the human blood-brain barrier will no doubt be required to achieve this. Although there has been significant recent progress, it appears that no perfect and robust in vitro model of the human blood-brain barrier has yet been developed and fully validated. Further developments and characterisation of in vitro models of the human blood-brain barrier will open new opportunities to study the structure and function of the blood-brain barrier, the effects of stress and disease conditions and exposure to drugs and xenobiotics and to develop their use in drug screening and discovery. This includes glucose transporters at the blood-brain barrier. Development of such improved in vitro models will bring together clinicians and researchers with expertise in cell and tissue engineering and in mechanical and electrical engineering. Structural biology and chemistry also have important roles to play. The crystal structure of GLUT1 could be employed in docking and modelling experiments to assist the in silico design of drugs, pro-drugs and drug delivery systems to target GLUT1. Compounds and drug delivery systems that show potential as therapeutic agents can then be chemically synthesised or produced and tested for binding and/or transport activities in biochemical and biophysical experiments with GLUT1 and in in vitro models of the blood-brain barrier. The recent crystal structures of GLUT3 and GLUT5 could be used in a similar manner as targets for inhibitors or for drug delivery to brain tumours. Hence, a further understanding and exploitation of glucose transporters at the blood-brain barrier requires the consolidation of a number of disparate clinical, scientific and engineering disciplines, which also applies to blood-brain barrier research in general.
References
Nicholson C (2001) Diffusion and related transport mechanisms in brain tissue. Rep Prog Phys 64(7):815–884
Avdeef A (2001) Physicochemical profiling (solubility, permeability and charge state). Curr Top Med Chem 1(4):277–351
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46(1–3):3–26
Hawkins RA, O'Kane RL, Simpson IA, Viña JR (2006) Structure of the blood-brain barrier and its role in the transport of amino acids. J Nutr 136(1):218S–226S
Zlokovic BV (2008) The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57(2):178–201
Engelhardt B, Sorokin L (2009) The blood-brain and the blood-cerebrospinal fluid barriers: function and dysfunction. Semin Immunopathol 31(4):497–511
Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ (2010) Structure and function of the blood-brain barrier. Neurobiol Dis 37(1):13–25
Redzic Z (2011) Molecular biology of the blood-brain and the blood-cerebrospinal fluid barriers: similarities and differences. Fluids Barriers CNS 8(1):3
Wong AD, Ye M, Levy AF, Rothstein JD, Bergles DE, Searson PC (2013) The blood-brain barrier: an engineering perspective. Front Neuroeng 6:7
Sanchez-Covarrubias L, Slosky LM, Thompson BJ, Davis TP, Ronaldson PT (2014) Transporters at CNS barrier sites: obstacles or opportunities for drug delivery? Curr Pharm Des 20(10):1422–1449
Keaney J, Campbell M (2015) The dynamic blood-brain barrier. FEBS J 282(21):4067–4079
Enerson BE, Drewes LR (2006) The rat blood-brain barrier transcriptome. J Cereb Blood Flow Metab 26(7):959–973
Kido Y, Tamai I, Okamoto M, Suzuki F, Tsuji A (2000) Functional clarification of MCT1-mediated transport of monocarboxylic acids at the blood-brain barrier using in vitro cultured cells and in vivo BUI studies. Pharm Res 17(1):55–62
Deane R, Bell RD, Sagare A, Zlokovic BV (2009) Clearance of amyloid-beta peptide across the blood-brain barrier: implication for therapies in Alzheimer's disease. CNS Neurol Disord Drug Targets 8(1):16–30
Zlokovic BV (2011) Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci 12(12):723–738
Pardridge WM (2012) Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab 32(11):1959–1972
Banks WA, Owen JB, Erickson MA (2012) Insulin in the brain: there and back again. Pharmacol Ther 136(1):82–93
Bien-Ly N, Yu YJ, Bumbaca D, Elstrott J, Boswell CA, Zhang Y, Luk W, Lu Y et al (2014) Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J Exp Med 211(2):233–244
Strazielle N, Ghersi-Egea JF (2015) Efflux transporters in blood-brain interfaces of the developing brain. Front Neurosci 9:21
Wittmann G, Szabon J, Mohácsik P, Nouriel SS, Gereben B, Fekete C, Lechan RM (2015) Parallel regulation of thyroid hormone transporters OATP1c1 and MCT8 during and after endotoxemia at the blood-brain barrier of male rodents. Endocrinology 156(4):1552–1564
Kuzawa CW, Chugani HT, Grossman LI, Lipovich L, Muzik O, Hof PR, Wildman DE, Sherwood CC et al (2014) Metabolic costs and evolutionary implications of human brain development. Proc Natl Acad Sci U S A 111(36):13010–13015
Benarroch EE (2014) Brain glucose transporters: implications for neurologic disease. Neurology 82(15):1374–1379
Dick APK, Harik SI, Klip A, Walker DM (1984) Identification and characterization of the glucose transporter of the blood-brain barrier by cytochalasin B binding and immunological reactivity. Proc Natl Acad Sci U S A 81(22):7233–7237
Joost HG, Thorens B (2001) The extended GLUT-family of sugar/polyol transport facilitators: nomenclature, sequence characteristics, and potential function of its novel members (review). Mol Membr Biol 18(4):247–256
Uldry M, Thorens B (2004) The SLC2 family of facilitated hexose and polyol transporters. Pflugers Arch 447(5):480–489
Zhao FQ, Keating AF (2007) Functional properties and genomics of glucose transporters. Curr Genomics 8(2):113–128
Manolescu AR, Witkowska K, Kinnaird A, Cessford T, Cheeseman C (2007) Facilitated hexose transporters: new perspectives on form and function. Physiology (Bethesda) 22:234–240
Carruthers A, DeZutter J, Ganguly A, Devaskar SU (2009) Will the original glucose transporter isoform please stand up! Am J Physiol Endocrinol Metab 297(4):E836–E848
Augustin R (2010) The protein family of glucose transport facilitators: it's not only about glucose after all. IUBMB Life 62(5):315–333
Thorens B, Mueckler M (2010) Glucose transporters in the 21st century. Am J Physiol Endocrinol Metab 298(2):E141–E145
Cura AJ, Carruthers A (2012) Role of monosaccharide transport proteins in carbohydrate assimilation, distribution, metabolism, and homeostasis. Compr Physiol 2(2):863–914
Mueckler M, Thorens B (2013) The SLC2 (GLUT) family of membrane transporters. Mol Asp Med 34(2–3):121–138
Madej MG, Sun L, Yan N, Kaback HR (2014) Functional architecture of MFS D-glucose transporters. Proc Natl Acad Sci U S A 111(7):E719–E727
Pao SS, Paulsen IT, Saier MH Jr (1998) Major facilitator superfamily. Microbiol Mol Biol Rev 62(1):1–34
Saier MH Jr, Beatty JT, Goffeau A, Harley KT, Heijne WH, Huang SC, Jack DL, Jähn PS et al (1999) The major facilitator superfamily. J Mol Microbiol Biotechnol 1(2):257–279
Reddy VS, Shlykov MA, Castillo R, Sun EI, Saier MH Jr (2012) The major facilitator superfamily (MFS) revisited. FEBS J 279(111):2022–2035
Iancu CV, Zamoon J, Woo SB, Aleshin A, Choe JY (2013) Crystal structure of a glucose/H+ symporter and its mechanism of action. Proc Natl Acad Sci U S A 110(44):17862–17867
Wright EM, Loo DD, Hirayama BA (2011) Biology of human sodium glucose transporters. Physiol Rev 91(2):733–794
Wright EM (2013) Glucose transport families SLC5 and SLC50. Mol Asp Med 34(2–3):183–196
Kasahara M, Hinkle PC (1997) Reconstitution and purification of the D-glucose transporter from human erythrocytes. J Biol Chem 252(20):7384–7390
Zoccoli MA, Baldwin SA, Lienhard GE (1978) The monosaccharide transport system of the human erythrocyte. Solubilization and characterization on the basis of cytochalasin B binding. J Biol Chem 253(19):6923–6930
Mueckler M, Caruso C, Baldwin SA, Panico M, Blench I, Morris HR, Allard WJ, Lienhard GE et al (1985) Sequence and structure of a human glucose transporter. Science 229(4717):941–945
Baldwin SA, Lienhard GE (1989) Purification and reconstitution of glucose transporter from human erythrocytes. Methods Enzymol 174:39–50
Deng D, Xu C, Sun P, Wu J, Yan C, Hu M, Yan N (2014) Crystal structure of the human glucose transporter GLUT1. Nature 510(7503):121–125
Sun L, Zeng X, Yan C, Sun X, Gong X, Rao Y, Yan N (2012) Crystal structure of a bacterial homologue of glucose transporters GLUT1-4. Nature 490(7420):361–366
Barnett JE, Holman GD, Munday KA (1973) Structural requirements for binding to the sugar transport system of the human erythrocyte. Biochem J 131(2):211–221
Leitch JM, Carruthers A (2009) Alpha- and beta-monosaccharide transport in human erythrocytes. Am J Physiol Cell Physiol 296(1):C151–C161
Lowe AG, Walmsley AR (1986) The kinetics of glucose transport in human red blood cells. Biochim Biophys Acta 857(2):146–154
Wheeler TJ, Hinkle PC (1981) Kinetic properties of the reconstituted glucose transporter from human erythrocytes. J Biol Chem 256(17):8907–8914
Wheeler TJ, Cole D, Hauck MA (1998) Characterization of glucose transport activity reconstituted from heart and other tissues. Biochim Biophys Acta 1414(1–2):217–230
Keller K, Strube M, Mueckler M (1989) Functional expression of the human HepG2 and rat adipocyte glucose transporters in Xenopus oocytes. Comparison of kinetic parameters. J Biol Chem 264(32):18884–18889
Gould GW, Lienhard GE (1989) Expression of a functional glucose transporter in Xenopus oocytes. Biochemistry 28(24):9447–9452
Vera JC, Rosen OM (1989) Functional expression of mammalian glucose transporters in Xenopus laevis oocytes: evidence for cell-dependent insulin sensitivity. Mol Cell Biol 9(10):4187–4195
Nishimura H, Pallardo FV, Seidner GA, Vannucci S, Simpson IA, Birnbaum MJ (1993) Kinetics of GLUT1 and GLUT4 glucose transporters expressed in Xenopus oocytes. J Biol Chem 268(12):8514–8520
Vera JC, Rivas CI, Fischbarg J, Golde DW (1993) Mammalian facilitative hexose transporters mediate the transport of dehydroascorbic acid. Nature 364(32):79–82
Sage JM, Carruthers A (2014) Human erythrocytes transport dehydroascorbic acid and sugars using the same transporter complex. Am J Physiol Cell Physiol 306(10):C910–C917
KC S, Cárcamo JM, Golde DW (2005) Vitamin C enters mitochondria via facilitative glucose transporter 1 (Glut1) and confers mitochondrial protection against oxidative injury. FASEB J 19(12):1657–1667
Jiang X, McDermott JR, Ajees AA, Rosen BP, Liu Z (2010) Trivalent arsenicals and glucose use different translocation pathways in mammalian GLUT1. Metallomics 2(3):211–219
Patching SG (2015) Roles of facilitative glucose transporter GLUT1 in [18F]FDG positron emission tomography (PET) imaging of human diseases. J Diagn Imaging Ther 2(1):30–102
Bloch R (1973) Inhibition of glucose transport in the human erythrocyte by cytochalasin B. Biochemistry 12(23):4799–4801
Basketter DA, Widdas WF (1978) Asymmetry of the hexose transfer system in human erythrocytes. Comparison of the effects of cytochalasin B, phloretin and maltose as competitive inhibitors. J Physiol 278:389–401
Sergeant S, Kim HD (1985) Inhibition of 3-O-methylglucose transport in human erythrocytes by forskolin. J Biol Chem 260(27):14677–14682
Martin HJ, Kornmann F, Fuhrmann GF (2003) The inhibitory effects of flavonoids and antiestrogens on the Glut1 glucose transporter in human erythrocytes. Chem Biol Interact 146(3):225–235
Robichaud T, Appleyard AN, Herbert RB, Henderson PJ, Carruthers A (2011) Determinants of ligand binding affinity and cooperativity at the GLUT1 endofacial site. Biochemistry 50(15):3137–3148
Farrell CL, Pardridge WM (1991) Blood-brain barrier glucose transporter is asymmetrically distributed on brain capillary endothelial lumenal and ablumenal membranes: an electron microscopic immunogold study. Proc Natl Acad Sci U S A 88(13):5779–5783
Hawkins RA, Peterson DR, Viña JR (2002) The complementary membranes forming the blood-brain barrier. IUBMB Life 54(3):101–107
Gerhart DZ, LeVasseur RJ, Broderius MA, Drewes LR (1989) Glucose transporter localization in brain using light and electron immunocytochemistry. J Neurosci Res 22(4):464–472
Cornford EM, Hyman S, Swartz BE (1994) The human brain glut1 glucose transporter: ultrastructural localization to the blood-brain barrier endothelia. J Cereb Blood Flow Metab 14(1):106–112
Duelli R, Maurer MH, Staudt R, Heiland S, Duembgen L, Kuschinsky W (2000) Increased cerebral glucose utilization and decreased glucose transporter Glut1 during chronic hyperglycemia in rat brain. Brain Res 858(2):338–347
Duelli R, Kuschinsky W (2001) Brain glucose transporters: relationship to local energy demand. News Physiol Sci 16:71–76
McAllister MS, Krizanac-Bengez L, Macchia F, Naftalin RJ, Pedley KC, Mayberg MR, Marroni M, Leaman S et al (2001) Mechanisms of glucose transport at the blood-brain barrier: an in vitro study. Brain Res 904(1):20–30
Simpson IA, Vannucci SJ, DeJoseph MR, Hawkins RA (2001) Glucose transporter asymmetries in the bovine blood-brain barrier. J Biol Chem 276(16):12725–12729
Cornford EM, Hyman S (2005) Localization of brain endothelial luminal and abluminal transporters with immunogold electron microscopy. NeuroRx 2(1):27–43
Kubo Y, Ohtsuki S, Uchida Y, Terasaki T (2015) Quantitative determination of luminal and abluminal membrane distributions of transporters in porcine brain capillaries by plasma membrane fractionation and quantitative targeted proteomics. J Pharm Sci 104(9):3060–3068
Devraj K, Klinger ME, Myers RL, Mokashi A, Hawkins RA, Simpson IA (2011) GLUT-1 glucose transporters in the blood-brain barrier: differential phosphorylation. J Neurosci Res 89(12):1913–1925
Barros LF, Bittner CX, Loaiza A, Porras OH (2007) A quantitative overview of glucose dynamics in the gliovascular unit. Glia 55(12):1222–1237
Kreft M, Lukšič M, Zorec TM, Prebil M, Zorec R (2013) Diffusion of D-glucose measured in the cytosol of a single astrocyte. Cell Mol Life Sci 70(8):1483–1492
Pardridge WM (1983) Brain metabolism: a perspective from the blood-brain barrier. Physiol Rev 63(4):1481–1535
Aleshin AE, Zeng C, Bourenkov GP, Bartunik HD, Fromm HJ, Honzatko RB (1998) The mechanism of regulation of hexokinase: new insights from the crystal structure of recombinant human brain hexokinase complexed with glucose and glucose-6-phosphate. Structure 6(1):39–50
Lund-Andersen H (1979) Transport of glucose from blood to brain. Physiol Rev 59(2):305–352
Blomqvist G, Grill V, Ingvar M, Widén L, Stone-Elander S (1998) The effect of hyperglycaemia on regional cerebral glucose oxidation in humans studied with [1-11C]-D-glucose. Acta Physiol Scand 163(4):403–415
Gruetter R, Novotny EJ, Boulware SD, Rothman DL, Mason GF, Shulman GI, Shulman RG, Tamborlane WV (1992) Direct measurement of brain glucose concentrations in humans by 13C NMR spectroscopy. Proc Natl Acad Sci U S A 89(3):1109–1112
Gruetter R, Ugurbil K, Seaquist ER (1998) Steady-state cerebral glucose concentrations and transport in the human brain. J Neurochem 70(1):397–408
McNay EC, Gold PE (1999) Extracellular glucose concentrations in the rat hippocampus measured by zero-net-flux: effects of microdialysis flow rate, strain, and age. J Neurochem 72(2):785–790
Reinstrup P, Stahl N, Mellergard P, Uski T, Ungerstedt U, Nordstrom CH (2000) Intracerebral microdialysis in clinical practice: baseline values for chemical markers during wakefulness, anesthesia, and neurosurgery. Neurosurgery 47(3):701–709
Wilson JE (1980) Brain hexokinase, the prototype ambiquitous enzyme. Curr Top Cell Regul 16:1–54
McGowan KM, Long SD, Pekala PH (1995) Glucose transporter gene expression: regulation of transcription and mRNA stability. Pharmacol Ther 66(3):465–505
Dwyer KJ, Boado RJ, Pardridge WM (1996) Cis-element/cytoplasmic protein interaction within the 3'-untranslated region of the glut1 glucose transporter mRNA. J Neurochem 66(2):449–458
Devaskar S, Zahm DS, Holtzclaw L, Chundu K, Wadzinski BE (1991) Developmental regulation of the distribution of rat brain insulin-insensitive (Glut 1) glucose transporter. Endocrinology 129(3):1530–1540
Kaiser N, Sasson S, Feener EP, Boukobza-Vardi N, Higashi S, Moller DE, Davidheiser S, Przybylski RJ et al (1993) Differential regulation of glucose transport and transporters by glucose in vascular endothelial and smooth muscle cells. Diabetes 42(1):80–89
Zheng PP, Romme E, van der Spek PJ, Dirven CM, Willemsen R, Kros JM (2010) Glut1/SLC2A1 is crucial for the development of the blood-brain barrier in vivo. Ann Neurol 68(6):835–844
Vannucci SJ, Seaman LB, Brucklacher RM, Vannucci RC (1994) Glucose transport in developing rat brain: glucose transporter proteins, rate constants and cerebral glucose utilization. Mol Cell Biochem 140(2):177–184
Nualart F, Godoy A, Reinicke K (1999) Expression of the hexose transporters GLUT1 and GLUT2 during the early development of the human brain. Brain Res 824(1):97–104
Virgintino D, Robertson D, Benagiano V, Errede M, Bertossi M, Ambrosi G, Roncali L (2000) Immunogold cytochemistry of the blood-brain barrier glucose transporter GLUT1 and endogenous albumin in the developing human brain. Brain Res Dev Brain Res 123(1):95–101
Vannucci SJ, Simpson IA (2003) Developmental switch in brain nutrient transporter expression in the rat. Am J Physiol Endocrinol Metab 285(5):E1127–E1134
Dormire SL (2009) The potential role of glucose transport changes in hot flash physiology: a hypothesis. Biol Res Nurs 10(3):241–247
Morgello S, Uson RR, Schwartz EJ, Haber RS (1995) The human blood-brain barrier glucose transporter (GLUT1) is a glucose transporter of gray matter astrocytes. Glia 14(1):43–54
Yu S, Ding WG (1998) The 45 kDa form of glucose transporter 1 (glut1) is localized in oligodendrocyte and astrocyte but not in microglia in the rat brain. Brain Res 797(1):65–72
Birnbaum MJ, Haspel HC, Rosen OM (1986) Cloning and characterization of a cDNA encoding the rat brain glucose-transporter protein. Proc Natl Acad Sci U S A 83(16):5784–5788
Asano T, Takata K, Katagiri H, Ishihara H, Inukai K, Anai M, Hirano H, Yazaki Y et al (1993) The role of N-glycosylation in the targeting and stability of GLUT1 glucose transporter. FEBS Lett 324(3):258–261
Asano T, Katagiri H, Takata K, Lin JL, Ishihara H, Inukai K, Tsukuda K, Kikuchi M et al (1991) The role of N-glycosylation of GLUT1 for glucose transport activity. J Biol Chem 266(36):24632–24636
Onetti R, Baulida J, Bassols A (1997) Increased glucose transport in ras-transformed fibroblasts: a possible role for N-glycosylation of GLUT1. FEBS Lett 407(3):267–270
McMahon RJ, Hwang JB, Frost SC (2000) Glucose deprivation does not affect GLUT1 targeting in 3T3-L1 adipocytes. Biochem Biophys Res Commun 273(3):859–864
Maher F, Vannucci SJ, Simpson IA (1994) Glucose transporter proteins in brain. FASEB J 8(13):1003–1011
Maher F (1995) Immunolocalization of GLUT1 and GLUT3 glucose transporters in primary cultured neurons and glia. J Neurosci Res 42(4):459–469
Maher F, Davies-Hill TM, Lysko PG, Henneberry RC, Simpson IA (1991) Expression of two glucose transporters, GLUT1 and GLUT3, in cultured cerebellar neurons: evidence for neuron-specific expression of GLUT3. Mol Cell Neurosci 2(4):351–360
Mantych GJ, James DE, Chung HD, Devaskar SU (1992) Cellular localization and characterization of glut3 glucose transporter isoform in human brain. Endocrinology 131(3):1270–1278
Gerhart DZ, Broderius MA, Borson ND, Drewes LR (1992) Neurons and microvessels express the brain glucose transporter protein glut3. Proc Natl Acad Sci U S A 89(2):733–737
Maher F, Davies-Hill TM, Simpson IA (1996) Substrate specificity and kinetic parameters of glut3 in rat cerebellar granule neurons. Biochem J 315(3):827–831
Simpson IA, Carruthers A, Vannucci SJ (2007) Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J Cereb Blood Flow Metab 27(11):1766–1791
Maher F, Simpson IA (1994) The GLUT3 glucose transporter is the predominant isoform in primary cultured neurons: assessment by biosynthetic and photoaffinity labelling. Biochem J 301(2):379–384
Vannucci SJ, Maher F, Simpson IA (1997) Glucose transporter proteins in brain: delivery of glucose to neurons and glia. Glia 21(1):2–21
Simpson IA, Dwyer D, Malide D, Moley KH, Travis A, Vannucci SJ (2008) The facilitative glucose transporter GLUT3: 20 years of distinction. Am J Physiol Endocrinol Metab 295(2):E242–E253
Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE, Weil RJ, Nakano I et al (2013) Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci 16(10):1373–1382
Deng D, Sun P, Yan C, Ke M, Jiang X, Xiong L, Ren W, Hirata K et al (2015) Molecular basis of ligand recognition and transport by glucose transporters. Nature 526(7573):391–396
Payne J, Maher F, Simpson I, Mattice L, Davies P (1997) Glucose transporter glut 5 expression in microglial cells. Glia 21(3):327–331
Horikoshi Y, Sasaki A, Taguchi N, Maeda M, Tsukagoshi H, Sato K, Yamaguchi H (2003) Human GLUT5 immunolabeling is useful for evaluating microglial status in neuropathological study using paraffin sections. Acta Neuropathol 105(2):157–162
Mantych GJ, James DE, Devaskar SU (1993) Jejunal/kidney glucose transporter isoform (Glut-5) is expressed in the human blood-brain barrier. Endocrinology 132(1):35–40
Burant CF, Takeda J, Brot-Laroche E, Bell GI, Davidson NO (1992) Fructose transporter in human spermatozoa and small intestine is GLUT5. J Biol Chem 267(21):14523–14526
Kane S, Seatter MJ, Gould GW (1997) Functional studies of human GLUT5: effect of pH on substrate selection and an analysis of substrate interactions. Biochem Biophys Res Commun 238(2):503–505
Douard V, Ferraris RP (2008) Regulation of the fructose transporter GLUT5 in health and disease. Am J Physiol Endocrinol Metab 295(2):E227–E237
Barone S, Fussell SL, Singh AK, Lucas F, Xu J, Kim C, Wu X, Yu Y et al (2009) Slc2a5 (Glut5) is essential for the absorption of fructose in the intestine and generation of fructose-induced hypertension. J Biol Chem 284(8):5056–5066
Nomura N, Verdon G, Kang HJ, Shimamura T, Nomura Y, Sonoda Y, Hussien SA, Qureshi AA et al (2015) Structure and mechanism of the mammalian fructose transporter GLUT5. Nature 526(7573):397–401
Leloup C, Arluison M, Lepetit N, Cartier N, Marfaing-Jallat P, Ferre P, Penicaud L (1994) Glucose transporter 2 (GLUT 2): expression in specific brain nuclei. Brain Res 638(1–2):221–226
Leloup C, Allard C, Carneiro L, Fioramonti X, Collins S, Pénicaud L (2015) Glucose and hypothalamic astrocytes: more than a fueling role? Neuroscience. doi:10.1016/j.neuroscience.2015.06.007
Arluison M, Quignon M, Nguyen P, Thorens B, Leloup C, Penicaud L (2004) Distribution and anatomical localization of the glucose transporter 2 (GLUT2) in the adult rat brain–an immunohistochemical study. J Chem Neuroanat 28(3):117–136
Arluison M, Quignon M, Thorens B, Leloup C, Penicaud L (2004) Immunocytochemical localization of the glucose transporter 2 (GLUT2) in the adult rat brain. II. Electron microscopic study. J Chem Neuroanat 28(3):137–146
Jurcovicova J (2014) Glucose transport in brain–effect of inflammation. Endocr Regul 48(1):35–48
Marín-Juez R, Rovira M, Crespo D, van der Vaart M, Spaink HP, Planas JV (2015) GLUT2-mediated glucose uptake and availability are required for embryonic brain development in zebrafish. J Cereb Blood Flow Metab 35(1):74–85
Forsyth R, Fray A, Boutelle M, Fillenz M, Middleditch C, Burchell A (1996) A role for astrocytes in glucose delivery to neurons? Dev Neurosci 18(5–6):360–370
McCall AL, van Bueren AM, Huang L, Stenbit A, Celnik E, Charron MJ (1997) Forebrain endothelium expresses GLUT4, the insulin-responsive glucose transporter. Brain Res 744(2):318–326
Ngarmukos C, Baur EL, Kumagai AK (2001) Co-localization of glut1 and glut4 in the blood-brain barrier of the rat ventromedial hypothalamus. Brain Res 900(1):1–8
Kobayashi M, Nikami H, Morimatsu M, Saito M (1996) Expression and localization of insulin-regulatable glucose transporter (glut4) in rat brain. Neurosci Lett 213(2):103–106
El Messari S, Leloup C, Quignon M, Brisorgueil MJ, Penicaud L, Arluison M (1998) Immunocytochemical localization of the insulin-responsive glucose transporter 4 (glut4) in the rat central nervous system. J Comp Neurol 399(4):492–512
Apelt J, Mehlhorn G, Schliebs R (1999) Insulin-sensitive glut4 glucose transporters are colocalized with glut3-expressing cells and demonstrate a chemically distinct neuron-specific localization in rat brain. J Neurosci Res 57(5):693–705
Reagan LP, Rosell DR, Alves SE, Hoskin EK, McCall AL, Charron MJ, McEwen BS (2002) Glut8 glucose transporter is localized to excitatory and inhibitory neurons in the rat hippocampus. Brain Res 932(1–2):129–134
Sankar R, Thamotharan S, Shin D, Moley KH, Devaskar SU (2002) Insulin-responsive glucose transporters-glut8 and glut4 are expressed in the developing mammalian brain. Brain Res Mol Brain Res 107(2):157–165
Gomez O, Ballester-Lurbe B, Mesonero JE, Terrado J (2011) Glucose transporters glut4 and glut8 are upregulated after facial nerve axotomy in adult mice. J Anat 219(4):525–530
Schmidt S, Joost HG, Schürmann A (2009) GLUT8, the enigmatic intracellular hexose transporter. Am J Physiol Endocrinol Metab 296(4):E614–E618
Doege H, Bocianski A, Joost HG, Schürmann A (2000) Activity and genomic organization of human glucose transporter 9 (GLUT9), a novel member of the family of sugar-transport facilitators predominantly expressed in brain and leucocytes. Biochem J 350(3):771–776
Stuart CA, Ross IR, Howell ME, McCurry MP, Wood TG, Ceci JD, Kennel SJ, Wall J (2011) Brain glucose transporter (Glut3) haploinsufficiency does not impair mouse brain glucose uptake. Brain Res 1384:15–22
Nishizaki T, Kammesheidt A, Sumikawa K, Asada T, Okada Y (1995) A sodium- and energy-dependent glucose transporter with similarities to sglt1-2 is expressed in bovine cortical vessels. Neurosci Res 22(1):13–22
Nishizaki T, Matsuoka T (1998) Low glucose enhances Na+/glucose transport in bovine brain artery endothelial cells. Stroke 29(4):844–849
Elfeber K, Köhler A, Lutzenburg M, Osswald C, Galla HJ, Witte OW, Koepsell H (2004) Localization of the Na+-D-glucose cotransporter SGLT1 in the blood-brain barrier. Histochem Cell Biol 121(3):201–207
Vemula S, Roder KE, Yang T, Bhat GJ, Thekkumkara TJ, Abbruscato TJ (2009) A functional role for sodium-dependent glucose transport across the blood-brain barrier during oxygen glucose deprivation. J Pharmacol Exp Ther 328(2):487–495
Poppe R, Karbach U, Gambaryan S, Wiesinger H, Lutzenburg M, Kraemer M, Witte OW, Koepsell H (1997) Expression of the Na+-D-glucose cotransporter SGLT1 in neurons. J Neurochem 69(1):84–94
O'Malley D, Reimann F, Simpson AK, Gribble FM (2006) Sodium-coupled glucose cotransporters contribute to hypothalamic glucose sensing. Diabetes 55(12):3381–3386
Yu AS, Hirayama BA, Timbol G, Liu J, Diez-Sampedro A, Kepe V, Satyamurthy N, Huang SC et al (2013) Regional distribution of SGLT activity in rat brain in vivo. Am J Physiol Cell Physiol 304(3):C240–C247
Wright EM, Hirayama BA, Loo DF (2007) Active sugar transport in health and disease. J Intern Med 261(1):32–43
Chen XZ, Coady MJ, Jackson F, Berteloot A, Lapointe JY (1995) Thermodynamic determination of the Na+: glucose coupling ratio for the human SGLT1 cotransporter. Biophys J 69(6):2405–2414
Mackenzie B, Loo DD, Wright EM (1998) Relationships between Na+/glucose cotransporter (SGLT1) currents and fluxes. J Membr Biol 162(2):101–106
Lee WJ, Peterson DR, Sukowski EJ, Hawkins RA (1997) Glucose transport by isolated plasma membranes of the bovine blood-brain barrier. Am J Physiol 272(5):C1552–C1557
Kanai Y, Lee WS, You G, Brown D, Hediger MA (1994) The human kidney low affinity Na+/glucose cotransporter SGLT2. Delineation of the major renal reabsorptive mechanism for D-glucose. J Clin Invest 93(1):397–404
You G, Lee WS, Barros EJ, Kanai Y, Huo TL, Khawaja S, Wells RG, Nigam SK et al (1995) Molecular characteristics of Na+-coupled glucose transporters in adult and embryonic rat kidney. J Biol Chem 270(49):29365–29371
Yu AS, Hirayama BA, Timbol G, Liu J, Basarah E, Kepe V, Satyamurthy N, Huang SC et al (2010) Functional expression of SGLTs in rat brain. Am J Physiol Cell Physiol 299(6):C1277–C1284
Kumagai AK, Kang YS, Boado RJ, Pardridge WM (1995) Upregulation of bloodbrain barrier GLUT1 glucose transporter protein and mRNA in experimental chronic hypoglycemia. Diabetes 44(12):1399–1404
Simpson IA, Appel NM, Hokari M, Oki J, Holman GD, Maher F, Koehler-Stec EM, Vannucci SJ et al (1999) Blood-brain barrier glucose transporter: effects of hypo- and hyperglycemia revisited. J Neurochem 72(1):238–247
Sajja RK, Prasad S, Cucullo L (2014) Impact of altered glycaemia on blood-brain barrier endothelium: an in vitro study using the hCMEC/D3 cell line. Fluids Barriers CNS 11:8
Uehara Y, Nipper V, McCall AL (1997) Chronic insulin hypoglycemia induces GLUT-3 protein in rat brain neurons. Am J Physiol 272(4):E716–E719
Lee DH, Chung MY, Lee JU, Kang DG, Paek YW (2000) Changes of glucose transporters in the cerebral adaptation to hypoglycemia. Diabetes Res Clin Pract 47(1):15–23
Sahin K, Tuzcu M, Orhan C, Ali S, Sahin N, Gencoglu H, Ozkan Y, Hayirli A et al (2013) Chromium modulates expressions of neuronal plasticity markers and glial fibrillary acidic proteins in hypoglycemia-induced brain injury. Life Sci 93(25–26):1039–1048
Zhao F, Deng J, Yu X, Li D, Shi H, Zhao Y (2015) Protective effects of vascular endothelial growth factor in cultured brain endothelial cells against hypoglycemia. Metab Brain Dis 30(4):999–1007
(2012) Standards of medical care in diabetes. Diabetes Care 35(1):S11–S63
Pardridge WM, Triguero D, Farrell CR (1990) Downregulation of blood-brain barrier glucose transporter in experimental diabetes. Diabetes 39(9):1040–1044
Hou WK, Xian YX, Zhang L, Lai H, Hou XG, Xu YX, Yu T, Xu FY et al (2007) Influence of blood glucose on the expression of glucose trans-porter proteins 1 and 3 in the brain of diabetic rats. Chin Med J (Engl) 120(19):1704–1709
Hasselbalch SG, Knudsen GM, Capaldo B, Postiglione A, Paulson OB (2001) Blood-brain barrier transport and brain metabolism of glucose during acute hyperglycemia in humans. J Clin Endocrinol Metab 86(5):1986–1990
Jacob RJ, Fan X, Evans ML, Dziura J, Sherwin RS (2002) Brain glucose levels are elevated in chronically hyperglycemic diabetic rats: no evidence for protective adaptation by the blood brain barrier. Metabolism 51(12):1522–1524
Seaquist ER, Tkac I, Damberg G, Thomas W, Gruetter R (2005) Brain glucose concentrations in poorly controlled diabetes mellitus as measured by high-field magnetic resonance spectroscopy. Metabolism 54(8):1008–1013
www.alz.org/downloads/ facts_Fig.s_2012.pdf
Luchsinger JA, Reitz C, Patel B, Tang MX, Manly JJ, Mayeux R (2007) Relation of diabetes to mild cognitive impairment. Arch Neurol 64(4):570–575
Iadecola C, Davisson RL (2008) Hypertension and cerebrovascular dysfunction. Cell Metab 7(6):476–484
Whitmer RA, Gustafson DR, Barrett-Connor E, Haan MN, Gunderson EP, Yaffe K (2008) Central obesity and increased risk of dementia more than three decades later. Neurology 71(14):1057–1064
Knopman DS, Roberts R (2010) Vascular risk factors: imaging and neuropathologic correlates. J Alzheimers Dis 20(3):699–709
Naderali EK, Ratcliffe SH, Dale MC (2009) Obesity and Alzheimer's disease: a link between body weight and cognitive function in old age. Am J Alzheimers Dis Other Demen 24(6):445–449
Dolan H, Crain B, Troncoso J, Resnick SM, Zonderman AB, O’brien RJ (2010) Atherosclerosis, dementia, and Alzheimer’s disease in the BLSA cohort. Ann Neurol 68(2):231–240
Suemoto CK, Nitrini R, Grinberg LT, Ferretti RE, Farfel JM, Leite RE, Menezes PR, Fregni F et al (2011) Atherosclerosis and dementia: a cross-sectional study with pathological analysis of the carotid arteries. Stroke 42(12):3614–3615
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297(5580):353–356
Karran E, Mercken M, De Strooper B (2011) The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov 10(9):698–712
Reitz C (2012) Alzheimer's disease and the amyloid cascade hypothesis: a critical review. Int J Alzheimers Dis 2012:369808
Armstrong RA (2014) A critical analysis of the 'amyloid cascade hypothesis'. Folia Neuropathol 52(3):211–225
Drachman DA (2014) The amyloid hypothesis, time to move on: amyloid is the downstream result, not cause, of Alzheimer's disease. Alzheimers Dement 10(3):372–380
Morris GP, Clark IA, Vissel B (2014) Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer's disease. Acta Neuropathol Commun 2:135
Zlokovic BV (2005) Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci 28(4):202–208
Zlokovic BV (2010) Neurodegeneration and the neurovascular unit. Nat Med 16(12):1370–1371
de la Torre JC (2010) Vascular risk factor detection and control may prevent Alzheimer’s disease. Aging Res Rev 9(3):218–225
Marchesi VT (2011) Alzheimer’s dementia begins as a disease of small blood vessels, damaged by oxidative-induced inflammation and dysregulated amyloid metabolism: implications for early detection and therapy. FASEB J 25(1):5–13
Sagare AP, Bell RD, Zlokovic BV (2012) Neurovascular dysfunction and faulty amyloid β-peptide clearance in Alzheimer disease. Cold Spring Harb Perspect Med 2(10):a011452
Lyros E, Bakogiannis C, Liu Y, Fassbender K (2014) Molecular links between endothelial dysfunction and neurodegeneration in Alzheimer's disease. Current Alzheimer Res 11(1):18–26
Harik SI, LaManna JC (1991) Altered glucose metabolism in microvessels from patients with Alzheimer’s disease. Ann Neurol 29(5):573
Meneilly GS, Hill A (1993) Alterations in glucose metabolism in patients with Alzheimer’s disease. J Am Geriatr Soc 41(7):710–714
Casadesus G, Moreira PI, Nunomura A, Siedlak SL, Bligh-Glover W, Balraj E, Petot G, Smith MA et al (2007) Indices of metabolic dysfunction and oxidative stress. Neurochem Res 32(4–5):717–722
Hunt A, Schonknecht P, Henze M, Seidl U, Haberkorn U, Schroder J (2007) Reduced cerebral glucose metabolism in patients at risk for Alzheimer’s disease. Psychiatry Res 155(2):147–154
Mosconi L, Pupi A, De Leon MJ (2008) Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann N Y Acad Sci 1147:180–195
Furst AJ, Lal RA (2011) Amyloid-beta and glucose metabolism in Alzheimer’s disease. J Alzheimers Dis 26(3):105–116
Chen Z, Zhong C (2013) Decoding Alzheimer's disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies. Prog Neurobiol 108:21–43
Friedland RP, Jagust WJ, Huesman RH, Koss E, Knittel B, Mathis CA, Ober BA, Mazoyer BM et al (1989) Regional cerebral glucose transport and utilization in Alzheimer’s disease. Neurology 39(11):1427–1434
Jagust WJ, Seab JP, Huesman RH, Valk PE, Mathis CA, Reed BR, Coxson PG, Budinger TF (1991) Diminished glucose transport in Alzheimer’s disease: dynamic pet studies. J Cereb Blood Flow Metab 11(2):323–330
Piert M, Koeppe RA, Giordani B, Berent S, Kuhl DE (1996) Diminished glucose transport and phosphorylation in Alzheimer’s disease determined by dynamic FDG-PET. J Nucl Med 37(2):201–208
Samuraki M, Matsunari I, Chen WP, Yajima K, Yanase D, Fujikawa A, Takeda N, Nishimura S et al (2007) Partial volume effect-corrected FDG PET and grey matter volume loss in patients with mild Alzheimer’s disease. Eur J Nucl Med Mol Imaging 34(10):1658–1669
Nobili F, Morbelli S (2010) [18F]FDG-PET as a biomarker for early Alzheimer’s disease. Open Nucl Med J 2:46–52
Mosconi L, Berti V, Glodzik L, Pupi A, De Santi S, de Leon MJ (2010) Pre-clinical detection of Alzheimer's disease using FDG-PET, with or without amyloid imaging. J Alzheimers Dis 20(3):843–854
Mosconi L, McHugh PF (2011) FDG- and amyloid-PET in Alzheimer's disease: is the whole greater than the sum of the parts? Q J Nucl Med Mol Imaging 55(3):250–264
Ishii K (2014) PET approaches for diagnosis of dementia. AJNR Am J Neuroradiol 35(11):2030–2038
Perani D, Schillaci O, Padovani A, Nobili FM, Iaccarino L, Della Rosa PA, Frisoni G, Caltagirone C (2014) A survey of FDG- and amyloid-PET imaging in dementia and GRADE analysis. Biomed Res Int 2014:785039
Shokouhi S, Claassen D, Riddle W (2014) Imaging brain metabolism and pathology in Alzheimer's disease with positron emission tomography. J Alzheimers Dis Parkinsonism 4(2):143
Kalaria RN, Harik SI (1989) Reduced glucose transporter at the blood-brain barrier and in cerebral cortex in Alzheimer disease. J Neurochem 53(4):1083–1088
Simpson IA, Chundu KR, Davies-Hill T, Honer WG, Davies P (1994) Decreased concentrations of glut1 and glut3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann Neurol 35(5):546–551
Harr SD, Simonian NA, Hyman BT (1995) Functional alterations in Alzheimer’s disease: decreased glucose transporter 3 immunoreactivity in the perforant pathway terminal zone. J Neuropathol Exp Neurol 54(1):38–41
Mooradian AD, Chung HC, Shah GN (1997) Glut-1 expression in the cerebra of patients with Alzheimer’s disease. Neurobiol Aging 18(5):469–474
Bailey TL, Rivara CB, Rocher AB, Hof PR (2004) The nature and effects of cortical microvascular pathology in aging and Alzheimer’s disease. Neurol Res 26(5):573–578
Liu Y, Liu F, Iqbal K, Grundke-Iqbal I, Gong CX (2008) Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett 582(2):359–364
Li X, Lu F, Wang JZ, Gong CX (2006) Concurrent alterations of O-GlcNAcylation and phosphorylation of tau in mouse brains during fasting. Eur J Neurosci 23(8):2078–2086
Dias WB, Hart GW (2007) O-GlcNAc modification in diabetes and Alzheimer's disease. Mol Biosyst 3(11):766–772
Liu F, Shi J, Tanimukai H, Gu J, Gu J, Grundke-Iqbal I, Iqbal K, Gong CX (2009) Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer's disease. Brain 132(7):1820–1832
Liu Y, Liu F, Grundke-Iqbal I, Iqbal K, Gong CX (2009) Brain glucose transporters, O-GlcNAcylation and phosphorylation of tau in diabetes and Alzheimer’s disease. J Neurochem 111(1):242–249
Lee CW, Shih YH, Wu SY, Yang T, Lin C, Kuo YM (2013) Hypoglycemia induces tau hyperphosphorylation. Curr Alzheimer Res 10(3):298–308
Yuzwa SA, Vocadlo DJ (2014) O-GlcNAc and neurodegeneration: biochemical mechanisms and potential roles in Alzheimer's disease and beyond. Chem Soc Rev 43(19):6839–6858
Zhu Y, Shan X, Yuzwa SA, Vocadlo DJ (2014) The emerging link between O-GlcNAc and Alzheimer disease. J Biol Chem 289(50):34472–34481
Winkler EA, Nishida Y, Sagare AP, Rege SV, Bell RD, Perlmutter D, Sengillo JD, Hillman S et al (2015) GLUT1 reductions exacerbate Alzheimer's disease vasculo-neuronal dysfunction and degeneration. Nat Neurosci 18(4):521–530
Akter K, Lanza EA, Martin SA, Myronyuk N, Rua M, Raffa RB (2011) Diabetes mellitus and Alzheimer’s disease: shared pathology and treatment? Br J Clin Pharmacol 71(3):365–376
Correia SC, Santos RX, Carvalho C, Cardoso S, Candeias E, Santos MS, Oliveira CR, Moreira PI (2012) Insulin signaling, glucose metabolism and mitochondria: major players in Alzheimer's disease and diabetes interrelation. Brain Res 1441:64–78
Adeghate E, Donáth T, Adem A (2013) Alzheimer disease and diabetes mellitus: do they have anything in common? Curr Alzheimer Res 10(6):609–617
Ahmad W (2013) Overlapped metabolic and therapeutic links between Alzheimer and diabetes. Mol Neurobiol 47(1):399–424
De Felice FG (2013) Connecting type 2 diabetes to Alzheimer's disease. Expert Rev Neurother 13(12):1297–1299
Jayaraman A, Pike CJ (2014) Alzheimer's disease and type 2 diabetes: multiple mechanisms contribute to interactions. Curr Diab Rep 14(4):476
Mushtaq G, Khan JA, Kamal MA (2014) Biological mechanisms linking Alzheimer's disease and type-2 diabetes mellitus. CNS Neurol Disord Drug Targets 13(7):1192–1201
Verdile G, Fuller SJ, Martins RN (2015) The role of type 2 diabetes in neurodegeneration. Neurobiol Dis. doi:10.1016/j.nbd.2015.04.008
de la Monte SM, Wands JR (2008) Alzheimer's disease is type 3 diabetes-evidence reviewed. J Diabetes Sci Technol 2(6):1101–1113
Kroner Z (2009) The relationship between Alzheimer’s disease and diabetes: type 3 diabetes? Altern Med Rev 14(4):373–379
Accardi G, Caruso C, Colonna-Romano G, Camarda C, Monastero R, Candore G (2012) Can Alzheimer disease be a form of type 3 diabetes? Rejuvenation Res 15(2):217–221
Ahmed S, Mahmood Z, Zahid S (2015) Linking insulin with Alzheimer's disease: emergence as type III diabetes. Neurol Sci 36(10):1763–1769
Shah K, Desilva S, Abbruscato T (2012) The role of glucose transporters in brain disease: diabetes and Alzheimer’s disease. Int J Mol Sci 13(10):12629–12655
Thompson BJ, Ronaldson PT (2014) Drug delivery to the ischemic brain. Adv Pharmacol 71:165–202
Shah K, Abbruscato T (2014) The role of blood-brain barrier transporters in pathophysiology and pharmacotherapy of stroke. Curr Pharm Des 20(10):1510–1522
Alluri H, Anasooya Shaji C, Davis ML, Tharakan B (2015) Oxygen-glucose deprivation and reoxygenation as an in vitro ischemia-reperfusion injury model for studying blood-brain barrier dysfunction. J Vis Exp 99:e52699
McCall AL, Van Bueren AM, Nipper V, Moholt-Siebert M, Downes H, Lessov N (1996) Forebrain ischemia increases GLUT1 protein in brain microvessels and parenchyma. J Cereb Blood Flow Metab 16(1):69–76
Vannucci SJ, Seaman LB, Vannucci RC (1996) Effects of hypoxia-ischemia on GLUT1 and GLUT3 glucose transporters in immature rat brain. J Cereb Blood Flow Metab 16(1):77–81
Vannucci SJ, Reinhart R, Maher F, Bondy CA, Lee WH, Vannucci RC, Simpson IA (1998) Alterations in GLUT1 and GLUT3 glucose transporter gene expression following unilateral hypoxia-ischemia in the immature rat brain. Brain Res Dev Brain Res 107(2):255–264
Yeh WL, Lin CJ, Fu WM (2008) Enhancement of glucose transporter expression of brain endothelial cells by vascular endothelial growth factor derived from glioma exposed to hypoxia. Mol Pharmacol 73(1):170–177
Cura AJ, Carruthers A (2010) Acute modulation of sugar transport in brain capillary endothelial cell cultures during activation of the metabolic stress pathway. J Biol Chem 285(20):15430–15439
Zhang WW, Zhang L, Hou WK, Xu YX, Xu H, Lou FC, Zhang Y, Wang Q (2009) Dynamic expression of glucose transporters 1 and 3 in the brain of diabetic rats with cerebral ischemia reperfusion. Chin Med J (Engl) 122(17):1996–2001
Zovein A, Flowers-Ziegler J, Thamotharan S, Shin D, Sankar R, Nguyen K, Gambhir S, Devaskar SU (2004) Postnatal hypoxic-ischemic brain injury alters mechanisms mediating neuronal glucose transport. Am J Physiol Regul Integr Comp Physiol 286(2):R273–R282
Iwabuchi S, Kawahara K (2011) Inducible astrocytic glucose transporter-3 contributes to the enhanced storage of intracellular glycogen during reperfusion after ischemia. Neurochem Int 59(2):319–325
Espinoza-Rojo M, Iturralde-Rodríguez KI, Chánez-Cárdenas ME, Ruiz-Tachiquín ME, Aguilera P (2010) Glucose transporters regulation on ischemic brain: possible role as therapeutic target. Cent Nerv Syst Agents Med Chem 10(4):317–325
Zhang S, Zuo W, Guo XF, He WB, Chen NH (2014) Cerebral glucose transporter: the possible therapeutic target for ischemic stroke. Neurochem Int 70:22–29
Shah KK, Boreddy PR, Abbruscato TJ (2015) Nicotine pre-exposure reduces stroke-induced glucose transporter-1 activity at the blood-brain barrier in mice. Fluids Barriers CNS 12:10
Yamazaki Y, Harada S, Tokuyama S (2012) Post-ischemic hyperglycemia exacerbates the development of cerebral ischemic neuronal damage through the cerebral sodium-glucose transporter. Brain Res 1489:113–120
Yamazaki Y, Harada S, Tokuyama S (2014) Sodium-glucose transporter type 3-mediated neuroprotective effect of acetylcholine suppresses the development of cerebral ischemic neuronal damage. Neuroscience 269:134–142
Yamazaki Y, Harada S, Tokuyama S (2015) Relationship between cerebral sodium-glucose transporter and hyperglycemia in cerebral ischemia. Neurosci Lett 604:134–139
De Vivo DC, Trifiletti RR, Jacobson RI, Ronen GM, Behmand RA, Harik SI (1991) Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. New Eng J Med 325(10):703–709
Klepper J, Willemsen M, Verrips A, Guertsen E, Herrmann R, Kutzick C, Flörcken A, Voit T (2001) Autosomal dominant transmission of GLUT1 deficiency. Hum Mol Genet 10(1):63–68
Klepper J, Scheffer H, Elsaid MF, Kamsteeg EJ, Leferink M, Ben-Omran T (2009) Autosomal recessive inheritance of GLUT1 deficiency syndrome. Neuropediatrics 40(5):207–210
Rotstein M, Engelstad K, Yang H, Wang D, Levy B, Chung WK, De Vivo DC (2010) Glut1 deficiency: inheritance pattern determined by haploinsufficiency. Ann Neurol 68(6):955–958
Wang BY, Kalir T, Sabo E, Sherman DE, Cohen C, Burstein DE (2000) Immunohistochemical staining of GLUT1 in benign, hyperplastic and malignant endometrial epithelia. Cancer 88(12):2774–2781
Klepper J, Leiendecker B, Bredahl R, Athanassopoulos S, Heinen F, Gertsen E, Flörcken A, Metz A et al (2002) Introduction of a ketogenic diet in young infants. J Inherit Metab Dis 25(6):449–460
Brockmann K (2009) The expanding phenotype of GLUT1-deficiency syndrome. Brain Dev 31(7):545–552
De Giorgis V, Veggiotti P (2013) GLUT1 deficiency syndrome 2013: current state of the art. Seizure 22(10):803–811
Pearson TS, Akman C, Hinton VJ, Engelstad K, De Vivo DC (2013) Phenotypic spectrum of glucose transporter type 1 deficiency syndrome (Glut1 DS). Curr Neurol Neurosci Rep 13(4):342
Gras D, Roze E, Caillet S, Méneret A, Doummar D, Billette de Villemeur T, Vidailhet M, Mochel F (2014) GLUT1 deficiency syndrome: an update. Rev Neurol (Paris) 170(2):91–99
Leen WG, Taher M, Verbeek MM, Kamsteeg EJ, van de Warrenburg BP, Willemsen MA (2014) GLUT1 deficiency syndrome into adulthood: a follow-up study. J Neurol 261(3):589–599
Ragona F, Matricardi S, Castellotti B, Patrini M, Freri E, Binelli S, Granata T (2014) Refractory absence epilepsy and glut1 deficiency syndrome: a new case report and literature review. Neuropediatrics 45(5):328–332
Tzadok M, Nissenkorn A, Porper K, Matot I, Marcu S, Anikster Y, Menascu S, Bercovich D et al (2014) The many faces of Glut1 deficiency syndrome. J Child Neurol 29(3):349–359
Suls A, Mullen SA, Weber YG, Verhaert K, Ceulemans B, Guerrini R, Wuttke TV, Salvo-Vargas A et al (2009) Early-onset absence epilepsy caused by mutations in the glucose transporter GLUT1. Ann Neurol 66(3):415–419
Mullen SA, Suls A, De Jonghe P, Berkovic SF, Scheffer IE (2010) Absence epilepsies with widely variable onset are a key feature of familial GLUT1 deficiency. Neurology 75(5):432–440
Arsov T, Mullen SA, Damiano JA, Lawrence KM, Huh LL, Nolan M, Young H, Thouin A et al (2012) Early onset absence epilepsy: 1 in 10 cases is caused by GLUT1 deficiency. Epilepsia 53(12):e204–e207
Arsov T, Mullen SA, Rogers S, Phillips AM, Lawrence KM, Damiano JA, Goldberg-Stern H, Afawi Z et al (2012) Glucose transporter 1 deficiency in the idiopathic generalized epilepsies. Ann Neurol 72(5):807–815
Striano P, Weber YG, Toliat MR, Schubert J, Leu C, Chaimana R, Baulac S, Guerrero R et al (2012) GLUT1 mutations are a rare cause of familial idiopathic generalized epilepsy. Neurology 78(8):557–562
Weber YG, Storch A, Wuttke TV, Brockmann K, Kempfle J, Maljevic S, Margari L, Kamm C et al (2008) GLUT1 mutations are a cause of paroxysmal exertion-induced dyskinesias and induce hemolytic anemia by a cation leak. J Clin Invest 118(6):2157–2168
Suls A, Dedeken P, Goffin K, Van Esch H, Dupont P, Cassiman D, Kempfle J, Wuttke TV et al (2008) Paroxysmal exercise-induced dyskinesia and epilepsy is due to mutations in SLC2A1, encoding the glucose transporter GLUT1. Brain 131(7):1831–1844
Schneider SA, Paisan-Ruiz C, Garcia-Gorostiaga I, Quinn NP, Weber YG, Lerche H, Hardy J, Bhatia KP (2009) GLUT1 gene mutations cause sporadic paroxysmal exercise-induced dyskinesias. Mov Disord 24(11):1684–1688
Weber YG, Kamm C, Suls A, Kempfle J, Kotschet K, Schüle R, Wuttke TV, Maljevic S et al (2011) Paroxysmal choreoathetosis/spasticity (DYT9) is caused by a GLUT1 defect. Neurology 77(10):959–964
Verrotti A, D'Egidio C, Agostinelli S, Gobbi G (2012) Glut1 deficiency: when to suspect and how to diagnose? Eur J Paediatr Neurol 16(1):3–9
Leen WG, Wevers RA, Kamsteeg EJ, Scheffer H, Verbeek MM, Willemsen MA (2013) Cerebrospinal fluid analysis in the workup of GLUT1 deficiency syndrome: a systematic review. JAMA Neurol 70(11):1440–1444
Klepper J, Garcia-Alvarez M, O'Driscoll KR, Parides MK, Wang D, Ho YY, De Vivo DC (1999) Erythrocyte 3-O-methyl-D-glucose uptake assay for diagnosis of glucose-transporter-protein syndrome. J Clin Lab Anal 13(3):116–121
Yang H, Wang D, Engelstad K, Bagay L, Wei Y, Rotstein M, Aggarwal V, Levy B et al (2011) Glut1 deficiency syndrome and erythrocyte glucose uptake assay. Ann Neurol 70(6):996–1005
Wang D, Pascual JM, De Vivo D. Glucose transporter type 1 deficiency syndrome. In Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K (eds). GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2015
Pascual JM, Van Heertum RL, Wang D, Engelstad K, De Vivo DC (2002) Imaging the metabolic footprint of Glut1 deficiency on the brain. Ann Neurol 52(4):458–464
Klepper J, Diefenbach S, Kohlschütter A, Voit T (2004) Effects of the ketogenic diet in the glucose transporter 1 deficiency syndrome. Prostaglandins Leukot Essent Fatty Acids 70(3):321–327
Klepper J (2008) Glucose transporter deficiency syndrome (GLUT1DS) and the ketogenic diet. Epilepsia 49(8):46–49
Klepper J, Leiendecker B (2013) Glut1 deficiency syndrome and novel ketogenic diets. J Child Neurol 28(8):1045–1048
Bertoli S, Trentani C, Ferraris C, De Giorgis V, Veggiotti P, Tagliabue A (2014) Long-term effects of a ketogenic diet on body composition and bone mineralization in GLUT-1 deficiency syndrome: a case series. Nutrition 30(6):726–728
Veggiotti P, De Giorgis V (2014) Dietary treatments and new therapeutic perspective in GLUT1 deficiency syndrome. Curr Treat Options Neurol 16(5):291
Chenouard A, Vuillaumier-Barrot S, Seta N, Kuster A (2015) A cause of permanent ketosis: GLUT-1 deficiency. JIMD Rep 18:79–83
Singh LD, Singh SP, Handa RK, Ehmann S, Snyder AK (1996) Effects of ethanol on GLUT1 protein and gene expression in rat astrocytes. Metab Brain Dis 11(4):343–357
Hu IC, Singh SP, Snyder AK (1995) Effects of ethanol on glucose transporter expression in cultured hippocampal neurons. Alcohol Clin Exp Res 19(6):1398–1402
Handa RK, DeJoseph MR, Singh LD, Hawkins RA, Singh SP (2000) Glucose transporters and glucose utilization in rat brain after acute ethanol administration. Metab Brain Dis 15(3):211–222
Abdul Muneer PM, Alikunju S, Szlachetka AM, Haorah J (2011) Inhibitory effects of alcohol on glucose transport across the blood-brain barrier leads to neurodegeneration: preventive role of acetyl-L-carnitine. Psychopharmacology (Berlin) 214(3):707–718
Duelli R, Staudt R, Grünwald F, Kuschinsky W (1998) Increase of glucose transporter densities (Glut1 and Glut3) during chronic administration of nicotine in rat brain. Brain Res 782(1–2):36–42
Canis M, Mack B, Gires O, Maurer MH, Kuschinsky W, Duembgen L, Duelli R (2009) Increased densities of monocarboxylate transport protein MCT1 after chronic administration of nicotine in rat brain. Neurosci Res 64(4):429–435
Abdul Muneer PM, Alikunju S, Szlachetka AM, Murrin LC, Haorah J (2011) Impairment of brain endothelial glucose transporter by methamphetamine causes blood-brain barrier dysfunction. Mol Neurodegener 6:23
Abdul Muneer PM, Alikunju S, Szlachetka AM, Haorah J (2011) Methamphetamine inhibits the glucose uptake by human neurons and astrocytes: stabilization by acetyl-L-carnitine. PLoS One 6(4):e19258
Rask-Andersen M, Masuram S, Fredriksson R, Schiöth HB (2013) Solute carriers as drug targets: current use, clinical trials and prospective. Mol Asp Med 34(2–3):702–710
Pardridge WM (2003) Blood-brain barrier drug targeting: the future of brain drug development. Mol Interv 3(2):90–105
Pardridge WM (2005) Molecular biology of the blood-brain barrier. Mol Biotechnol 30(1):57–70
Pardridge WM (2007) Blood-brain barrier delivery. Drug Discov Today 12(1–2):54–61
Ohtsuki S, Terasaki T (2007) Contribution of carrier-mediated transport systems to the blood-brain barrier as a supporting and protecting interface for the brain; importance for CNS drug discovery and development. Pharm Res 24(9):1745–1758
Patel MM, Goyal BR, Bhadada SV, Bhatt JS, Amin AF (2009) Getting into the brain: approaches to enhance brain drug delivery. CNS Drugs 23(1):35–58
Stenehjem DD, Hartz AM, Bauer B, Anderson GW (2009) Novel and emerging strategies in drug delivery for overcoming the blood-brain barrier. Future Med Chem 1(9):1623–1641
Ronaldson PT (2014) Targeting transporters for CNS drug delivery. Curr Pharm Des 20(10):1419–1421
Alyautdin R, Khalin I, Nafeeza MI, Haron MH, Kuznetsov D (2014) Nanoscale drug delivery systems and the blood-brain barrier. Int J Nanomedicine 9:795–811
On NH, Miller DW (2014) Transporter-based delivery of anticancer drugs to the brain: improving brain penetration by minimizing drug efflux at the blood-brain barrier. Curr Pharm Des 20(10):1499–1509
Tashima T (2015) Intriguing possibilities and beneficial aspects of transporter-conscious drug design. Bioorg Med Chem 23(15):4119–4131
Pardridge WM (2015) Blood-brain barrier endogenous transporters as therapeutic targets: a new model for small molecule CNS drug discovery. Expert Opin Ther Targets 19(8):1059–1072
Yang C, Tirucherai GS, Mitra AK (2001) Prodrug based optimal drug delivery via membrane transporter/receptor. Expert Opin Biol Ther 1(2):159–175
Prokai-Tatrai K, Prokai L (2003) Modifying peptide properties by prodrug design for enhanced transport into the CNS. Prog Drug Res 61:155–188
Stella VJ, Nti-Addae KW (2007) Prodrug strategies to overcome poor water solubility. Adv Drug Deliv Rev 59(7):677–694
Huttunen KM, Rautio J (2011) Prodrugs–an efficient way to breach delivery and targeting barriers. Curr Top Med Chem 11(18):2265–2287
Pavan B, Dalpiaz A, Ciliberti N, Biondi C, Manfredini S, Vertuani S (2008) Progress in drug delivery to the central nervous system by the prodrug approach. Molecules 13(5):1035–1065
Pavan B, Dalpiaz A (2011) Prodrugs and endogenous transporters: are they suitable tools for drug targeting into the central nervous system? Curr Pharm Des 17(32):3560–3576
Peura L, Malmioja K, Huttunen K, Leppänen J, Hämäläinen M, Forsberg MM, Gynther M, Rautio J et al (2013) Design, synthesis and brain uptake of LAT1-targeted amino acid prodrugs of dopamine. Pharm Res 30(10):2523–2537
Tamai I, Tsuji A (2000) Transporter-mediated permeation of drugs across the blood-brain barrier. J Pharm Sci 89(11):1371–1388
Guo X, Geng M, Du G (2005) Glucose transporter 1, distribution in the brain and in neural disorders: its relationship with transport of neuroactive drugs through the blood-brain barrier. Biochem Genet 43(3–4):175–187
Halmos T, Santarromana M, Antonakis K, Scherman D (1996) Synthesis of glucose-chlorambucil derivatives and their recognition by the human GLUT1 glucose transporter. Eur J Pharmacol 318(2–3):477–484
Halmos T, Santarromana M, Antonakis K, Scherman D (1997) Synthesis of O-methylsulfonyl derivatives of D-glucose as potential alkylating agents for targeted drug delivery to the brain. Evaluation of their interaction with the human erythrocyte GLUT1 hexose transporter. Carbohydr Res 299(1–2):15–21
Bilsky EJ, Egleton RD, Mitchell SA, Palian MM, Davis P, Huber JD, Jones H, Yamamura HI et al (2000) Enkephalin glycopeptide analogues produce analgesia with reduced dependence liability. J Med Chem 43(13):2586–2590
Polt R, Porreca F, Szabò LZ, Bilsky EJ, Davis P, Abbruscato TJ, Davis TP, Harvath R et al (1994) Glycopeptide enkephalin analogues produce analgesia in mice: evidence for penetration of the blood-brain barrier. Proc Natl Acad Sci U S A 91(15):7114–7118
Elmagbari NO, Egleton RD, Palian MM, Lowery JJ, Schmid WR, Davis P, Navratilova E, Dhanasekaran M et al (2004) Antinociceptive structure-activity studies with enkephalin-based opioid glycopeptides. J Pharmacol Exp Ther 311(1):290–297
Battaglia G, La Russa M, Bruno V, Arenare L, Ippolito R, Copani A, Bonina F, Nicoletti F (2000) Systematically administered D-glucose conjugates of 7-chlorokynurenic acid are centrally available and exert anticonvulsant activity in rodents. Brain Res 860(1–2):149–156
Bonina F, Arenare L, Ippolito R, Boatto G, Battaglia G, Bruno V, de Caparariis P (2000) Synthesis, pharmacokinetics and anticonvulsant activity of 7-chlorokynurenic acid prodrugs. Int J Pharm 202(1–2):79–88
Bonina F, Puglia C, Rimoli MG, Melisi D, Boatto G, Nieddu M, Malignano A, La Rana G et al (2003) Glycosyl derivatives of dopamine and L-dopa as antiparkinson prodrugs: synthesis, pharmacological activity and in vitro stability studies. J Drug Target 11(1):25–36
Dalpiaz A, Filosa R, de Caprariis P, Conte G, Bortolotti F, Biondi C, Scatturin A, Prasad PD et al (2007) Molecular mechanism involved in the transport of a prodrug dopamine glycosyl conjugate. Int J Pharm 336(1):133–139
Gynther M, Ropponen J, Laine K, Leppänen J, Haapakoski P, Peura L, Järvinen T, Rautio J (2009) Glucose promoiety enables glucose transporter mediated brain uptake of ketoprofen and indomethacin prodrugs in rats. J Med Chem 52(10):3348–3353
Chen Q, Gong T, Liu J, Wang X, Fu H, Zhang Z (2009) Synthesis, in vitro and in vivo characterization of glycosyl derivatives of ibuprofen as novel prodrugs for brain drug delivery. J Drug Target 17(4):318–328
Zhao Y, Qu B, Wu X, Li X, Liu Q, Jin X, Guo L, Hai L et al (2014) Design, synthesis and biological evaluation of brain targeting l-ascorbic acid prodrugs of ibuprofen with "lock-in" function. Eur J Med Chem 82:314–323
Jain KK (2012) Nanobiotechnology-based strategies for crossing the blood-brain barrier. Nanomedicine (London) 7(8):1225–1233
Costantino L, Boraschi D, Eaton M (2014) Challenges in the design of clinically useful brain-targeted drug nanocarriers. Curr Med Chem 21(37):4227–4246
Zhou X, Porter AL, Robinson DK, Shim MS, Guo Y (2014) Nano-enabled drug delivery: a research profile. Nanomedicine 10(5):889–896
Hwang SR, Kim K (2014) Nano-enabled delivery systems across the blood-brain barrier. Arch Pharm Res 37(1):24–30
Ma J, Porter AL, Aminabhavi TM, Zhu D (2015) Nano-enabled drug delivery systems for brain cancer and Alzheimer's disease: research patterns and opportunities. Nanomedicine 11(7):1763–1771
Qin Y, Fan W, Chen H, Yao N, Tang W, Tang J, Yuan W, Kuai R et al (2010) In vitro and in vivo investigation of glucose-mediated brain-targeting liposomes. J Drug Target 18(7):536–549
Lei F, Fan W, Li XK, Wang S, Hai L, Wu Y (2011) Design, synthesis and preliminary bio-evaluation of glucose-cholesterol derivatives as ligands for brain targeting liposomes. Chin Chem Lett 22(7):831–834
Xie F, Yao N, Qin Y, Zhang Q, Chen H, Yuan M, Tang J, Li X et al (2012) Investigation of glucose-modified liposomes using polyethylene glycols with different chain lengths as the linkers for brain targeting. Int J Nanomedicine 7:163–175
Qu B, Li X, Guan M, Li X, Hai L, Wu Y (2014) Design, synthesis and biological evaluation of multivalent glucosides with high affinity as ligands for brain targeting liposomes. Eur J Med Chem 72:110–118
Hao ZF, Cui YX, Li MH, Du D, Liu MF, Tao HQ, Li S, Cao FY et al (2013) Liposomes modified with P-aminophenyl-α-D-mannopyranoside: a carrier for targeting cerebral functional regions in mice. Eur J Pharm Biopharm 84(3):505–516
Du D, Chang N, Sun S, Li M, Yu H, Liu M, Liu X, Wang G et al (2014) The role of glucose transporters in the distribution of p-aminophenyl-α-d-mannopyranoside modified liposomes within mice brain. J Control Release 182:99–110
Zidan AS, Aldawsari H (2015) Ultrasound effects on brain-targeting mannosylated liposomes: in vitro and blood-brain barrier transport investigations. Drug Des Devel Ther 9:3885–3898
Shao K, Zhang Y, Ding N, Huang S, Wu J, Li J, Yang C, Leng Q et al (2015) Functionalized nanoscale micelles with brain targeting ability and intercellular microenvironment biosensitivity for anti-intracranial infection applications. Adv Healthc Mater 4(2):291–300
Shao K, Ding N, Huang S, Ren S, Zhang Y, Kuang Y, Guo Y, Ma H et al (2014) Smart nanodevice combined tumor-specific vector with cellular microenvironment-triggered property for highly effective antiglioma therapy. ACS Nano 8(2):1191–1203
Guo Y, Zhang Y, Li J, Zhang Y, Lu Y, Jiang X, He X, Ma H et al (2015) Cell microenvironment-controlled antitumor drug releasing-nanomicelles for GLUT1-targeting hepatocellular carcinoma therapy. ACS Appl Mater Interfaces 7(9):5444–5453
Niu J, Wang A, Ke Z, Zheng Z (2014) Glucose transporter and folic acid receptor-mediated Pluronic P105 polymeric micelles loaded with doxorubicin for brain tumor treating. J Drug Target 22(8):712–723
Gromnicova R, Davies HA, Sreekanthreddy P, Romero IA, Lund T, Roitt IM, Phillips J, Male DK (2013) Glucose-coated gold nanoparticles transfer across human brain endothelium and enter astrocytes in vitro. PLoS One 8(12):e81043
Jiang X, Xin H, Ren Q, Gu J, Zhu L, Du F, Feng C, Xie Y et al (2014) Nanoparticles of 2-deoxy-D-glucose functionalized poly(ethylene glycol)-co-poly(trimethylene carbonate) for dual-targeted drug delivery in glioma treatment. Biomaterials 35(1):518–529
Naik P, Cucullo L (2012) In vitro blood-brain barrier models: current and perspective technologies. J Pharm Sci 101(4):1337–1354
Abbott NJ (2002) Astrocyte‐endothelial interactions and the blood‐brain barrier permeability. J Anat 200(6):629–638
Parkinson FE, Hacking C (2005) Pericyte abundance affects sucrose permeability in cultures of rat brain microvascular endothelial cells. Brain Res 1049(1):8–14
He Y, Yao Y, Tsirka SE, Cao Y (2014) Cell-culture models of the blood-brain barrier. Stroke 45(8):2514–2526
Weksler BB, Subileau EA, Perriere N, Charneau P, Holloway K, Leveque M, Tricoire-Leignel H, Nicotra A et al (2005) Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J 19(13):1872–1874
Tarbell JM (2010) Shear stress and the endothelial transport barrier. Cardiovasc Res 87(2):320–330
Bussolari SR, Dewey CF Jr, Gimbrone MA Jr (1982) Apparatus for subjecting living cells to fluid shear stress. Rev Sci Instrum 53(12):1851–1854
Stanness KA, Guatteo E, Janigro D (1996) A dynamic model of the blood-brain barrier “in vitro”. Neurotoxicology 17(2):481–496
Janigro D, Leaman SM, Stanness KA (1999) Dynamic modeling of the blood-brain barrier: a novel tool for studies of drug delivery to the brain. Pharm Sci Technol Today 2(1):7–12
Cucullo L, McAllister MS, Kight K, Krizanac-Bengez L, Marroni M, Mayberg MR, Stanness KA, Janigro D (2002) A new dynamic in vitro model for the multidimensional study of astrocyte-endothelial cell interactions at the blood-brain barrier. Brain Res 951(2):243–254
Santaguida S, Janigro D, Hossain M, Oby E, Rapp E, Cucullo L (2006) Side by side comparison between dynamic versus static models of blood-brain barrier in vitro: a permeability study. Brain Res 1109(1):1–13
Cucullo L, Couraud PO, Weksler B, Romero IA, Hossain M, Rapp E, Janigro D (2008) Immortalized human brain endothelial cells and flow-based vascular modeling: a marriage of convenience for rational neurovascular studies. J Cereb Blood Flow Metab 28(2):312–328
Booth R, Kim H (2011) A multi-layered microfluidic device for in vitro bloodbrain barrier permeability studies. In: International conference on miniaturized systems for chemistry and life sciences. Red Hook: Curran Associates, Inc. pp 1388–1390
Booth R, Kim H (2012) Characterization of a microfluidic in vitro model of the blood-brain barrier (μBBB). Lab Chip 12(10):1784–1792
Yeon JH, Na D, Choi K, Ryu SW, Choi C, Park JK (2012) Reliable permeability assay system in a microfluidic device mimicking cerebral vasculatures. Biomed Microdevices 14(6):1141–1148
Griep LM, Wolbers F, de Wagenaar B, ter Braak PM, Weksler BB, Romero IA, Couraud PO, Vermes I et al (2013) BBB on chip: microfluidic platform to mechanically and biochemically modulate blood-brain barrier function. Biomed Microdevices 15(1):145–150
Prabhakarpandian B, Shen MC, Nichols JB, Mills IR, Sidoryk-Wegrzynowicz M, Aschner M, Pant K (2013) SyM-BBB: a microfluidic blood brain barrier model. Lab Chip 13(6):1093–1101
Gumbleton M, Audus KL (2001) Progress and limitations in the use of in vitro cell cultures to serve as a permeability screen for the blood-brain barrier. J Pharm Sci 90(11):1681–1698
Deli MA, Abrahám CS, Kataoka Y, Niwa M (2005) Permeability studies on in vitro blood-brain barrier models: physiology, pathology, and pharmacology. Cell Mol Neurobiol 25(1):59–127
Deli E (2007) Blood–brain barrier models (chapter 2). In: Abel L (ed) Handbook of neurochemistry and molecular neurobiology (3rd edition). Springer, Berlin, pp 29–55
Ogunshola OO (2011) In vitro modeling of the blood-brain barrier: simplicity versus complexity. Curr Pharm Des 17(26):2755–2761
Tóth A, Veszelka S, Nakagawa S, Niwa M, Deli MA (2011) Patented in vitro blood-brain barrier models in CNS drug discovery. Recent Pat CNS Drug Discov 6(2):107–118
Wilhelm I, Fazakas C, Krizbai IA (2011) In vitro models of the blood-brain barrier. Acta Neurobiol Exp (Wars) 71(1):113–128
Shawahna R, Decleves X, Scherrmann JM (2013) Hurdles with using in vitro models to predict human blood-brain barrier drug permeability: a special focus on transporters and metabolizing enzymes. Curr Drug Metab 14(1):120–136
Shityakov S, Salvador E, Förster C (2013) In silico, in vitro and in vivo methods to analyse drug permeation across the blood brain barrier: a critical review. OA Anaesthetics 1:13
Abbott NJ, Dolman DEM, Yusof SR, Reichel A (2014) In vitro models of CNS barriers. In Drug delivery to the brain. New York: Springer pp 163–197
Bicker J, Alves G, Fortuna A, Falcão A (2014) Blood-brain barrier models and their relevance for a successful development of CNS drug delivery systems: a review. Eur J Pharm Biopharm 87(3):409–432
Czupalla CJ, Liebner S, Devraj K (2014) In vitro models of the blood-brain barrier. Methods Mol Biol 1135:415–437
Palmiotti CA, Prasad S, Naik P, Abul KM, Sajja RK, Achyuta AH, Cucullo L (2014) In vitro cerebrovascular modeling in the 21st century: current and prospective technologies. Pharm Res 31(12):3229–3250
Wilhelm I, Krizbai IA (2014) In vitro models of the blood–brain barrier for the study of drug delivery to the brain. Mol Pharm 11(7):1949–1963
Wolff A, Antfolk M, Brodin B, Tenje M (2015) In vitro blood-brain barrier models–an overview of established models and new microfluidic approaches. J Pharm Sci 104(9):2727–2746
Weksler B, Romero IA, Couraud PO (2013) The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS 10(1):16
Poller B, Gutmann H, Krähenbühl S, Weksler B, Romero I, Couraud PO, Tuffin G, Drewe J et al (2008) The human brain endothelial cell line hCMEC/D3 as a human blood-brain barrier model for drug transport studies. J Neurochem 107(5):1358–1368
Carl SM, Lindley DJ, Das D, Couraud PO, Weksler BB, Romero I, Mowery SA, Knipp GT (2010) ABC and SLC transporter expression and proton oligopeptide transporter (POT) mediated permeation across the human blood-brain barrier cell line, hCMEC/D3 [corrected]. Mol Pharm 7(4):1057–1068
Eigenmann DE, Xue G, Kim KS, Moses AV, Hamburger M, Oufir M (2013) Comparative study of four immortalized human brain capillary endothelial cell lines, hCMEC/D3, hBMEC, TY10, and BB19, and optimization of culture conditions, for an in vitro blood-brain barrier model for drug permeability studies. Fluids Barriers CNS 10(1):33
Urich E, Lazic SE, Molnos J, Wells I, Freskgård PO (2012) Transcriptional profiling of human brain endothelial cells reveals key properties crucial for predictive in vitro blood-brain barrier models. PLoS One 7(5):e38149
Ohtsuki S, Ikeda C, Uchida Y, Sakamoto Y, Miller F, Glacial F, Decleves X, Scherrmann JM et al (2013) Quantitative targeted absolute proteomic analysis of transporters, receptors and junction proteins for validation of human cerebral microvascular endothelial cell line hCMEC/D3 as a human blood-brain barrier model. Mol Pharm 10(1):289–296
Meireles M, Martel F, Araújo J, Santos-Buelga C, Gonzalez-Manzano S, Dueñas M, de Freitas V, Mateus N et al (2013) Characterization and modulation of glucose uptake in a human blood-brain barrier model. J Membr Biol 246(9):669–677
Lippmann ES, Azarin SM, Kay JE, Nessler RA, Wilson HK, Al-Ahmad A, Palecek SP, Shusta EV (2012) Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat Biotechnol 30(8):783–791
Cecchelli R, Aday S, Sevin E, Almeida C, Culot M, Dehouck L, Coisne C, Engelhardt B et al (2014) A stable and reproducible human blood-brain barrier model derived from hematopoietic stem cells. PLoS One 9(6):e99733
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The author declares that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Patching, S.G. Glucose Transporters at the Blood-Brain Barrier: Function, Regulation and Gateways for Drug Delivery. Mol Neurobiol 54, 1046–1077 (2017). https://doi.org/10.1007/s12035-015-9672-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9672-6