
Abstract
Among K2P channels, a few of them turned out to be difficult to express in heterologous systems and were coined “silent subunits”. Recent studies have shed light on the mechanisms behind this apparent lack of channel activity at the plasma membrane. For TWIK1 and THIK2 channels, silence is related to a combination of intracellular retention and low intrinsic activity. TWIK1 is constitutively endocytosed from the plasma membrane before being transported to recycling endosomes, whereas THIK2 is restricted to endoplasmic reticulum. These intracellular localizations are related to trafficking signals located in the cytoplasmic parts of the channels. When these motifs are mutated or masked, channels are redistributed at the plasma membrane and produce measurable currents. However, these currents are of modest amplitude. This weak basal activity is due to a hydrophobic barrier in the deep pore that limits water and ions in the conduction pathway. Other silent channels KCNK7, TWIK2, and TASK5 are still under study. Expression and characterization of these K2P channels pave the way for a better understanding of the mechanisms controlling intracellular trafficking of membrane proteins, ion conduction, and channel gating.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
K2P channels produce background K+ conductances that set the negative membrane resting potential of many cell types [16, 21, 26, 29]. Tandem of pore domains in a weak inward rectifying K+ channel 1 (TWIK1, KCNK1, K2P1.1) [27, 28] is the prototype of a family comprising 15 members in mammals. These K2P channels are classified into six subfamilies according to their sequence conservation and functional properties: the weak rectifying group (TWIK1, TWIK2, and KCNK7), the mechano-gated group (TREK1, TREK2, and TRAAK), the acid-sensing group (TASK1, TASK3, and TASK5), the alkaline-sensitive group (TALK1, TALK2, and TASK2), the halothane-sensitive group (THIK1 and THIK2), and the spinal cord-expressed TRESK channel. Each K2P subunit contains four membrane-spanning helices (M1 to M4) and two pore-forming domains (P1 and P2). Each pore domain comprises a pore-helix (PH) and a pore loop containing the K+-selectivity signature sequence (GY/FG in P1 and GF/LG in P2) that is present in all K+ channels cloned from bacteria to mammals. K2P channels assemble as dimers with 4 P-loops forming the selectivity filter and inner helices M2 and M4 delineating the central pore cavity. Another feature is the M1P1 extracellular loop that is not present in the other families of K+ channels [30]. In many K2P subunits, this self-interacting domain contains a cysteine residue involved in the formation of a covalent disulfide bridge between two subunits. Proposed in 1996 [30], this overall arrangement was nicely validated in 2012 when 3D crystal structures of TWIK1 and TRAAK were solved [7, 37].
K2P channels have been cloned by sequence homology. They are present in many different tissues. The first step toward deciphering their functional roles was to characterize their electrophysiological properties in heterologous expression systems. Unfortunately, TWIK1 only gave modest currents in oocytes, and no current in transfected mammalian cell lines [27]. Then, most of the initial findings on electrophysiological and pharmacological properties as well as regulations of K2P channels came from the expression of K2P channels that generate much higher levels of current, especially TREK1 and TASK1 [15, 19]. These seminal studies revealed that K2P channels display only very limited voltage and time-dependence, designing them as leak or background K+ channels. These electrophysiological studies also revealed that K2P channels were finely tuned by a remarkable diversity of stimuli including chemical and physical parameters (stretch, temperature, pH, stimulation of neurotransmitter and hormone receptors, bioactive lipids) (for recent reviews, see [16, 26]). These channels proved also to be targets of drugs of therapeutic interest including volatile anesthetics, local anesthetics, and antidepressants. The next logical step was to develop animal models to understand the physiological roles of these channels. Mainly based on knockout mice, this approach is still under progress but has already shown the implication of K2P channels in functions as diverse as aldosterone secretion by adrenal glands, breathing control at the central and peripheral levels, immunity, heart function, general anesthesia, and neuroprotection against ischemia, and pain detection (for recent reviews, see [16, 26]).
Another important point that has been addressed was to understand why the expression levels of K2P channels were so variable between channels. Figure 1 illustrates this heterogeneity. Whereas 2/3 of the mammalian K2P channels are active and produce strong currents in oocytes (>1 μA), the remaining 1/3 are poorly active (TWIK1 < 1 μA) or silent in oocytes (TWIK2, KCNK7, TASK5, and THIK2 < 0.5 μA). These observations raise intriguing questions regarding the mechanisms restricting the functional expression of these subunits, and the physiological reasons and consequences of such cellular control. In this article, we will review the data related to TWIK1 and THIK2. We will also discuss the other silent K2P channels TWIK2, KCNK7, and TASK5.

K2P channels exhibit various expression levels in Xenopus oocytes. Displayed values are mean amplitudes ± SEM at +50 mV collected in the literature or in our personal data for similar conditions of injection and recording. The pH value of the extracellular solution was 7.4, except for TALK1 and TALK2 that were recorded at pH 8.9. K2P channels generating significant currents are shown in grey and channels generating very modest current or no current in red
TWIK1 channels traffic from the plasma membrane to recycling endosomes
Soon after cloning, TWIK1 was localized in different tissues and cells by immunochemistry. In mouse kidney and cochlea, TWIK1 is highly expressed in secretory epithelial cells where its distribution is limited to cytoplasm and apical membrane [1, 13]. In proximal tubules of the kidney, TWIK1 is mainly restricted to a subapical compartment below the α-actin-rich microvilli that line the lumen [14]. Intracellular localization of TWIK1 was further studied in stably transfected MDCK cells [14]. In this kidney cell line, TWIK1 staining is perinuclear and vesiculotubular. TWIK1 co-localizes perfectly with Vamp8 (Fig. 2a) and Rab11 (unpublished result), two proteins commonly used as markers of the recycling endosomal compartment. This compartment is dependent on intact microtubules for its integrity, and a treatment with nocodazole, a microtubule-depolymerizing agent, disperses TWIK1-containing vesicles (Fig. 2a). In polarized MDCK cells, TWIK1 is present in the subapical recycling compartment like in native kidney cells (Fig. 2d). In non-polarized cells, antibody-feeding experiments have shown that TWIK1 is initially present at the cell surface but constitutively internalized and rapidly redistributed into vesicles (Fig. 2b) [18]. TWIK1 endocytosis is dependent on dynamin, as co-expression of a dominant-negative form of dynamin prevents its internalization (Fig. 2b). We also found that TWIK1 co-localizes with clathrin pits at the plasma membrane (unpublished data).

TWIK1 traffics between the plasma membrane and the recycling endosomal compartment. a Co-localization of TWIK1 (in red) with Vamp8 (in green), a marker of recycling endosomes in transfected MDCK cells. Overlapping red and green are in yellow. Cell nuclei are in blue. Right panel: scattering of TWIK1-containing vesicles after a treatment with nocodazole, a tubulin-depolymerizing agent. Adapted from [14]. b Illustration of the dynamin-dependent endocytosis of TWIK1 by an antibody feeding experiment. MDCK cells expressing TWIK1 were first incubated at 4 °C with anti-TWIK1 antibody and then incubated at 37 °C for 0, 10, or 20 min before fixation and labeling with a fluorescent secondary antibody. Right panel: co-expression of a dominant negative mutant of dynamin (K44) prevents TWIK1 internalization. Adapted from [18]. c TWIK1 (in red) forms a complex with the small G protein ARF6 in the GTP-bound form (ARF6DN, in blue) and its nucleotide exchange factor EFA6 (in green) at the plasma membrane of transfected BHK cells. Overlapping red, green, and blue are in white. Adapted from [14]. d A di-isoleucine motif controls TWIK1 endocytosis. Left schema: position of the endocytosis motif in the cytoplasmic C-terminus of TWIK1. Right panels: distribution in polarized MDCK cells of TWIK1 and TWIK1-AA, in which isoleucines 293,294 were replaced by alanines. Actin is in green and ZO-1, a marker for tight junctions in blue. Adapted from [18]. e Schematic representation of TWIK1 trafficking in cells
Together, these results suggest that TWIK1 follows the regular synthetic pathway from the endoplasmic reticulum (ER) to the plasma membrane, where the channel is endocytosed and targeted to the endosomal compartment to be ultimately stored in recycling endosomes. How is TWIK1 internalized and addressed to recycling endosomes? Cellular trafficking of membrane proteins often relies on specific signals, which function as an address code to target proteins to different intracellular organelles or plasma membrane domains. K+ channels provide good examples of proteins containing such sorting signals. ER retention/retrieval signals have been implicated in the control of KATP channel assembly [49, 55], whereas ER export signals facilitate the sorting of Kir2 [32] and TASK [58] channels from the ER to the plasma membrane. Progressive deletion of the cytoplasmic C-ter of TWIK1 and site-directed mutagenesis allowed the identification of an isoleucine repeat (I293/I294) reminiscent of the classical di-leucine-based endocytosis motif [18]. This type of motif promotes interaction with adaptor protein complexes, which regulate the assembly of clathrin-coated vesicles, and their endocytosis. When this di-isoleucine motif is mutated, the channel redistributes to the plasma membrane (Fig. 2d). This localization is associated with the expression of measurable K+ currents. Interestingly, TWIK1 was shown to interact with the nucleotide exchange factor EFA6 when this factor interacts with the small G protein ARF6. Co-expression of EFA6 and of a mutant of ARF6 mimicking the GDP-bound form abolishes TWIK1 endocytosis (Fig. 2c). ARF6 is known to regulate internalization of the ß2 adrenergic receptor (ß2AR) by directly interacting in an agonist-dependent manner with ß-arrestins [12]. Once activated by its ligands, ß2AR becomes phosphorylated and recruits ß-arrestin. Then, ß-arrestin interacts simultaneously with ARF6GDP and EFA6, promoting ARF6 activation and GDP/GTP exchange, recruitment of adaptor proteins and clathrin, and vesicular endocytosis [34]. By analogy, it is tempting to propose that activity-independent binding of TWIK1 to EFA6/ARF6GDP promotes activation of ARF6 and the recruitment of adaptor proteins via the di-isoleucine motif of TWIK1. Such a mechanism would explain the rapid and constitutive internalization of TWIK1 in many cell types. Despite repeated attempts, we were unable to find a specific signal for TWIK1 addressing to the recycling endosomes, suggesting that the di-isoleucine motif may also be a signal for this particular targeting.
Figure 2e summarizes our current knowledge on cellular trafficking of TWIK1. At the plasma membrane, TWIK1 interacts with ARF6 and EFA6. This interaction favors a rapid and constitutive internalization via clathrin-coated vesicles, in a dynamin-dependent manner. Following endocytosis, TWIK1 concentrates in the endosomal recycling compartment. Little is known about recycling of TWIK1 channels to the plasma membrane. We found that activation of two different Gi-coupled receptors, ß2AR and the serotonin receptor 5-HT1R, promotes a current increase in MDCK and CHO cells expressing TWIK1 channels. This effect is not observed in cells transfected with the mutant TWIK1-I293,294A that is locked at the cell surface [18]. Moreover, the kinetics of the effect are very slow and compatible with a modification of the vesicular recycling of TWIK1. From these results, intracellular retention of TWIK1 appears as a major mechanism that explains the weakness of its current expression level in heterologous systems. An alternative explanation was proposed: TWIK1 would be modified by addition of a SUMO peptide to lysine 274 [42, 44]. Mutation of this lysine into a glutamate, which prevents sumoylation, produces mutated channels that give more currents. However, this “sumo” hypothesis would imply that TWIK1 is sumoylated in the many cell types in which it does not give current. We have observed the increase of current produced by the mutation of lysine 274 [17]. However, how to explain that TWIK1-I293,293A or TWIK1-L146D (see the next paragraph) produce more currents when these mutated channels have an intact sumoylation site? Furthermore, experiments with dominant-negative dynamin shows that it is possible to record a current increase without modifying the channel sequence [18]. At this point, a parallel effect of sumoylation is not excluded, but this post-translational modification cannot be the main regulatory mechanism explaining TWIK1 silencing.
What is the physiological significance of the endosomal recycling of TWIK1? TWIK1 displays a unique feature that may be related to its recycling. It exhibits an ionic selectivity that changes in response to pathological or physiological conditions [8, 33]. During this process—detailed in [11] and in a review of this special issue—TWIK1 switches from a K+-selective state to a state in which it becomes permeable to Na+. This dynamic alteration is slow and reversible: several tens of minutes are necessary for a complete shift between the two states. This change is induced by pathological hypokalemia and contributes to a cardiac paradoxical depolarization that increases the risk for lethal arrhythmia in humans [33]. This change is also induced by low pH values. Astrocytes and pancreatic and renal cells from TWIK1−/− knockout mice display a resting membrane potential more hyperpolarized than that of wild-type animals [8, 36, 53], suggesting that TWIK1 behaves as a depolarizing channel in these cells. How could TWIK1 produce depolarizing currents at physiological pH and K+ concentrations? Along the biosynthetic pathway and in the endosomal compartment, the intravesicular pH is acid. This condition is expected to promote a conformational rearrangement of TWIK1 toward the non-selective state. When TWIK1 arrives at the cell surface, it acts as a depolarizing channel permeable to Na+ rather than as a hyperpolarizing K+-selective channel. If TWIK1 spends enough time at the plasma membrane before endocytosis, it can switch back to its K+ selective form and produces K+ currents. This is certainly the case in human cardiomyocytes and Xenopus oocytes, explaining the original description of TWIK1 as a K+-selective channel [27, 28]. However, if TWIK1 is rapidly internalized, it does not spend enough time at the plasma membrane to recover its K+ selectivity. Then, its influence would be depolarizing as observed in astrocytes and renal and pancreatic cells [8, 36, 53]. If true, this hypothesis implies that the recycling rate between endosomes and the plasma membrane sets the depolarizing or hyperpolarizing influence of TWIK1 in the many tissues and cell types that express this channel. Hormones or neuromodulators acting through Gi-coupled receptors may regulate this recycling and influence cell excitability. Because of its abundance in endosomes, TWIK1 may also play a role in these organelles. As a cargo protein, TWIK1 may affect the organization of the endosomal compartment. As a channel, TWIK1 may participate in the acidification of early and recycling endosomes. The loss of selectivity of TWIK1 would facilitate the exchange of intravesicular Na+ ions against protons by a positive feedback mechanism based on its capacity to transport Na+ ions in acidic conditions.
Retention of the THIK2 channels in the endoplasmic reticulum
Tandem pore domain halothane-inhibited K+ channel subunits THIK1 (K2P13.1, KCNK13) and THIK2 (K2P12.1, KCNK12) have been cloned in 2001 [20, 46]. Despite a strong sequence homology, only THIK1 produces current upon heterologous expression. Human and rat THIK2 are silent in Xenopus oocytes (Fig. 1). Because rat THIK2 was originally shown to be present at the plasma membrane in oocytes, it has been classified as a non-functional subunit. THIK2 is expressed in many tissues. In renal epithelial cells, it is present in intracellular and basolateral membranes [51]. A GFP-tagged version of rat THIK2, which again failed to generate current in HEK293 and COS-7 cells, was reported to be intracellular [22]. Recently, we re-examined the subcellular localization of human THIK1 and THIK2 in transfected cells. Whereas THIK1 is present at the cell surface of MDCK cells, THIK2 is mainly detected in a large intracellular and reticular network extending through the cell [9]. THIK2 co-localizes perfectly with calreticulin, an ER resident protein (Fig. 3a). Similar observations were reported for the EGFP-tagged rat version of THIK2 expressed in HeLa cells [47].

Retention of THIK2 in the endoplasmic reticulum (ER). a In transfected MDCK cells, THIK2 (in green) colocalizes with calreticulin (in red), an endogenous marker of ER. Overlapping red and green are in yellow. Adapted from [9]. b The N-terminus of THIK2 contains an ER-retention signal (R11,12,14,15,16 in bold underlined) that includes a phosphorylation motif for protein kinase A (S13, in red). c Deletion or mutation of the ER retention motif promotes a re-localization of THIK2 (in green) at the cell surface. Adapted from [9]. d Serine 13 modulates surface expression of CD161/THIK2 chimera. Cell surface expression of the chimeras was quantified by flow cytometry. Adapted from [9]
Looking for a sorting signal, we progressively deleted the cytoplasmic N-ter region of THIK2. We identified an arginine-rich motif (RRSRRR) responsible for human THIK2 localization in the ER (Fig. 3b and c). The same motif was identified in the rat THIK2 [47]. When fused to the surface-expressed CD161 protein, the N-ter of THIK2 is sufficient to redistribute the chimeric protein to the ER (Fig. 3d). Replacing the five arginines of this motif by alanines (AASAAA) produces a mutated THIK2 channel relocated at the plasma membrane and able to generate modest but significant current in Xenopus oocytes (Fig. 3c). This arginine-rich motif resembles the well-characterized arginine-based ER localization signal first identified in the early 90s and found now in a growing list of membrane proteins. Extensive mutagenesis on arginine-based signals suggests that the consensus sequence is RXR with X representing any amino acid. The increasing number of arginines in the vicinity of the signal, notably before RXR, determines the strength of the retention [54]. With five arginines, the ER-retention signal of THIK2 is particularly strong. In KATP channels, GABAB R1, NMDA NR1 channels, the arginine-based motif is located in the cytoplasmic C-ter whereas in Nav1.8, it is in the intracellular loop connecting transmembrane segments I and II [35, 50, 55, 56]. THIK2 is the first example of ion channel with an ariginine-based signal located in the N-ter [9, 47].
Other motifs are implicated in ER-retention of membrane proteins. Because it is difficult to distinguish between strict retention in the ER and retrieval from the Golgi to the ER, those motifs are commonly called ER-retention/retrieval motifs. Di-lysine (KKxx), di-basic (KR), and tri-basic (KRR) motifs are responsible for the complex trafficking of TASK1 and TASK3 channels—detailed in another review of this special issue. Briefly, a di-basic motif (KR) in TASK1 N-ter reduces its surface expression, whereas a tri-basic retention signal (KRR) in the C-ter of both TASK1 and TASK3 can bind protein 14-3-3 [40, 45, 57]. In the absence of 14-3-3, the signal is exposed to the COP-I machinery and channels are sent from the Golgi to the ER by retrograde transport. Phosphorylation of serine near the tri-basic signal (KRRxS) increases the affinity for 14-3-3, preventing the retrieval by COP-I. Interestingly, the arginine-based retention/retrieval motif of THIK2 contains a serine residue that is in a consensus site for phosphorylation by protein kinase A (RRSRRR). Replacing this serine by aspartate to mimic phosphorylation decreases ER distribution of the CD161/THIK2 N-ter fusion protein (Fig. 3d). The same mutation introduced in THIK2 (THIK2-S13D) increases current amplitude when compared to THIK2 or to a mutant channel containing a neutral residue (THIK2-S13A) [9]. These results suggest that phosphorylation regulates cellular trafficking of human THIK2 [9]. It should be noted that serine 13 is replaced by a cysteine residue in rodents, suggesting other mechanisms of regulations.
What is the physiological significance of THIK2 retention in the ER? ER-retention signals participate in the quality control of membrane proteins, a necessary checkpoint in the cell. Exposition of arginine-based motifs can reflect improper folding of a protein. For multimeric proteins, exhibition of the ER-retention/retrieval motif can prevent ER exit of unassembled, incompletely, or improperly assembled protein complex. This function has been largely illustrated by studies on KATP channels and GABABR1 [35, 55]. In those cases, arginine-based motifs are exposed in monomeric proteins and act as ER-localization signal but are hidden in the protein complexes that can therefore traffic to the plasma membrane. One way to the mask ER-retention/retrieval signal of THIK2 could be the assembly with other K2P channel subunits. We have recently shown that THIK1 and THIK2 form active heterodimeric channels [6]. This heterodimer is located at the plasma membrane, suggesting that THIK1 is able to mask the retention/retrieval signal present in THIK2. Alternatively, we have also shown that THIK2 forms active homodimers [9]. Then, beside a role at the plasma membrane, a role of THIK2 homodimers in the ER is not excluded. In the ER, Ca2+ uptake and release are accompanied by counterion movements across the ER membrane, which prevent or reduce the generation of membrane potentials and balance for electroneutrality in the lumen. THIK2 activity may be important for ER Ca2+ uptake, as has been shown for the small conductance calcium-sensitive K+ channels [25].
The deep pore of TWIK1 and THIK2 contains a hydrophobic barrier that limits current amplitude
As described in the previous paragraphs, the low level of activity of TWIK1 and THIK2 can chiefly be attributed to their peculiar cellular trafficking. Mutated versions of TWIK1 and THIK2 that redistribute to the cell surface produce measurable currents, but of limited amplitudes compared to TREK or TASK currents. These results suggest that TWIK1 and THIK2 have low intrinsic activities at rest, also contributing to their silencing in the plasma membrane. Several studies based on mutagenesis inside or in close proximity of their pore domains provide new clues to understand the low basal activity of TWIK1 and THIK2.
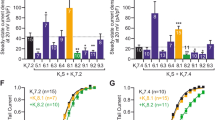
Highly active K2P channels KCNK0 and TREK1 contain three glycine residues in their M2 inner helix (Fig. 4a). In KCNK0, replacement of these glycines by larger hydrophobic residues (G134A, G139V, G141V) dramatically decreases open probability [5]. TWIK1 contains large hydrophobic residues at these positions and replacing them by glycines (L146G-A151G-V153G) produces a threefold increase of TWIK1 current in Xenopus oocytes [8]. Substitution of the first hydrophobic residue of TWIK1 by an aspartate (L146D) results in a 16-fold increase in current amplitude (Fig. 4b). By analogy with the big conductance K+ channels (BK channels), this may be explained by an electrostatic effect, where the negative charge of aspartate alters the electrical potential and increases local K+ concentration [39]. Very recently, molecular simulations combined with mutagenesis suggested an alternative mechanism [2]. This is described in a review of this special issue. Briefly, the authors of this study found that any hydrophilic substitution of L146 increased TWIK1 channel activity (L146S, L146T, and L146N) (Fig. 4b). The same holds true for other pore-facing residues in the spatial vicinity of L146, namely residues L261 and L264 in the inner helix M4. These hydrophobic pore-facing residues of M2 and M4 form a hydrophobic barrier, which contributes to the low basal activity of TWIK1. Molecular dynamic simulations show that water molecules are more excluded in the hydrophobic pore of TWIK1 than in the hydrophilic pores of the TWIK1 mutants. This “dewetting zone” in wild-type TWIK1 stands just below the selectivity filter precisely at the L146/L261 level on ∼5 Å depth and is called the hydrophobic cuff [2]. Although additional effect of these pore mutations on channel properties cannot be excluded, it is likely that polar mutations in the hydrophobic cuff increase K+ flux by disrupting the hydrophobic barrier to water molecules and hydrated K+ ions.

Mutations in the inner helices of TWIK1 and THIK2 stimulate channel activity. a Sequence alignment of M2 inner helix of active (KCNK0 and TREK1) and silent (TWIK1 and THIK2) K2P channels. Glycine residues in KCNK0 and TREK1 and the corresponding larger hydrophobic residues in TWIK1 and THIK2 are shown in red boxes. A proline residue conserved among all K2P channels, except in THIK channels, is highlighted with yellow. b Molecular determinants of the low intrinsic activity of TWIK1. Left: localization of L146 and L261 in the TWIK1 crystal structure (PDB ID:3UKM). Side view of TWIK1 including the selectivity filter and the pore helix (in grey), the inner helices M2 (in blue) and M4 followed by the C-helix (in brown). Cytoplasmic N- and C-terminus domains, outer helices M1 and M3, and extracellular domains are not depicted. Middle: average current–voltage relationships of TWIK1 and TWIK1-L146D, recorded in Xenopus oocytes. These channels contain also the di-isoleucine mutation for a stable expression at the cell surface. L146D mutation produces a 16-fold increase in current amplitude. Adapted from [8]. Right: mean current amplitude at 0 mV for oocytes expressing TWIK1-AA channels with a range of different hydrophobic (grey) and hydrophilic (red) substitutions at leucine 146. Adapted from [2]. c Molecular determinants of the low intrinsic activity of THIK2. Left: localization of residues A155 and I158 in a structural modeling of a THIK2 based on the TWIK1 crystal structure. Side view of THIK2 including the outer helices M1 (in white), the selectivity filter and the pore helix (in brown) and the inner helices M2 (in blue). Cytoplasmic N- and C-terminus domains, transmembrane segments M3 and M4 and extracellular domains are not depicted. Middle: average current–voltage relationships of THIK2, single mutant THIK2-A155P and double mutant THIK2-A155P-I158D, recorded in Xenopus oocytes. Adapted from [9]. Right: mean current amplitude at 0 mV from oocytes expressing THIK2, THIK2-A155P-I158D, or THIK2-5RA-A155P-I158D. The graph illustrates the additive effect of gating (A155P-I158D) and trafficking mutations (5RA, mutation of the five arginines of the ER-retention signal). Adapted from [9]
Like TWIK1, THIK2 harbors hydrophobic residues (I158, L163, L165) in place of the three glycines in KCNK0 and TREK1 (Fig. 4a). Substitution of the first isoleucine by glycine (I158G) or aspartate (I158D) greatly increases current amplitude of THIK2 in Xenopus oocytes [9, 47], suggesting that THIK2 also contains a hydrophobic barrier. Another mutation stimulates THIK2 activity: A155P in M2 (Fig. 4c). Proline 155 is present at an equivalent position in all K2P channels, except THIK1 and THIK2 (Fig. 4a). Moreover, X-ray structures of TWIK and TRAAK suggest that the inner helix M2 is kinked at this proline, widening the pore and leaving it more accessible to the cytoplasmic solution [7, 37]. Combining the two mutations in THIK2 (A155P-I158D) produces a stronger current than each individual mutation (Fig. 4c), suggesting that these mutations stimulate channel activity by distinct mechanisms. Whereas introduction of a proline (A155P) might favor the bending of M2 and the widening of the conduction pathway, introduction of an aspartate (I158D) might disrupt the hydrophobic barrier favoring water and hydrated ions to flow through THIK2 channel.
What happens when mutations affecting channel gating and channel trafficking are combined? As expected, combining gating mutations (A155P-I158D) with mutation of the ER-retention motif in human THIK2 (5-RA) gives a channel producing 140 times more current (Fig. 4c). Similar results were obtained with the rat THIK2 [47]. The same holds true for TWIK1 when combining the mutation of the hydrophobic barrier (L146D) to the mutation of the endocytosis signal (I293/294A) (Fig. 4c) [8]. In both cases, the current levels of these mutated channels are now equivalent to the currents produced by TREK and TASK channels. The regulation of these “silent” channels in native cells and tissues remains an open question. They have low activity at rest but some yet unidentified conditions may favor a stimulation of their activity, not only by favoring their trafficking to the cell surface but also by directly acting on their gating. Further experiments using mutated TWIK1 and THIK2 locked at the plasma membrane should help to identify the latter ones. The hydrophobic barrier may constitute a powerful gate if coupled to the outer gate that has been identified in the other K2P channels [4].
Other silent K2P channels
Beside TWIK1 and THIK2, three other K2P subunits produce no current in Xenopus oocytes (Fig. 1). Two of them, TWIK2 and KCNK7 belong to the same subfamily than TWIK1, and the third one is TASK5. Expression of human TWIK2 in oocytes gives currents hardly distinct from endogenous background conductances [10, 43] (Fig. 1). In COS7 cells, rat TWIK2, which does not generate current in oocytes, produces 15-fold more current than human TWIK2 [41]. These differences are certainly related to differences between expression systems as well as variability in the TWIK2 sequences of different species. Inhibitory proteins may silence rat TWIK2 in oocytes but not in COS7. Human TWIK2 protein shares 84 % identity with rat TWIK2 protein but this does not rule out the possibility that forward trafficking signals may be present in rat TWIK2 but absent in its human counterpart. Alternatively, endocytic or ER-retention motifs can be hidden or inactivated in rat TWIK2 but effective in its human ortholog. At the physiological level, TWIK2 is highly expressed in the systemic and pulmonary vasculature. TWIK2 gene inactivation leads to systemic hypertension in the mouse, indicating that this channel is important in regulating pulmonary vascular tone [31].
All our attempts to record mouse or human KCNK7 channels in oocytes, mammalian, and insect cell lines were unsuccessful [48]. The main reason for this lack of channel activity seems to be its intracellular distribution. KCNK7 is not detectable in the plasma membrane but strictly localizes to the ER. As for THIK2, ER-retention/retrieval signals were searched in the cytoplasmic regions of KCNK7. Progressive deletions of the cytoplasmic C-ter of KCNK7 and construction of chimeras between KCNK7 and TASK1, or between KCNK7 and TREK1, were unable to rescue surface expression and current recording ([48] and personal data). Attempts to stimulate channel activity by pharmacological and cellular activators (pH, Ca2+, PMA,…) or by introducing mutations at key positions in the pore domains were unsuccessful ([48] and personal data). KCNK7 may be an intracellular channel involved in ER Ca2+ uptake or release as postulated for THIK2. Interestingly, human KCNK7 contains in its C-ter an EF-hand domain that is a Ca2+ binding motif, making this channel a possible candidate for ER Ca2+ sensing. Other approaches have to be undertaken, such as reconstitution into lipid bilayers or protein partner screening, to obtain current expression from KCNK7. It should be noted that human KCNK7 contains a GLE sequence in its second pore-domain whereas its mouse paralog has the classical GLG motif. The GLE sequence is not compatible with a K+-selective pore. It would be highly improbable that KCNK7 exhibits radically different ion selectivity in the two mammalian lineages, suggesting that KCNK7 exhibits a permanently altered selectivity. Reconstitution of KCNK7 should help to study its ionic selectivity.
The last silent K2P subunit that needs to be investigated is TASK5 [3, 23, 24, 52]. Unlike the related isoforms TASK1 and TASK3 that elicit large K+ currents in oocytes, TASK5 fails to generate measurable currents in heterologous expression systems (Fig. 1). Chimeric channels containing various parts of TASK5 and TASK3 produce currents suggesting that TASK5 is a functional subunit [23, 24]. The reasons for the TASK5 lack of function are purely speculative. TASK5 channels may not reach the plasma membrane due to improper folding of incomplete assembly [23]. An interesting feature of human TASK5 is a polymorphism occurring at high frequency (60 %) that gives a TASK5 protein containing an unusual EYG sequence in its first pore-domain instead of the classical GYG motif [23, 24]. A similar mutation in the pore-domain of Kir3.4 channel leads to loss of selectivity and to primary aldosteronism in human [38]. As it is difficult to envisage that TASK5 behaves as a K+-selective channel in some individuals and as a Na+-permeable channel in others (depending on the expressed alleles), TASK5 is either an inactive subunit or a channel with a permanently altered selectivity. As for KCNK7, approaches based on purification and reconstitution should be undertaken to record currents and to study the ion selectivity of TASK5.
Conclusion and perspectives
We and others have demonstrated that TWIK1 and THIK2 are functional K2P channels. Because of unique properties of their trafficking and gating that are summarized in Fig. 5, they do not produce high levels of currents. A number of questions remain about their regulations and their physiological roles in native tissues, both in intracellular compartments and at the cell surface. By analogy with TWIK1 and THIK2, KCNK7 and TASK5 are very unlikely to be non-functional subunits. Their genes have not accumulated mutations, premature stop codons, or frame shifts in their coding region. Moreover, they are widely expressed in many tissues and abundantly found in some specific structures like the retinal ganglion cell layer for KCNK7 and the central auditory system for TASK5 [23, 48]. Often, their expression is regulated during development, after particular treatment or during certain pathological condition (inflammation, deafness). Understanding the molecular regulation of all these channels will not only provide insight into their physiological function but will also give fundamental information on cell mechanisms regulating membrane protein trafficking as well as new concepts for ion conduction and gating.

Molecular mechanisms that “silence” TWIK1 and THIK2 current expression. a Hydrophobic residues (L146, V153, L261, and L264) are facing the channel inner cavity forming a hydrophobic barrier to water molecules and hydrated K+ limiting ion permeation. Their substitution by glycine or hydrophilic residues increases channel activity. The stimulating effect of K274E mutation in the C-helix is still unclear. Mutation of the di-isoleucine motif in the C-terminus domain prevents channel endocytosis and increases TWIK1 current at the cell surface. b Mutating the arg-based ER-retention signal in the N-ter domain of THIK2 increases cell surface delivery. Converting the serine embedded in this motif to aspartate (S13D) also promotes channel expression suggesting a phosphorylation-dependent regulation of THIK2 trafficking. Relative to the gating of THIK2, introduction of a proline in M2 increases channel activity probably by widening the ion conduction pathway. Like TWIK1, THIK2 possesses a hydrophobic barrier in its inner cavity: replacement of the hydrophobic isoleucine 158 by a hydrophilic residue (I158D) greatly enhances channel activity
References
Arrighi I, Lesage F, Scimeca JC, Carle GF, Barhanin J (1998) Structure, chromosome localization, and tissue distribution of the mouse twik K+ channel gene. FEBS Lett 425(2):310–316
Aryal P, Abd-Wahab F, Bucci G, Sansom MS, Tucker SJ (2014) A hydrophobic barrier deep within the inner pore of the TWIK-1 K2P potassium channel. Nat Commun 5:4377. doi:10.1038/ncomms5377
Ashmole I, Goodwin PA, Stanfield PR (2001) TASK-5, a novel member of the tandem pore K+ channel family. Pflugers Arch 442(6):828–833
Bagriantsev SN, Peyronnet R, Clark KA, Honore E, Minor DL Jr (2011) Multiple modalities converge on a common gate to control K2P channel function. The EMBO J 30(17):3594–3606. doi:10.1038/emboj.2011.230
Ben-Abu Y, Zhou Y, Zilberberg N, Yifrach O (2009) Inverse coupling in leak and voltage-activated K+ channel gates underlies distinct roles in electrical signaling. Nat Struct Mol Biol 16(1):71–79. doi:10.1038/nsmb.1525
Blin S, Chatelain FC, Feliciangeli S, Kand D, Lesage F, Bichet D (2014) Tandem pore domain halothane-inhibited K+ channel subunits THIK1 and THIK2 assemble and form active channels. J Biol Chem (in press)
Brohawn SG, del Marmol J, MacKinnon R (2012) Crystal structure of the human K2P TRAAK, a lipid- and mechano-sensitive K+ ion channel. Science 335(6067):436–441. doi:10.1126/science.1213808
Chatelain FC, Bichet D, Douguet D, Feliciangeli S, Bendahhou S, Reichold M, Warth R, Barhanin J, Lesage F (2012) TWIK1, a unique background channel with variable ion selectivity. Proc Natl Acad Sci U S A 109(14):5499–5504. doi:10.1073/pnas.1201132109
Chatelain FC, Bichet D, Feliciangeli S, Larroque MM, Braud VM, Douguet D, Lesage F (2013) Silencing of the tandem pore domain halothane-inhibited K+ channel 2 (THIK2) relies on combined intracellular retention and low intrinsic activity at the plasma membrane. J Biol Chem 288(49):35081–35092. doi:10.1074/jbc.M113.503318
Chavez RA, Gray AT, Zhao BB, Kindler CH, Mazurek MJ, Mehta Y, Forsayeth JR, Yost CS (1999) TWIK-2, a new weak inward rectifying member of the tandem pore domain potassium channel family. J Biol Chem 274(12):7887–7892
Chen H, Chatelain FC, Lesage F (2014) Altered and dynamic ion selectivity of K channels in cell development and excitability. Trends Pharmacol Sci 35(9):461–469. doi:10.1016/j.tips.2014.06.002
Claing A, Chen W, Miller WE, Vitale N, Moss J, Premont RT, Lefkowitz RJ (2001) beta-Arrestin-mediated ADP-ribosylation factor 6 activation and beta 2-adrenergic receptor endocytosis. J Biol Chem 276(45):42509–42513. doi:10.1074/jbc.M108399200
Cluzeaud F, Reyes R, Escoubet B, Fay M, Lazdunski M, Bonvalet JP, Lesage F, Farman N (1998) Expression of TWIK-1, a novel weakly inward rectifying potassium channel in rat kidney. Am J Physiol 275(6 Pt 1):C1602–C1609
Decressac S, Franco M, Bendahhou S, Warth R, Knauer S, Barhanin J, Lazdunski M, Lesage F (2004) ARF6-dependent interaction of the TWIK1 K+ channel with EFA6, a GDP/GTP exchange factor for ARF6. EMBO Rep 5(12):1171–1175
Duprat F, Lesage F, Fink M, Reyes R, Heurteaux C, Lazdunski M (1997) TASK, a human background K+ channel to sense external pH variations near physiological pH. EMBO J 16(17):5464–5471
Enyedi P, Czirjak G (2010) Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev 90(2):559–605. doi:10.1152/physrev.00029.2009
Feliciangeli S, Bendahhou S, Sandoz G, Gounon P, Reichold M, Warth R, Lazdunski M, Barhanin J, Lesage F (2007) Does sumoylation control K2P1/TWIK1 background K+ channels? Cell 130(3):563–569
Feliciangeli S, Tardy MP, Sandoz G, Chatelain FC, Warth R, Barhanin J, Bendahhou S, Lesage F (2010) Potassium channel silencing by constitutive endocytosis and intracellular sequestration. J Biol Chem 285(7):4798–4805. doi:10.1074/jbc.M109.078535
Fink M, Duprat F, Lesage F, Reyes R, Romey G, Heurteaux C, Lazdunski M (1996) Cloning, functional expression and brain localization of a novel unconventional outward rectifier K+ channel. EMBO J 15(24):6854–6862
Girard C, Duprat F, Terrenoire C, Tinel N, Fosset M, Romey G, Lazdunski M, Lesage F (2001) Genomic and functional characteristics of novel human pancreatic 2P domain K(+) channels. Biochem Biophys Res Commun 282(1):249–256. doi:10.1006/bbrc.2001.4562
Goldstein SA, Bayliss DA, Kim D, Lesage F, Plant LD, Rajan S (2005) International Union of Pharmacology. LV. Nomenclature and molecular relationships of two-P potassium channels. Pharmacol Rev 57(4):527–540
Kang D, Hogan JO, Kim D (2014) THIK-1 (K2P13.1) is a small-conductance background K(+) channel in rat trigeminal ganglion neurons. Pflugers Arch 466(7):1289–1300. doi:10.1007/s00424-013-1358-1
Karschin C, Wischmeyer E, Preisig-Muller R, Rajan S, Derst C, Grzeschik KH, Daut J, Karschin A (2001) Expression pattern in brain of TASK-1, TASK-3, and a tandem pore domain K(+) channel subunit, TASK-5, associated with the central auditory nervous system. Mol Cell Neurosci 18(6):632–648. doi:10.1006/mcne.2001.1045
Kim D, Gnatenco C (2001) TASK-5, a new member of the tandem-pore K(+) channel family. Biochem Biophys Res Commun 284(4):923–930. doi:10.1006/bbrc.2001.5064
Kuum M, Kaasik A, Joubert F, Ventura-Clapier R, Veksler V (2009) Energetic state is a strong regulator of sarcoplasmic reticulum Ca2+ loss in cardiac muscle: different efficiencies of different energy sources. Cardiovasc Res 83(1):89–96. doi:10.1093/cvr/cvp125
Lesage F, Barhanin J (2011) Molecular physiology of pH-sensitive background K(2P) channels. Physiology (Bethesda) 26(6):424–437. doi:10.1152/physiol.00029.2011
Lesage F, Guillemare E, Fink M, Duprat F, Lazdunski M, Romey G, Barhanin J (1996) TWIK-1, a ubiquitous human weakly inward rectifying K+ channel with a novel structure. EMBO J 15(5):1004–1011
Lesage F, Lauritzen I, Duprat F, Reyes R, Fink M, Heurteaux C, Lazdunski M (1997) The structure, function and distribution of the mouse TWIK-1 K+ channel. FEBS Lett 402(1):28–32
Lesage F, Lazdunski M (2000) Molecular and functional properties of two-pore-domain potassium channels. Am J Physiol 279(5):F793–F801
Lesage F, Reyes R, Fink M, Duprat F, Guillemare E, Lazdunski M (1996) Dimerization of TWIK-1 K+ channel subunits via a disulfide bridge. EMBO J 15(23):6400–6407
Lloyd EE, Pandit LM, Crossland RF, Marrelli SP, Bryan RM Jr (2013) Endothelium-dependent relaxations in the aorta from K(2p)6.1 knockout mice. Am J Physiol Regul Integr Comp Physiol 305(1):R60–R67. doi:10.1152/ajpregu.00126.2013
Ma D, Jan LY (2002) ER transport signals and trafficking of potassium channels and receptors. Curr Opin Neurobiol 12(3):287–292
Ma L, Zhang X, Chen H (2011) TWIK-1 two-pore domain potassium channels change ion selectivity and conduct inward leak sodium currents in hypokalemia. Sci Signal 4(176):ra37. doi:10.1126/scisignal.2001726
Macia E, Partisani M, Paleotti O, Luton F, Franco M (2012) Arf6 negatively controls the rapid recycling of the beta2 adrenergic receptor. J Cell Sci 125(Pt 17):4026–4035. doi:10.1242/jcs.102343
Margeta-Mitrovic M, Jan YN, Jan LY (2000) A trafficking checkpoint controls GABA(B) receptor heterodimerization. Neuron 27(1):97–106
Millar ID, Taylor HC, Cooper GJ, Kibble JD, Barhanin J, Robson L (2006) Adaptive downregulation of a quinidine-sensitive cation conductance in renal principal cells of TWIK-1 knockout mice. Pflugers Arch 453(1):107–116. doi:10.1007/s00424-006-0107-0
Miller AN, Long SB (2012) Crystal structure of the human two-pore domain potassium channel K2P1. Science 335(6067):432–436. doi:10.1126/science.1213274
Mulatero P, Tauber P, Zennaro MC, Monticone S, Lang K, Beuschlein F, Fischer E, Tizzani D, Pallauf A, Viola A, Amar L, Williams TA, Strom TM, Graf E, Bandulik S, Penton D, Plouin PF, Warth R, Allolio B, Jeunemaitre X, Veglio F, Reincke M (2012) KCNJ5 mutations in European families with nonglucocorticoid remediable familial hyperaldosteronism. Hypertension 59(2):235–240. doi:10.1161/HYPERTENSIONAHA.111.183996
Nimigean CM, Chappie JS, Miller C (2003) Electrostatic tuning of ion conductance in potassium channels. Biochemistry 42(31):9263–9268. doi:10.1021/bi0348720
O’Kelly I, Butler MH, Zilberberg N, Goldstein SA (2002) Forward transport. 14-3-3 binding overcomes retention in endoplasmic reticulum by dibasic signals. Cell 111(4):577–588
Patel AJ, Maingret F, Magnone V, Fosset M, Lazdunski M, Honore E (2000) TWIK-2, an inactivating 2P domain K+ channel. J Biol Chem 275(37):28722–28730. doi:10.1074/jbc.M003755200
Plant LD, Dementieva IS, Kollewe A, Olikara S, Marks JD, Goldstein SA (2010) One SUMO is sufficient to silence the dimeric potassium channel K2P1. Proc Natl Acad Sci U S A 107(23):10743–10748. doi:10.1073/pnas.1004712107
Pountney DJ, Gulkarov I, Vega-Saenz de Miera E, Holmes D, Saganich M, Rudy B, Artman M, Coetzee WA (1999) Identification and cloning of TWIK-originated similarity sequence (TOSS): a novel human 2-pore K+ channel principal subunit. FEBS Lett 450(3):191–196
Rajan S, Plant LD, Rabin ML, Butler MH, Goldstein SA (2005) Sumoylation silences the plasma membrane leak K+ channel K2P1. Cell 121(1):37–47
Rajan S, Preisig-Muller R, Wischmeyer E, Nehring R, Hanley PJ, Renigunta V, Musset B, Schlichthorl G, Derst C, Karschin A, Daut J (2002) Interaction with 14-3-3 proteins promotes functional expression of the potassium channels TASK-1 and TASK-3. J Physiol 545(Pt 1):13–26
Rajan S, Wischmeyer E, Karschin C, Preisig-Muller R, Grzeschik KH, Daut J, Karschin A, Derst C (2001) THIK-1 and THIK-2, a novel subfamily of tandem pore domain K+ channels. J Biol Chem 276(10):7302–7311. doi:10.1074/jbc.M008985200
Renigunta V, Zou X, Kling S, Schlichthorl G, Daut J (2013) Breaking the silence: functional expression of the two-pore-domain potassium channel THIK-2. Pflugers Arch. doi:10.1007/s00424-013-1404-z
Salinas M, Reyes R, Lesage F, Fosset M, Heurteaux C, Romey G, Lazdunski M (1999) Cloning of a new mouse two-P domain channel subunit and a human homologue with a unique pore structure. J Biol Chem 274(17):11751–11760
Schwappach B, Zerangue N, Jan YN, Jan LY (2000) Molecular basis for K(ATP) assembly: transmembrane interactions mediate association of a K+ channel with an ABC transporter. Neuron 26(1):155–167
Standley S, Roche KW, McCallum J, Sans N, Wenthold RJ (2000) PDZ domain suppression of an ER retention signal in NMDA receptor NR1 splice variants. Neuron 28(3):887–898
Theilig F, Goranova I, Hirsch JR, Wieske M, Unsal S, Bachmann S, Veh RW, Derst C (2008) Cellular localization of THIK-1 (K(2P)13.1) and THIK-2 (K(2P)12.1) K channels in the mammalian kidney. Cell Physiol Biochem 21(1–3):63–74. doi:10.1159/000113748
Vega-Saenz de Miera E, Lau DH, Zhadina M, Pountney D, Coetzee WA, Rudy B (2001) KT3.2 and KT3.3, two novel human two-pore K(+) channels closely related to TASK-1. J Neurophysiol 86(1):130–142
Wang W, Putra A, Schools GP, Ma B, Chen H, Kaczmarek LK, Barhanin J, Lesage F, Zhou M (2013) The contribution of TWIK-1 channels to astrocyte K(+) current is limited by retention in intracellular compartments. Front Cell Neurosci 7:246. doi:10.3389/fncel.2013.00246
Zerangue N, Malan MJ, Fried SR, Dazin PF, Jan YN, Jan LY, Schwappach B (2001) Analysis of endoplasmic reticulum trafficking signals by combinatorial screening in mammalian cells. Proc Natl Acad Sci U S A 98(5):2431–2436. doi:10.1073/pnas.051630198
Zerangue N, Schwappach B, Jan YN, Jan LY (1999) A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron 22(3):537–548
Zhang ZN, Li Q, Liu C, Wang HB, Wang Q, Bao L (2008) The voltage-gated Na+ channel Nav1.8 contains an ER-retention/retrieval signal antagonized by the beta3 subunit. J Cell Sci 121(Pt 19):3243–3252. doi:10.1242/jcs.026856
Zuzarte M, Heusser K, Renigunta V, Schlichthorl G, Rinne S, Wischmeyer E, Daut J, Schwappach B, Preisig-Muller R (2009) Intracellular traffic of the K+ channels TASK-1 and TASK-3: role of N- and C-terminal sorting signals and interaction with 14-3-3 proteins. J Physiol 587(Pt 5):929–952. doi:10.1113/jphysiol.2008.164756
Zuzarte M, Rinne S, Schlichthorl G, Schubert A, Daut J, Preisig-Muller R (2007) A di-acidic sequence motif enhances the surface expression of the potassium channel TASK-3. Traffic 8(8):1093–1100. doi:10.1111/j.1600-0854.2007.00593.x
Acknowledgments
Authors are supported by the Fondation pour la Recherche Médicale (Equipe labellisée FRM 2011) and by the Agence Nationale de la Recherche (Laboratory of Excellence “Ion Channel Science and Therapeutics”, grant ANR-11-LABX-0015-01).
Author information
Authors and Affiliations
Corresponding author
Additional information
“This article is published as part of a Special Issue on K2P channels.”
Rights and permissions
About this article

Cite this article
Bichet, D., Blin, S., Feliciangeli, S. et al. Silent but not dumb: how cellular trafficking and pore gating modulate expression of TWIK1 and THIK2. Pflugers Arch - Eur J Physiol 467, 1121–1131 (2015). https://doi.org/10.1007/s00424-014-1631-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-014-1631-y