
Abstract
TWIK-1 two-pore domain K+ channels are highly expressed in mature hippocampal astrocytes. While the TWIK-1 activity is readily detectable on astrocyte membrane, the majority of channels are retained in the intracellular compartments, which raises an intriguing question of whether the membrane TWIK-1 channels could be dynamically regulated for functions yet unknown. Here, the regulation of TWIK-1 membrane expression by Gi/Go-coupled metabotropic glutamate receptor 3 (mGluR3) and its functional impact on ammonium uptake has been studied. Activation of mGluR3 induced a marked translocation of TWIK-1 channels from the cytoplasm to the membrane surface. Consistent with our early observation that membrane TWIK-1 behaves as nonselective monovalent cation channel, mGluR3-mediated TWIK-1 membrane expression was associated with a depolarizing membrane potential (V M). As TWIK-1 exhibits a discernibly high permeability to ammonium (NH4 +), a critical substrate in glutamate-glutamine cycle for neurotransmitter replenishment, regulation of NH4 + uptake capacity by TWIK-1 membrane expression was determined by response of astrocyte V M to bath application of 5 mM NH4Cl. Stimulation of mGluR3 potentiated NH4 +-induced V M depolarization by ∼30 % in wild type, but not in TWIK-1 knockout astrocytes. Furthermore, activation of mGluR3 mediated a coordinated translocation of TWIK-1 channels with recycling endosomes toward astrocyte membrane and the mGluR3-mediated potentiation of NH4 + uptake required a functional Rab-mediated trafficking pathway. Altogether, we demonstrate that the activation of mGluR3 up-regulates the membrane expression of TWIK-1 that in turn enhances NH4 + uptake in astrocytes, a mechanism potentially important for functional coupling of astrocyte glutamate-glutamine cycle with the replenishment of neurotransmitters in neurons.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The TWIK-1 two-pore domain K+ channel shows the highest mRNA expression level among other known K+ channels in mature astrocytes [1, 2]. However, astrocytes in TWIK-1 gene knockout mice show only a mildly altered membrane potential and K+ conductance attributable to retention of large amount of channels in the intracellular compartments [3]. Interestingly, the membrane TWIK-1 exhibits unique ion permeability. First, the membrane TWIK-1 is a nonselective monovalent cation channel with high permeability to Na+ in astrocytes, kidney tubular, and pancreatic β cells [3–5]. Second, the TWIK-1 preferentially conducts inward NH4 + currents that is 40-fold higher than the K+ currents in heterologous systems [6]; this implies that TWIK-1 may function as a highly efficient NH4 + uptake channel in astrocytes. Nevertheless, owing to the aforementioned biophysical properties, TWIK-1 unlikely serves as a conventional K+ channel; thus, the role of TWIK-1 in astrocytes and the mechanisms regulating its membrane expression need to be determined.
A predominant cytoplasmic location of TWIK-1 has been revealed by immunocytochemical studies from several native and transfected cells [7, 8]. Association of TWIK-1 with recycling endosomes has been further identified as a responsible mechanism for a lack of functional membrane currents in cultured kidney cells [9]. In the event of a fall in extracellular K+ (hypokalemia) or pH, TWIK-1 switches to conduct Na+ [4, 10]; thus, the leak Na+ behavior of TWIK-1 in astrocyte and other native cells is likely attributable to a transient presence and rapid retrieval of membrane channels to the acidic environment inside recycling endosomes [11].
In the study by Feliciangeli and colleagues, activation of several Gi/Go-coupled receptors, such 5-HT and α2-adrenergic receptors, has led to an upregulation of functional TWIK-1 expression [9]. Astrocytes express several Gi-coupled receptors, such as metabotropic glutamate receptor 3 (mGluR3), GABAB receptor, and endocannabinoid receptor CB1 [1]; therefore, it is possible that similar regulatory pathways may exist in astrocytes for dynamic regulation of membrane TWIK-1 expression. Additionally, mGluR3 has been revealed as the only mGluR isoform in cortical and hippocampal astrocytes in adult mice [12, 13], though little is known about the basic function of mGluR3 in astrocytes. In the present study, we tested a hypothesis that activation of mGluR3 recruits cytoplasmic TWIK-1 channels to membrane in hippocampal astrocytes. We show that the activation of mGluR3 increases membrane TWIK-1 expression through a Rab-mediated endosomal recycling pathway. Functionally, an increasing membrane TWIK-1 expression is associated with an elevated NH4 + uptake, a substrate critical in the glutamate-glutamine cycle for the repletion of neurotransmitter glutamate and GABA in neurons.
Materials and Methods
Animals
All the experimental procedures were performed in accordance with a protocol approved by the Animal Care and Use Committees of the Ohio State University. TWIK-1 knockout mice were created from mice of C57BL/6J genetic background, where the exon 2 gene was deleted [3, 8]. All the experiments were performed from the littermates that contained wild-type (WT), TWIK-1 heterozygous (TWIK-1+/−), and knockout (TWIK-1−/−) male and female mice at postnatal day (P) 21–28 unless indicated otherwise. The genotypes of the used mice were individually confirmed by a polymerase chain reaction (PCR) genotype procedure we have reported [3].
Fresh Dissociation of Single Astrocytes
After anesthesia and decapitation, the brains were rapidly removed from skulls and placed in oxygenated (95 % O2/5 % CO2) Ca2+-free artificial cerebral spinal fluid (Ca2+-free aCSF) (in mM): 125 NaCl, 25 NaHCO3, 1.25 NaH2PO4, 3.5 KCl, 1 MgCl2, 1 Na-pyruvate, and 10 glucose [14, 15]. Coronal hippocampal slices at 400-μm thickness were sectioned and transferred to Ca2+-free aCSF at 34 °C supplemented with astrocytic marker sulforhodamine101 (SR-101, 1 μM) [16]. After SR-101 incubation, the CA1 regions were dissected out from slices and placed in standard aCSF containing 24 U/ml papain and 0.8 mg/ml L-cysteine for 7 min. The standard aCSF contained (in mM): 125 NaCl, 25 NaHCO3, 1.25 NaH2PO4, 3.5 KCl, 2 CaCl2, 1 MgCl2, and 10 glucose. After papain digestion, the loosened slices were gently triturated five to seven times into a cell suspension and transferred into a petri dish mounted on a motorized inverted fluorescent microscope (Leica DMIRE2, Leica Microsystems Inc., USA) with constant aCSF perfusion. Single freshly isolated astrocytes were identified by SR101 fluorescent staining and harvested for the following quantitative RT-PCR (qRT-PCR) analysis.
Quantitative RT-PCR
RNAs were extracted from 30 freshly isolated astrocytes by using RNeasy mini kit (Qiagen, CA) immediately after cell harvesting, and RNAs were converted into cDNAs using Applied Biosystems’s High Capacity cDNA Reverse Transcription Kit (Grand Islands, NY). The gene transcripts of interest were mGluR2, mGluR3, mGluR5, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and the primers used to identify the transcripts are as follows: mGluR2 5′-TCAATGTCAAGTTTGATGCC-3′, 5′-GGATGATGCTAGTATCCAGAG-3′; mGluR3 5′-ACATATTCTCAGTCCTCTGC-3′, 5′-AGAAGGTACTAGGGTTGTTG-3′; mGluR5 5′-AAGGACAGATAAAGGTGATCC-3′; 5′-TCAAATCACAACCTGTCAAG-3′; and GAPDH 5′-CGTGGAGTCTACTGGTGT-3′, 5′-TGTCATATTTCTCGTGGT-3′. Each pair of primers was tested by conventional PCR that yielded a single band at the anticipated molecular weight. qRT-PCR was run using the SYBR® Select Master Mix (Invitrogen, NY) and the StepOnePlus™ Real-Time PCR system (Life Technologies, NY). Each target gene was assayed in triplicate from a single mouse sample, and three mice samples were used in total. GAPDH was used as an internal reference for each sample. Quantification of each target gene was acquired as cycle threshold (Ct) values and normalized to the Ct value of GAPDH (target Ct − GAPDH Ct) as ∆Ct. The expression levels of the target genes were quantified as 2−∆Ct.
Preparation of Acute Hippocampal Slices
Hippocampal slices were prepared as described previously [3]. Briefly, brains were rapidly removed from skulls and placed into ice-cold slice cutting aCSF with reduced Ca2+ and increased Mg2+ (in mM: 125 NaCl, 3.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 0.1 CaCl2, 3 MgCl2, and 10 glucose). Coronal hippocampal slices (250 μm) were cut at 4 °C with a Vibratome (Pelco 1500) and transferred to the oxygenated normal aCSF (osmolality, 295 ± 5 mOsm; pH 7.3–7.4), recovering at room temperature for at least 1 h before recording or immunostaining.
Immunohistochemistry
After treated with mGluR2/3 agonist or agonist plus antagonist, the hippocampal slices were washed with PBS and fixed in 4 % paraformaldehyde for 1 h at room temperature. Permeabilization was then followed in 0.2 % Triton X-100 PBS for 20 min. The slices were then incubated with a blocking solution consisting of 5 % normal donkey serum (DNS) and 0.01 %Triton X-100 in PBS for 4 h. The primary antibodies were diluted into a 10 % DNS/0.005 %Triton X-100 solution and applied to slices at 4 °C overnight. They were rabbit anti-TWIK-1 (1:400, Alomone Labs, Jerusalem, Israel), guinea pig anti-GLT1 (1:1000, Millipore, Billerica, MA), goat anti-GFAP (1:1000, Abcam, Cambridge, MA), or rat anti-Rab11 (1: 500, Abcam, Cambridge, MA). Followed by rise of slices with blocking solution, the secondary antibodies were applied for 1 h at room temperature. They were Cy3 donkey anti-rabbit (1:1000), Cy5 donkey anti-guinea pig (1:1000), and Alexa Fluor 488-conjugated donkey anti-goat antibody (1:1000) (Jackson ImmunoResearch Laboratories). Immunofluorescence images were obtained from a confocal microscope (LSM510, Carl Zeiss). Astrocytes were identified by glial fibrillary acidic protein (GFAP) staining and glutamate transporter GLT-1 was used as astrocyte membrane surface maker [13]. The Pearson’s correlation coefficient (PCC) was used for quantification of membrane TWIK-1 expression from astrocytic soma and processes based on TWIK-1/GLT-1 colocalization. The “Co-localization Analysis” plugin in NIH ImageJ software was used in these analyses. Only the cells showing a clearly outlined soma and bushy processes in GFAP staining were selected. As for the colocalization analysis in astrocyte processes, three to four process areas extending from soma (marked as rectangles in Fig. 2c) were used in each cell to obtain an average PCC value. Each PCC was further tested by Costes Randomization of the “Co-localization Test” plugin to confirm that the observed PCC value was not generated by random chance.
Electrophysiology
Individual hippocampal slices were transferred to the recording chamber mounted on an Olympus BX51WI microscope, with constant perfusion of oxygenated aCSF (2.0 ml/min). Astrocytes located in the CA1 region were visualized using an infrared differential interference contrast (IR-DIC) video camera. Whole-cell patch clamp recordings were performed using a MultiClamp 700A amplifier and pClamp 9.2 software (Molecular Devices, Sunnyvale, CA). Borosilicate glass pipettes (Warner Instrument) were pulled from a Micropipette Puller (Model P-87, Sutter Instrument). The recording electrode had a resistance of 5–7 MΩ when filled with the electrode solution containing (in mM) 140 K-gluconate, 13.4 Na-gluconate, 0.5 CaCl2, 1.0 MgCl2, 5 EGTA, 10 HEPES, 3 Mg-ATP, and 0.3 Na-GTP (280 ± 5 mOsm). A minimum of 2 GΩ seal resistance was required before rupturing the membrane for whole-cell configuration.
The membrane potential (V M) was recorded under current clamp mode without applying any holding currents in PClamp 9.2 program. The liquid junction potential was compensated prior to form the cell-attached mode for all the recordings. In current clamp mode, the input resistance (Rin) was measured by injecting a pair of ±100 pA/100 ms pulse before and after recording. Recordings with an initial Rin greater than 25 MΩ or the Rin varied greater than 10 % during recording were discarded. To determine NH4 + uptake capacity, 5 mM NH4Cl was added to aCSF by equimolar substitution with NaCl. To maximally reduce Na+-K+ ATPase activity, 125 mM NaCl in aCSF and 14.5 mM Na-gluconate in electrode solution was substituted by equimolar LiCl. All the experiments were conducted at room temperature.
Western Blot Analysis
Hippocampal slices in control and with addition of mGluR2/3 agonist treatment were separately collected. The hydrophobic (cytoplasmic) and hydrophilic (membrane) proteins were then separated by Mem-PER Eukaryotic Membrane Protein Extraction Kit (Thermo Fisher Scientific, Rockford, IL) as we reported previously [3]. Fractioned protein samples were then cleaned up by SDS-PAGE Sample Prep Kit (Thermo Fisher Scientific, Rockford, IL), to remove excessive detergents that may interfere the following test for protein concentration by BCA assay and SDS-PAGE.
When investigating the age-dependent expression of proteins of interest, the total protein of hippocampi was extracted from mice of different postnatal days. Hippocampal tissues were rapidly removed and homogenized by a Pro Homogenizer (Oxford, CT) in ice-cold lysis buffer containing 1 % Triton X-100, 25 mM Tris–HCl (pH = 7.2), 150 mM NaCl, 1 mM EDTA and protease inhibitor cocktails (Sigma-Aldrich, St. Louis, MO). Homogenates were laid on ice for 30 min and then centrifuged (3000×g, 15 min at 4 °C). The supernatants were transferred to new tubes and stored as aliquots at −80 °C until use. Protein concentration was determined by BCA assay. To perform SDS-PAGE, samples were mixed with a 5×reducing loading buffer containing 100 mM DTT (Thermo Fisher Scientific, Rockford, IL) and heated at 95 °C for 5 min. Equal amounts of proteins (25 μg/lane) were separated on a 4–12 % Tris-glycine gel (Bio-Rad, Hercules, CA, USA) and then transferred to a nitrocellulose membrane (Micron Separations Inc., Westborough, MA). The membranes were blocked with 5 % nonfat milk for 1 h at room temperature. The membranes were incubated with rabbit anti-TWIK-1 antibodies (1:2000) at 4 °C overnight. After secondary antibody incubation (Jackson ImmunoResearch Laboratories, Maine, USA), immunoreactivity was detected with an enhanced chemiluminescent detection (Thermo Fisher Scientific, Rockford, IL). After detecting TWIK-1 immunoreactivity, the membranes were stripped with stripping buffer (0.4 M glycine, 0.2 % SDS, and 2 % Tween 20, pH 2.0) and re-probed with the following rabbit primary antibodies sequentially: anti-Kir4.1 (1:600, Alomone Labs, Jerusalem, Israel), anti-Na+/K+ ATPase α2 (+) polypeptide (ATP1α2) (1:1000; Abgent, San Diego, CA), anti-GAPDH (1:8000, Sigma-Aldrich, St. Louis, MO), and anti-GFAP (1:500, DAKO, Carpinteria, CA). Anti-ATP1α2 and anti-GFAP were reliable protein fraction markers for membrane and cytoplasm, respectively [3]. The blots were scanned by the NIH ImageJ software.
Chemical Reagents
LY354740 hydrate, (2R, 4R)-APDC, and N-ethylmaleimide were obtained from Sigma-Aldrich (St. Louis, MO). LY 341495 and tetrodotoxin (TTX) were purchased from Tocris Bioscience (Bristol, UK). Psoromic acid was from Indofine Chemical Company, Inc. (Hillsborough, NJ). The above compounds were dissolved in preferred solvents and stored as aliquots at −80 °C until use. The final concentrations of the used stock solvents were less than 0.05 %. All other chemicals were purchased from Sigma-Aldrich.
Data Analyses
The patch clamp recording data were analyzed by Clampfit 9.0 (Molecular Devices, Sunnyvale, CA) and Origin 8.0 (OriginLab, Northhampton, MA). Results are given as means ± SEM. Statistical analysis was performed using Student’s t test or one-way ANOVA followed by post hoc Dunnett’s test on raw data. Significance level was set at P < 0.05.
Results
Mature Astrocytes Selectively Express mGluR3
To test our hypothesis that activation of Gi/Go-coupled mGluR3 recruits cytoplasmic TWIK-1 to astrocyte membrane, we first examined the expression profile of mGluRs from freshly dissociated hippocampal astrocytes by using qRT-PCR [15]. The mGluR5 in group I, and mGluR2 and mGluR3 in the group II were chosen as the candidate genes based on two recent reports [12, 13]. The results revealed that mGluR3 is the only highly expressed mGluR in astrocytes from mice older than P21 (Fig. 1), which confirms the results published by others noted above.

Metabotropic glutamate receptor 3 (mGluR3) mRNA is expressed highly in freshly dissociated hippocampal astrocytes. a Images of a freshly dissociated hippocampal astrocyte identified based on its well-maintained cell morphology and SR101-positive staining (middle). Scale bar, 10 μm. b qRT-PCR results from freshly dissociated astrocytes show a high-level expression of mGluR3, whereas the mGluR2 and mGluR5 mRNAs were barely detectable. The mRNAs were obtained and averaged from three independent cell harvests from three mice; each harvest contained 30 freshly dissociated astrocytes in qRT-PCR analyses. **P < 0.01
The mGluR3 Activation Increases Membrane TWIK-1 Expression in Hippocampal Astrocytes
We next asked whether activation of mGluR3 increases the membrane TWIK-1 expression in hippocampal astrocytes. Acutely prepared hippocampal slices were randomly divided into three groups: (1) control, (2) mGluR2/3 agonist treatment (LY354740, 2 μM/20 min), and (3) mGluR2/3 antagonist (LY341495, 10 μM, 10 min prior to and during agonist) + agonist (LY354740, 2 μM/20 min) treatment. The hippocampal slices were then subjected to GFAP/TWIK-1/GLT-1 triple immunostaining confocal imaging analysis. The individual astrocytes and the boundary of their soma were revealed by GFAP staining, and the levels of TWIK-1 membrane expression were analyzed based on the colocalization of TWIK-1 with membrane marker GLT-1 [13] (Fig. 2a). Although LY354740 is a common agonist for mGluR2 and mGluR3, the drug effect should be selective for mGluR3 as mGluR2 is absent in astrocytes (Fig. 1).

Activation of mGluR3 translocates intracellular TWIK-1 to astrocyte membrane. a Three panels representing three identified astrocytes in situ from three experimental groups as indicated. The cells in slices were subjected to GFAP (green)/TWIK-1 (red)/GLT-1 (magenta) triple immunostaining. LY354740: mGluR3 agonist (2 μM, 20 min). LY341495: mGluR3 antagonist (10 μM, 10 min). Scale bar, 10 μm. The somatic cytoplasm in all three cells is encircled by GFAP staining in the areas marked by squares. Topographic image illustrated the distribution and relative density of GFAP, TWIK-1, and GLT-1 positive signal before and after the agonist/antagonist application, obtained from soma area indicated in the column of merged image. A large amount of TWIK-1 punctate staining appeared in the cytoplasm in the control cell, but that was largely reduced after LY354740 treatment. The mGluR3 antagonist prevented agonist induced TWIK-1 translocation. b Fluorescence intensity profiles of TWIK-1 and GLT-1 in soma along the “a-b” lines indicated in the merged images in a, and only in the cell pretreated with mGlu3R agonist (middle panel), TWIK-1 fluorescence intensity shifted from cytoplasm to cell membrane. c The change in relative amount of membrane TWIK-1 is determined by the colocalization of TWIK-1 with astrocyte membrane marker GLT-1. Up panel, three subcellular regions along the primary processes (rectangles) were selected based on GFAP staining that extended from soma for TWIK-1/GLT-1 colocalization analysis. The somatic areas were also used (squares) for TWIK-1/GLT-1 colocalization analysis. Bottom panel, TWIK-1/GLT-1 colocalization in soma and process areas was analyzed by Pearson’s correlation coefficient (PCC). To achieve accurate PCC value for each astrocyte, three to four selected process areas, shown in the upper panel, were used and the obtained PCC values were averaged. Data are presented as mean ± SEM from indicated number of experiments. a.u. arbitrary unit, NS no significance; *P < 0.05; **P < 0.01 (color figure online)
The punctate TWIK-1 staining signals were concentrated inside the cytoplasm of somatic areas and distributed diffusely in processes in control astrocytes (Fig. 2, upper panel), whereas TWIK-1 staining shifted to somatic membrane in mGluR3 agonist treatment group (Fig. 2, middle panel), and this effect was largely inhibited by mGluR3 antagonist (Fig. 2a, bottom panel). The mGluR3-mediated change of TWIK-1 distribution pattern was further disclosed by 3D topographic images (Fig. 2a, right panel), and confocal line scanning (Fig. 2b); line scanning was conducted from the areas outlined by the white squares in Fig. 2a. Altogether, stimulation of mGluR3 translocates cytoplasmic TWIK-1 channels to membrane.
PCC analysis was performed to quantitatively analyze mGluR3-mediated change in membrane TWIK-1 expression in both soma and processes based on TWIK-1/GLT-1 colocalization (Fig. 2c, top). In each astrocyte, the TWIK-1 staining signals in somata and processes were quantified individually (see “Materials and Methods” for details). LY354740 treatment significantly increased the membrane TWIK-1 expression in soma; PCC 0.05 ± 0.03 (n = 24) in control versus 0.17 ± 0.01 (n = 42) in LY354740 (P = 0.002). LY354740 also significantly increased the TWIK-1 expression in the membrane of processes; PCC 0.08 ± 0.02 (n = 23) in control versus 0.14 ± 0.01 (n = 37) in LY354740 treatment (P = 0.013). The agonist effect was significantly inhibited by mGluR3 antagonist LY341495; PCC 0.09 ± 0.02 in soma (n = 16; P = 0.584 compared with control) and 0.07 ± 0.02 in processes (n = 16; P = 0.972, compared with control). These analyses further confirm that the mGluR3 activation increases membrane TWIK-1 expression in astrocytes.
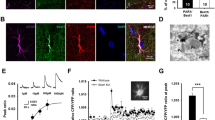
As TWIK-1 expression is highly astrocytic in mouse hippocampus [2, 3], we next examined the effect of mGluR3 activation on the redistribution of TWIK-1 channels between cytoplasm versus membrane using fractionation western blot analysis (Fig. 3). In the slices treated with LY354740 (2 μM, 20 min), the membrane TWIK-1 proteins increased significantly from the control level of 24.90 ± 1.89 to 33.78 ± 1.13 % (n = 4, P = 0.042) (Fig. 3b). Thus, at the tissue level, mGluR3 activation also induces translocation of TWIK-1 to astrocyte membrane.

Activation of mGluR3 translocates intracellular TWIK-1 to astrocyte membrane in hippocampal slice. a Western blots show the relative TWIK-1 distribution in cytoplasm (Cyto) versus transmembrane (Mem) from proteins isolated from control hippocampal slices and slices preincubated with mGluR3 agonist LY354740 (2 μM for 20 min). TWIK-1 proteins existed in both monomer and dimer forms in the plasma membrane and cytoplasmic fraction. GFAP (50 kDa) and ATP1α2 (112 kDa) are markers for cytoplasmic and membrane fractions, respectively. b Quantitative densitometric analysis of the relative Mem versus total TWIK-1 proteins, i.e., Cyto + Mem. mGluR3 agonist increases the membrane presence of TWIK-1 significantly compared to the control group. Data are presented as mean ± SEM from four independent western blot experiments. *P < 0.05
Elevated Membrane TWIK-1 Expression Is Associated with a Depolarization in Astrocyte V M
TWIK-1 is known to function as non-selective cation channel in astrocytes; thus, membrane TWIK-1 expression is expected to depolarize astrocyte V M [3]. It should be noted that, due to abundant and diverse expression of K+ conductances, inhibition of 45.5 % membrane K+ conductance mediated by Kir4.1 depolarized V M only by 2.6 mV [17]. Thus, a large amount of TWIK-1 membrane insertion is not expected to induce a large V M depolarization. To ensure that a small V M depolarization could be reliably detected in astrocytes, the stability of V M recording was analyzed over a 50-min recording time period (Fig. 4a); the V M fluctuated within a small 0.1 ± 0.02 mV with a small coefficient variance of 0.2 ± 0.02% (n = 10). In the presence of 0.5 μM TTX, astrocytic V M reached to a stable level at around 10 min and remained stable for at least 30 min.

Elevated membrane TWIK-1 expression is associated with astrocyte V M depolarization. a Whole-cell V M recording from WT and TWIK-1−/− astrocytes. The black trace shows a stable V M recording with negligible fluctuation over a 40-min duration. In the absence of 0.5 μM TTX, LY354740 induced an initial hyperpolarization and a following secondary depolarization in WT, but the second phase depolarization was significantly smaller in TWIK-1−/− astrocyte (upper panel). The initial hyperpolarization could be totally blocked by TTX treatment applied 5–10 min before a 20-min LY354740 (2 μM) incubation (lower panel). The amplitude of mGluR3-induced depolarization (ΔV) in the end of 20-min agonist treatment is summarized in b. Data are presented as mean ± SE from indicated number of recordings. *P < 0.05; **P < 0.01
Bath application of 2 μM LY354740 for 20 min induced an initial hyperpolarization and the following depolarization in wild type astrocytes. However, the second phase of depolarization was significantly less in TWIK-1−/− astrocytes (upper panel in Fig. 4a): the second phase depolarization (ΔV) was 1.20 ± 0.19 mV in WT (n = 9) and 0.55 ± 0.04 mV in TWIK-1−/− astrocytes (n = 4) (P = 0.009, Fig. 4b). The second mGluR3 agonist, 10 μM APDC, produced a similar effect in ΔV: 1.13 ± 0.23 mV in WT (n = 4) and 0.44 ± 0.15 mV in TWIK-1−/− (n = 6, P = 0.029, Fig. 4b). It has been shown that activation of presynaptic mGluR2/3 inhibits synaptic transmission [18], and we speculated that the first phase V M hyperpolarization could be a result of reduced extracellular K+ and glutamate concentrations following to the synaptic transmission inhibition. To test this possibility, basal neurotransmission was inhibited by 0.5 μM TTX 20 min prior to agonist application. This eliminated the initial V M hyperpolarization in both groups, whereas V M depolarization remained only in wild-type astrocytes (lower panel in Fig. 4a). In astrocytes treated with agonist 2 μM LY354740, the ΔV was 1.06 ± 0.15 mV (n = 17) in WT and 0.11 ± 0.25 mV (n = 8) in TWIK-1−/− astrocytes (P = 0.002; Fig. 4b). In astrocytes treated with agonist APDC, the ΔV was 1.41 ± 0.09 (n = 4) in WT and 0.69 ± 0.13 mV (n = 4) in TWIK-1−/− (P = 0.004, Fig. 4b). At the functional level, these results corroborated our observation that stimulation of mGluR3 readdresses the cytoplasmic TWIK-1 channels to astrocyte membrane.
Activation of mGluR3 Potentiates NH4 + Uptake in Astrocytes
The NH4 + can be taken up into astrocytes by inwardly rectifying K+ channel Kir4.1 [19], Na+-K+ ATPase (NKA) [20] and Na+-K+-2Cl− cotransporter (NKCC) [21]. Because membrane TWIK-1 expression appears to be dynamically regulated by mGluR3 and TWIK-1 is highly permeable to NH4 + [6], we next investigated whether membrane TWIK-1 represents a regulatory NH4 + uptake pathway that in turn would be expected to affect astrocyte functions, such as the glutamate-glutamine cycle [22]. The NH4 + concentration varies with 0.1∼1 mM in the normal brain [22–24]. However, it was reported that 1 mM NH4Cl induced V M depolarization was too small (∼2 mV) for reliable detection of Kir4.1-mediated NH4 + uptake [19]. Thus, we followed the same study by using 5 mM bath applied NH4 + in the following experiment.
Bath application of 5 mM NH4Cl for 10 min depolarized wild-type astrocytes with an initial peak amplitude of 8.28 ± 0.27 mV at 4 min and a following plateau (n = 10, Fig. 5a). Removal of bath NH4Cl hyperpolarized V M to below the control level by 2.06 ± 0.21 mV, reflecting an NH4 +-mediated overactivation of NKA [20] (more details in the following section and Fig. 7). When mGluR3 agonist LY354740 (2 μM) was applied for 10 min prior to and during 5 mM NH4Cl, the NH4 +-induced peak V M depolarization was potentiated by 26 % in 14 out of 16 cells compared to control (11.15 ± 0.28 mV, n = 14 cells). The second mGluR2/3 agonist APDC (10 μM) produced a similar potentiation (11.16 ± 0.42 mV) in 17 out of 19 astrocytes (Fig. 5c). The potentiation was inhibited by mGluR2/3 antagonist LY341495 (10 μM) (Fig. 5b, c). Thus activation of mGluR3 increases NH4 + uptake in astrocytes.

Activation of mGluR3 receptor potentiates NH4 +-induced depolarization in astrocytes. a Bath application of 5 mM NH4Cl for 10 min depolarized astrocyte V M by ∼8 mV (peak amplitude), and a 2–3 mV V M undershot, hyperpolarization to below the control, was followed upon NH4Cl withdrawal. b. Representative V M traces show the potentiation of NH4Cl-induced V M depolarization by mGluR3 agonist LY354740 (2 μM), and suppression of potentiation by addition of mGluR3 antagonist LY341495 (10 μM) coapplied with LY354740. The mGluR3 agonist was added to perfusate 10 min prior to and during NH4Cl application. The antagonist pretreatment time was 10 min. The second mGluR3 agonist, APDC (10 μM), showed a similar effect. The mGluR3 agonists and antagonist effects on NH4 +-induced V M depolarization are summarized in c. Data are presented as mean ± SEM from the indicated number of experiments. **P < 0.01; NS no significance
TWIK-1 Is Responsible for mGluR3-Induced V M Potentiation to NH4 + in Astrocytes
To determine if TWIK-1 is responsible for mGluR3-induced potentiation, the TWIK-1−/− mice was introduced into the following experiments. First, TWIK-1−/− astrocytes responded to 5 mM NH4Cl with a V M depolarization comparable to WT astrocytes (peak amplitude 8.63 ± 0.41 mV, n = 7, P = 0.998; Fig. 6a, b). When 100 μM BaCl2 was added in perfusate 5 min before and during NH4Cl perfusion, NH4 +-induced depolarization was significantly reduced in both WT and TWIK-1−/− astrocytes (6.60 ± 0.48 mV in WT, n = 12, P = 0.008 and 6.32 ± 0.26 mV in TWIK-1−/−, n = 5, P = 0.002; Fig. 6a, b), which was consistent with the notion that Kir4.1 is an important NH4 + uptake channel [19]. The result also indicated an unchanged expression of Kir4.1 in TWIK-1−/− astrocytes. Neither LY354740 nor APDC induced a potentiation in TWIK-1−/− astrocytes (Fig. 6d). Moreover, a TWIK-1 gene-dependent decrease in potentiation was evident in agonist LY354740 experiment: 11.15 ± 0.28 mV (n = 14) in WT, 9.56 ± 0.37 mV (n = 6) in TWIK-1+/−, and 8.4 ± 0.64 mV (n = 4) in TWIK-1−/− astrocytes (Fig. 6c, d). Thus, the TWIK-1 is the responsible channel for the mGluR3-mediated potentiation in NH4 + uptake.

mGluR3-mediated potentiation of NH4 + uptake depends on astrocyte TWIK-1 channels. a NH4 +-induced V M depolarization was comparable in amplitudes between WT and TWIK-1 KO astrocytes with or without BaCl2 application. Five millimolars of NH4Cl was applied for 10 min and 100 μM BaCl2 was added in bath 5 min before and during NH4Cl treatment. b The peak amplitudes of NH4 +-induced V M depolarization were summarized between the two genotypes. Ba2+ significantly reduced V M depolarization by 20 % for WT and 27 % for TWIK-1 KO astrocytes. c mGluR3-mediated potentiation of NH4 +-induced V M response reduced in a TWIK-1 gene dose-dependent manner. mGluR3 agonist LY354740 (2 μM) was used in these recordings, and a similar effect could be replicated by another mGluR3 agonist APDC (10 μM) shown in d; in both cases, agonists were added to bath 10 min prior to and during NH4 + application. Note that mGluR3-mediated potentiation was nearly absent in TWIK-1 KO astrocytes. e Representative western blot shows an age-dependent upregulation in TWIK-1 protein expression. The total hippocampal proteins were isolated from animals of different postnatal ages as indicated. GAPDH (36 kDa) and GFAP (50 kDa) were used as loading control. TWIK-1 protein is almost absent in P1 and expressed at very low level at P7 compared to those after P23. Kir4.1 is also expressed in an age-dependent manner, and the expression levels become evident from P13 on. f mGluR3-mediated potentiation of NH4 + uptake was absent in immature astrocytes (P7), which is summarized in g. h qRT-PCR results from freshly dissociated astrocytes show a comparable expression level of mGluR3 mRNA in P7 immature astrocytes to that of P21. The mRNAs were obtained and averaged from three independent cell harvests from three mice; each harvest contained 30 astrocytes. Data are presented as mean ± SEM from the indicated number of experiments. *P < 0.05; **P < 0.01; NS no significance
TWIK-1 expression reaches to an adult level at around P21 [1, 3]. Here, a low-level expression of TWIK-1 proteins is again evident in early postnatal developmental stages (Fig. 6e). We next explored a potential absence of potentiation of NH4 + uptake from P7 astrocytes. First, NH4 +-induced depolarization amounted only to 66 % of the adult level, 5.51 ± 0.33 mV (n = 5, P = 0.001) (Fig. 6f, g). A small depolarization can be explained in part by a low-level expression of Kir4.1 at this animal age (Fig. 6e) [19, 25–27]. Second, the low-level expression of TWIK-1 corresponded with an undetectable mGluR3-mediated potentiation: peak depolarization 5.71 ± 0.21 mV (n = 5; P = 0.874 compared to P7 control group). Meanwhile, the expression levels of mGluR3 were comparable between P7 and P21 astrocytes (Fig. 6h), excluding the possibility that the absence of potentiation was caused by the low expression levels of mGluR3 in immature astrocytes. Taken together, TWIK-1 functions to dynamically regulate NH4 + uptake under the control of mGluR3 in astrocytes.
Potentiation of mGluR3 Mediated NH4 + Uptake in the Absence of Na+-K+ ATPase Activity
Elevated NH4 + is known to increase NKA activity by competing with the extracellular K+ binding sites, resulting in electrogenic hyperpolarization of astrocytes [20, 24, 28]. This should partially mask NH4 +-induced basal V M depolarization primarily mediated by Kir4.1 and potentiation by TWIK-1 channels. To confirm this, the NKA activity was minimized by substitution of NaCl by LiCl in electrode and bath solutions [29, 30]. To prevent excessive accumulation of extracellular K+ resulting from NKA inhibition, 0.5 μM TTX was added for neurotransmission inhibition [31]. Inhibition of NKA eliminated an expected approximately 10 mV electrogenic pump potential in 10 min [32] (Fig. 7a, left). In the absence of NKA, 5 mM NH4 + induced a delayed time-to-peak (4.44 ± 0.34 min with pump vs 7.88 ± 0.59 min without pump, P = 0.001) and 22.0 % greater V M depolarization (8.28 ± 0.27 mV with pump vs 10.59 ± 0.36 mV without pump, P = 0.001) (Fig. 7a, right, b). Therefore, the electrogenic NKA NH4 + uptake does partially mask the basal NH4 + uptake.

Enhanced potentiation of mGluR3-mediated NH4 + uptake in the absence of Na+-K+ ATPase activity. a To eliminate Na+-K+ ATPase (NKA) activity, NaCl in the electrode solution (14 mM) and aCSF (125 mM) was substituted by equimolar LiCl, and the perfusate was switched from normal to high LiCl-containing aCSF (Li-aCSF) after whole-cell recording was established. These conditions depolarized V M by ∼10 mV resulting from inhibition of electrogenic NKA activity. NH4Cl (5 mM, 10 min) was applied 9∼10 min after NKA inhibition. Right panel, the NH4Cl response outlined in the dashed rectangle in left panel is displayed in an enlarged scale (green) and superimposed with a recording with intact NKA activity (black). The differences in NH4Cl response under these two conditions are summarized in b; elimination of NKA activity resulted in a (1) progressive increase in NH4 +-induced V M depolarization (left, quantified as “time to peak”), (2) an average of 22.0 % increase in peak amplitude (middle), and (3) a total abolishment of V M undershot upon withdrawal of NH4Cl (right). c Representative recording showing that mGluR3 activation induced potentiation was more pronounced in the absence of NKA activity. d mGluR3-induced potentiation of NH4 + response remained when NKA is inactive in WT but not in TWIK-1 KO astrocytes. Data are presented as mean ± SEM from the indicated number of experiments. **P < 0.01; NS no significance
It should be noted that a short-term NKA inhibition can overly stimulate NKA, resulting in a V M undershot to levels below the initial control upon restoration of NKA [33]. The inhibition of NKA, in our designed procedure, was indicated by the absence of V M undershot after NH4Cl withdrawal from bath (Fig. 7a, right, b, 2.06 ± 0.21 mV with pump vs −1.29 ± 0.66 mV without pump). A significantly larger V M undershot was observed after switch the bath from Li-aCSF to normal aCSF (Fig. 7a, b, 8.76 ± 1.41 mV, n = 5) compared to control (Fig. 5a, 2.06 ± 0.21 mV; P = 0.008). These results show that the NKA activity can be effectively inhibited in our experimental procedure.
We next sought to determine whether mGluR3-mediated potentiation remained in the absence of NKA activity. A net V M potentiation was induced by agonist LY354740 (∆3.26 mV, n = 6) and by agonist APDC (∆3.80 mV, n = 6). By both agonists, induced potentiation was greater than that of the control (∆2.88 mV, n = 14, Fig. 6d). Again, the mGluR3-induced potentiation was absent in TWIK-1−/− astrocytes (Fig. 7c, d), further supporting the notion that TWIK-1 serves as a regulatory channel for NH4 + uptake under the control of mGluR3.
Rab-Associated Endosome Recycling Pathway Is Required for Membrane TWIK-1 Expression
The newly inserted TWIK-1 channels stay on membrane only transiently; then, the channels are retrieved back to the recycling endosomes [11]. Thus, it is possible that a short-term inhibition of constitutive vesicle exocytosis may prevent mGluR3-mediated membrane accumulation of TWIK-1 channels and abolish the potentiation of NH4 + uptake, while leaving the function of the conventional K+ channels, such as Kir4.1, relatively intact in astrocytes.
To test this hypothesis, the membrane channel insertion through soluble N-ethylmaleimide (NEM)-sensitive factor attachment receptor (SNARE)-mediated fusion process was inhibited by 250 μM NEM added in the pipette solution [34, 35], NEM is an inhibitor for NEM-sensitive factor. In the control experiments, a 10-min NEM dialysis hyperpolarized the V M significantly compared with the control astrocytes (−76.63 ± 0.63 mV, n = 7, control is shown in Fig. 8c, P = 0.027). Moreover, the V M in NEM treatment group was comparable to TWIK-1−/− astrocytes (−76.45 ± 0.23 mV, n = 35; Fig. 8c, P = 0.535), indicating strongly that SNARE-mediated fusion process is constitutively active for membrane TWIK-1 insertion at resting state. A 10-min NEM dialysis prior to mGluR3 agonist treatment also completely eliminated mGluR3-mediated potentiation of NH4 + uptake (7.58 ± 0.47 mV, n = 7, P = 0.188 compared with control, Fig. 8a, d), indicating that the same SNARE-mediated fusion process is required for mGluR3-mediated potentiation of NH4 + uptake.

Exocytotic pathways underlying mGluR3-induced TWIK-1 membrane surface expression. a Inhibition of SNARE complex by N-ethylmaleimide (NEM, 250 μM) eliminated mGluR3-mediated potentiation of NH4 + response. NEM was applied intracellularly for 10 min prior to mGluR3 activation. The schematic illustrations of trafficking pathway and steps are shown in the left panel and recording traces are shown in the right panel. b Inhibition of RabGGTase by 5 μM psoromic acid (PA) abolished mGluR3-mediated potentiation of NH4 + uptake. RabGGTase is the enzyme that prenylates newly synthesized Rabs and delivers them to the membranes of donor compartments. Brian slices were incubated in 5 μM PA for 4 h before patch-clamp recording. c Comparison of the resting membrane potential of astrocyte under various conditions. Note that electrode dialysis of NEM and pretreatment of astrocytes with PA all hyperpolarized the V M to levels comparable to that of the TWIK-1−/− astrocytes. d Summary of the effect of NEM and PA on mGluR3-mediated potentiation of NH4 +. Data are presented as mean ± SEM from the indicated number of experiments. e Immunostaining showed that TWIK-1 and Rab11 were distributed coordinately in astrocyte soma in hippocampal slices. Both TWIK-1 and Rab11 signals were distributed evenly in astrocyte soma in resting state (control) and shifted to a polarized distribution toward to membrane after mGluR3 agonist treatment (LY354740). Scale bar, 5 μm. *P < 0.05; **P < 0.01; NS no significance. PM plasma membrane, NSF N-ethylmaleimide sensitive factor, EE early endosome, RE recycling endosome, LE late endosome, REP Rab escort protein, RabGGTase Rab geranylgeranyl transferase
We next explored whether Rab-mediated vesicle trafficking is required in mGluR3-induced TWIK-1 membrane expression and potentiation of NH4 + uptake. The proteins in Rab-GTPase family are critical regulators for vesicle trafficking, including endosomal trafficking, and prenylation of the Rab proteins is required for the newly synthesized Rabs to be delivered to the membranes of donor compartments in vesicle trafficking [36]. Thus, we incubated the brain slices with a selective RabGGTase inhibitor, psoromic acid (PA, 5 μM, 4 h), to prevent covalent Rab-prenylation [37]. This treatment resulted in a significant V M hyperpolarization, −76.29 ± 0.27 mV (n = 14, Fig. 8c), a V M level that was comparable to TWIK-1−/−astrocytes (Fig. 8c). The PA treatment also completely abolished mGluR3-mediated potentiation of NH4 + uptake (8.11 ± 0.41 mV, n = 4, P = 0.734, Fig. 8b, d). Thus, the Rab-mediated pathway appeared to be primarily responsible for mGluR3-induced membrane TWIK-1 expression.
Rab11 is a critical recycling endosome Rab protein required for transferring recycling endosome-associated proteins to cell membrane [36]. We then investigated whether Rab11-mediated recycling endosomal pathway was involved in TWIK-1 trafficking to membrane. Since no specific Rab11 inhibitor is yet available, colocalization of TWIK-1/Rab11 in astrocyte somatic area was analyzed by confocal immunocytochemistry in hippocampal slices (Fig. 8e). The results showed that TWIK-1 and Rab11 displayed a similar distribution pattern in the soma in control (n = 4), whereas the even TWIK-1/Rab11 distribution was altered by mGluR3 agonist that shifted both staining signals together toward the membrane, suggesting that TWIK-1 channels are associated with recycling endosomes and mGluR3 activation coordinately translocate TWIK-1/recycling endosomes to the membrane surface.
Discussion
Despite that TWIK-1 channels and mGluR3 are highly expressed in astrocytes, little is known about their function. In the present study, we show that membrane recruitment of TWIK-1 channels through mGluR3 activation underlies a regulatory mechanism for ammonium homeostasis in the CNS. Specifically, mGluR3 activation translocates recycling endosome-associated TWIK-1 channels to membrane that in turn increases NH4 + uptake in astrocytes.
The Characteristics of Astrocyte TWIK-1 Channels and mGluR3
Consistent with a general characteristic of TWIK-1 in heterologous expression systems and native cells [4, 8, 9, 38], TWIK-1 channels are predominantly retained in astrocyte intracellular compartments, and they behave as nonselective monovalent cation channels when expressed on the membrane [3]. These features indicate that TWIK-1 does not function as a classic K+ channel contributing to the highly negative and stable astrocyte V M. Consequently, the role of membrane TWIK-1 and the mechanisms that regulate the dynamic trafficking of TWIK-1 need to be determined.
The hydrophobic nature of the TWIK-1 pore has been revealed recently, which restricts ion permeation through these channels and contributes the low functional activity of the channels on the plasma membrane [39]. This unique feature however favors a high permeability of TWIK-1 for NH4 + in its hydrophobic form of NH3, and this view has been supported by a recent observation, where the inward NH4 + currents through TWIK-1 channels were 40-fold larger than that of the K+ currents in heterologous expression system [6].
In heterologous expression systems, activation of Gi/Go-coupled 5-HT1R and α2-adrenergic receptors increases membrane TWIK-1 expression [9]. In the present study, we show that mGluR3 is the only metabotropic glutamate receptor expressed in hippocampal astrocytes (Fig. 1). Importantly, hippocampal astrocytes do not express ionotropic glutamate receptors [14, 40], which leaves mGluR3 as the only astrocytic receptor to sense the dynamic change of glutamatergic transmission.
All the characteristics described in previous studies indicate that TWIK-1 would more likely function as an activity-dependent channel on the membrane, and its membrane expression could be regulated on demand through physiological stimuli. This has prompted us to test a hypothesis that glutamatergic transmission regulates astrocyte TWIK-1 membrane expression through mGluR3.
Activation of mGluR3 Promotes Recruitment of TWIK-1 in Astrocyte Membrane
The following evidences support the view that stimulation of astrocyte mGluR3 is a physiological signal that regulates the membrane TWIK-1 expression. First, treatment of hippocampal slices with mGluR3 agonist showed a significant translocation of intracellular TWIK-1 channels to astrocyte membrane (Figs. 2 and 3). Second, mGluR3 activation depolarized V M selectively in WT, but not TWIK-1−/− astrocytes (Fig. 4). It should be noted that significant amounts of TWIK-1 proteins still retained inside astrocytes after mGluR3 activation as observed in TWIK-1 immunostaining study (Fig. 2a), suggesting TWIK-1 may have other roles inside cytoplasm.
Association of TWIK-1 with Recycling Endosomes in Astrocytes
The vesicular membrane fusion is mediated by soluble NEM-sensitive factor attachment receptor (SNARE) proteins. As mGluR3 is not linked to intracellular Ca2+ stores, and a short-term 10-min NEM dialysis was sufficient to completely inhibit mGluR3-medited potentiation of NH4 + uptake, thus, constitutive exocytosis is a required final step for TWIK-1 membrane recruitment (Fig. 8a). Additionally, mGluR3 agonist effect occurred rapidly within 5–8 min (Figs. 4 and 6); thus, de novo synthesis of TWIK-1 channels unlikely plays a major role in mGluR3 action. Selective inhibition of covalent Rab-prenylation by PA also completely eliminated mGluR3-mediated potentiation of on NH4 + uptake (Fig. 8d); thus, the Rab-associated membrane trafficking is also required for mGluR3 action [37].
Interestingly, inhibition of exocytosis by NEM or covalent Rab-prenylation by PA all hyperpolarized V M to the levels comparable to TWIK-1−/− astrocytes (Fig. 8c); these together indicate strongly that constitutive TWIK-1 trafficking should be operating constantly at the resting condition, thus variation of ambient glutamate concentrations should be able to dynamically regulate TWIK-1 membrane insertion and internalization.
A diisoleucine repeat located in the cytoplasmic carboxyl-terminus of TWIK-1 has been identified as a critical retrieval motif in both cultured kidney cells and transfected oocytes [4, 9, 11]; currently, our results could not fully exclude the possibility that mGluR3 activation may also inhibit TWIK-1 retrieval through endocytosis as a second mechanism for the observed TWIK-1 accumulation in astrocyte membrane.
In kidney culture cells, recycling endosomes has been identified as the main TWIK-1 associated cytoplasm compartment [9]. Rab proteins, especially Rab11, are known to play critical roles in delivering proteins from recycling endosomes to the cell surface [36]. In this study, TWIK-1 and Rab11 appeared to be distributed evenly in astrocytes, whereas mGluR3 agonist drove TWIK-1 and Rab11 toward the membrane coordinately, suggesting TWIK-1 channels are likely mainly associated with this cytoplasm compartment in astrocytes.
Potentiation of TWIK-1-Medited NH4 + Uptake by mGluR3 Activation
The ammonia levels can be elevated up to 1∼5 mM in the brain during intense exercise or pathological hepatic encephalopathy [22, 41]. In the latter event, astrocytes are known to be the major cellular target of NH4 +. The NH4 + exhibits a similar hydrated radius and charge as that of the K+ so that can readily flow into astrocytes through K+ channels [19, 23]. Increases in the cellular influx of NH4 + accompanying changes in ion concentrations may contribute to disruptions in metabolism and neurotransmission [20]. For example, pathological accumulation of NH4 + is often followed by an elevated extracellular glutamate [42], which should in turn activate mGluR3 in astrocytes.
To explore a potential functional coupling between mGluR3 activation and TWIK-1 membrane expression, we used 5 mM NH4 + as a considerable V M depolarization could be induced (∼8 mV) in hippocampal astrocytes as reported previously [19]. In the present study, a similar V M depolarization was observed (Fig. 5a). Two mechanisms have been adduced to explain NH4 +-induced V M depolarization; a transient elevation of [K+]o and the entry of NH4 + through K+ channels. In hippocampal slices, a transient increase in [K+]o by ∼1 mM has been observed in the presence of 5 mM NH4 + in bath, and competitive binding of NH4 + and K+ at NKA K+ binding site has been shown as the underlying mechanism [19]. However, [K+]o increase by 1 mM only made a contribution of 30 % to NH4 +-induced depolarization [19]. Therefore, the channel-mediated NH4 +influx, primarily Kir4.1 in astrocytes, has been demonstrated as the major contributor to V M depolarization [19].
We show that mGluR3 activation enhances NH4 + uptake through increasing expression of membrane TWIK-1 channels in hippocampal astrocytes. This has been supported by the following evidences. First, mGluR3 agonist potentiated NH4 + uptake by 30 % in V M measurement. Second, the mGluR3-mediated potentiation was absent in TWIK-1−/− astrocytes. Third, the mGluR3-mediated potentiation was absent in immature astrocytes (P7) lacking of TWIK-1 expression. Fourth, mGluR3-mediated potentiation was further enhanced in the absence of NKA electrogenic NH4 + uptake potential (Fig. 7).
Functional Implication of Membrane Recruitment of TWIK-1 Channels by mGluR3 Activation
Dynamic change in glutamatergic transmission occurs under both physiological (e.g., arousal) and various pathological conditions in the CNS. In the hippocampus, mGluR3 appears to be the only glutamate receptor that enables astrocytes to listen to glutamatergic transmission [12]. We show that glutamatergic signaling and astrocyte TWIK-1 membrane expression are functionally coordinated, and one of the functional consequences identified in this study is the regulation of NH4 + uptake. Based on our findings, mGluR3 activation induces TWIK-1 membrane recruitment, leading to an increase of NH4 + uptake in astrocyte. Under physiological condition, NH4 + is an essential substrate in the glutamate-glutamine cycle [22]; thus, neuronal glutamate release may dynamically regulate TWIK-1 membrane expression and NH4 + uptake in astrocytes that in turn provide glutamine for neurotransmitter renewal on demand.
Under hyperammonemic conditions, elevated extracellular glutamate may overactive astrocytic mGluR3 that in turn increase TWIK-1 membrane insertion and NH4 + uptake. This may serve as a negative feedback mechanism to restore ammonium equilibrium. In the future, it would be important to examine the contribution of cell surface TWIK-1 readdressing in pathological regulation of ammonium homeostasis in pathological states.
References
Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA (2008) A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28:264–278
Zhou M, Xu G, Xie M, Zhang X, Schools GP, Ma L, Kimelberg HK, Chen H (2009) TWIK-1 and TREK-1 are potassium channels contributing significantly to astrocyte passive conductance in rat hippocampal slices. J Neurosci 29:8551–8564
Wang W, Putra A, Schools GP, Ma B, Chen H, Kaczmarek LK, Barhanin J, Lesage F, Zhou M (2013) The contribution of TWIK-1 channels to astrocyte K(+) current is limited by retention in intracellular compartments. Front Cell Neurosci 7:246
Chatelain FC, Bichet D, Douguet D, Feliciangeli S, Bendahhou S, Reichold M, Warth R, Barhanin J, Lesage F (2012) TWIK1, a unique background channel with variable ion selectivity. Proc Natl Acad Sci U S A 109:5499–5504
Millar ID, Taylor HC, Cooper GJ, Kibble JD, Barhanin J, Robson L (2006) Adaptive downregulation of a quinidine-sensitive cation conductance in renal principal cells of TWIK-1 knockout mice. Pflugers Arch 453:107–116
Ma L, Xie YP, Zhou M, Chen H (2012) Silent TWIK-1 potassium channels conduct monovalent cation currents. Biophys J 102:L34–L36
Decressac S, Franco M, Bendahhou S, Warth R, Knauer S, Barhanin J, Lazdunski M, Lesage F (2004) ARF6-dependent interaction of the TWIK1 K+ channel with EFA6, a GDP/GTP exchange factor for ARF6. EMBO Rep 5:1171–1175
Nie X, Arrighi I, Kaissling B, Pfaff I, Mann J, Barhanin J, Vallon V (2005) Expression and insights on function of potassium channel TWIK-1 in mouse kidney. Pflugers Arch 451:479–488
Feliciangeli S, Tardy MP, Sandoz G, Chatelain FC, Warth R, Barhanin J, Bendahhou S, Lesage F (2010) Potassium channel silencing by constitutive endocytosis and intracellular sequestration. J Biol Chem 285:4798–4805
Ma L, Zhang X, Chen H (2011) TWIK-1 two-pore domain potassium channels change ion selectivity and conduct inward leak sodium currents in hypokalemia. Sci Signal 4:ra37
Bichet D, Blin S, Feliciangeli S, Chatelain FC, Bobak N, Lesage F (2014) Silent but not dumb: how cellular trafficking and pore gating modulate expression of TWIK1 and THIK2. Pflugers Arch 467(5):1121–1131
Sun W, McConnell E, Pare JF, Xu Q, Chen M, Peng W, Lovatt D, Han X, Smith Y, Nedergaard M (2013) Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science 339:197–200
Morel L, Higashimori H, Tolman M, Yang Y (2014) VGluT1+ neuronal glutamatergic signaling regulates postnatal developmental maturation of cortical protoplasmic astroglia. J Neurosci 34:10950–10962
Zhou M, Kimelberg HK (2001) Freshly isolated hippocampal CA1 astrocytes comprise two populations differing in glutamate transporter and AMPA receptor expression. J Neurosci 21:7901–7908
Du Y, Ma B, Kiyoshi CM, Alford CC, Wang W, Zhou M (2015) Freshly dissociated mature hippocampal astrocytes exhibit similar passive membrane conductance and low membrane resistance as syncytial coupled astrocytes. J Neurophysiol 00206:02015
Nimmerjahn A, Kirchhoff F, Kerr JN, Helmchen F (2004) Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat Methods 1:31–37
Ma B, Xu G, Wang W, Enyeart JJ, Zhou M (2014) Dual patch voltage clamp study of low membrane resistance astrocytes in situ. Mol Brain 7:18
Price CJ, Karayannis T, Pal BZ, Capogna M (2005) Group II and III mGluRs-mediated presynaptic inhibition of EPSCs recorded from hippocampal interneurons of CA1 stratum lacunosum moleculare. Neuropharmacology 49(Suppl 1):45–56
Stephan J, Haack N, Kafitz KW, Durry S, Koch D, Hochstrate P, Seifert G, Steinhauser C, Rose CR (2012) Kir4.1 channels mediate a depolarization of hippocampal astrocytes under hyperammonemic conditions in situ. Glia 60:965–978
Kelly T, Rose CR (2010) Ammonium influx pathways into astrocytes and neurones of hippocampal slices. J Neurochem 115:1123–1136
Kelly T, Kafitz KW, Roderigo C, Rose CR (2009) Ammonium-evoked alterations in intracellular sodium and pH reduce glial glutamate transport activity. Glia 57:921–934
Felipo V, Butterworth RF (2002) Neurobiology of ammonia. Prog Neurobiol 67:259–279
Marcaggi P, Coles JA (2001) Ammonium in nervous tissue: transport across cell membranes, fluxes from neurons to glial cells, and role in signalling. Prog Neurobiol 64:157–183
Rangroo Thrane V, Thrane AS, Wang F, Cotrina ML, Smith NA, Chen M, Xu Q, Kang N, Fujita T, Nagelhus EA, Nedergaard M (2013) Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering. Nat Med 19:1643–1648
Seifert G, Huttmann K, Binder DK, Hartmann C, Wyczynski A, Neusch C, Steinhauser C (2009) Analysis of astroglial K+ channel expression in the developing hippocampus reveals a predominant role of the Kir4.1 subunit. J Neurosci 29:7474–7488
Nwaobi SE, Lin E, Peramsetty SR, Olsen ML (2014) DNA methylation functions as a critical regulator of Kir4.1 expression during CNS development. Glia 62:411–427
Olsen ML, Sontheimer H (2008) Functional implications for Kir4.1 channels in glial biology: from K+ buffering to cell differentiation. J Neurochem 107:589–601
Xue Z, Li B, Gu L, Hu X, Li M, Butterworth RF, Peng L (2010) Increased Na, K-ATPase alpha2 isoform gene expression by ammonia in astrocytes and in brain in vivo. Neurochem Int 57:395–403
Cho YW (1995) Lithium-induced inhibition of Na-K ATPase and Ca ATPase activities in rat brain synaptosome. J Korean Med Sci 10:7–13
Skou JC (1957) The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim Biophys Acta 23:394–401
Stelmashook EV, Weih M, Zorov D, Victorov I, Dirnagl U, Isaev N (1999) Short-term block of Na+/K+-ATPase in neuro-glial cell cultures of cerebellum induces glutamate dependent damage of granule cells. FEBS Lett 456:41–44
Glitsch HG (2001) Electrophysiology of the sodium-potassium-ATPase in cardiac cells. Physiol Rev 81:1791–1826
Xie M, Wang W, Kimelberg HK, Zhou M (2008) Oxygen and glucose deprivation-induced changes in astrocyte membrane potential and their underlying mechanisms in acute rat hippocampal slices. J Cereb Blood Flow Metab 28:456–467
Boycott HE, Barbier CS, Eichel CA, Costa KD, Martins RP, Louault F, Dilanian G, Coulombe A, Hatem SN, Balse E (2013) Shear stress triggers insertion of voltage-gated potassium channels from intracellular compartments in atrial myocytes. Proc Natl Acad Sci U S A 110:E3955–E3964
Butterworth MB, Frizzell RA, Johnson JP, Peters KW, Edinger RS (2005) PKA-dependent ENaC trafficking requires the SNARE-binding protein complexin. Am J Physiol Renal Physiol 289:F969–F977
Hutagalung AH, Novick PJ (2011) Role of Rab GTPases in membrane traffic and cell physiology. Physiol Rev 91:119–149
Deraeve C, Guo Z, Bon RS, Blankenfeldt W, DiLucrezia R, Wolf A, Menninger S, Stigter EA, Wetzel S, Choidas A, Alexandrov K, Waldmann H, Goody RS, Wu YW (2012) Psoromic acid is a selective and covalent Rab-prenylation inhibitor targeting autoinhibited RabGGTase. J Am Chem Soc 134:7384–7391
Lesage F, Guillemare E, Fink M, Duprat F, Lazdunski M, Romey G, Barhanin J (1996) TWIK-1, a ubiquitous human weakly inward rectifying K+ channel with a novel structure. EMBO J 15:1004–1011
Aryal P, Abd-Wahab F, Bucci G, Sansom MS, Tucker SJ (2014) A hydrophobic barrier deep within the inner pore of the TWIK-1 K2P potassium channel. Nat Commun 5:4377
Bergles DE, Jahr CE (1997) Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron 19:1297–1308
Nybo L, Dalsgaard MK, Steensberg A, Moller K, Secher NH (2005) Cerebral ammonia uptake and accumulation during prolonged exercise in humans. J Physiol 563:285–290
Hermenegildo C, Monfort P, Felipo V (2000) Activation of N-methyl-D-aspartate receptors in rat brain in vivo following acute ammonia intoxication: characterization by in vivo brain microdialysis. Hepatology 31:709–715
Acknowledgments
This work is sponsored by grants from the National Institute of Neurological Disorders and Stroke RO1NS062784 (MZ), start-up fund from the Ohio State University College of Medicine (MZ), the National Natural Science Foundation of China (No. 81400973) (WW), and the Fundamental Research Funds for the Central Universities of China, HUST (2015QN146) (WW).
Authors’ contributions
WW and MZ conceived the project. WW, CMK, YD, BM, CCA, and HC conducted the research or assisted the research, discussed the project, and assisted the manuscript preparation. WW and MZ wrote the manuscript. MZ supervised the project.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Wang, W., Kiyoshi, C.M., Du, Y. et al. mGluR3 Activation Recruits Cytoplasmic TWIK-1 Channels to Membrane that Enhances Ammonium Uptake in Hippocampal Astrocytes. Mol Neurobiol 53, 6169–6182 (2016). https://doi.org/10.1007/s12035-015-9496-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9496-4