
Abstract
Recent evidence suggests that nerve growth factor IB (Nur77) and nuclear receptor related1 (Nurr1) are differentially involved in dopaminergic neurodegeneration. Since memantine has shown clinically relevant efficacy in Parkinson’s disease (PD) and displayed a potent protective effect on dopaminergic neurons in experimental PD models, we asked if it exerts its neuroprotection by regulating Nur77 and Nurr1 signaling. We adopted a well-established in vitro PD model, 6-hydroxydopamine (OHDA)-lesioned PC12 cells, to test our hypothesis. Different concentrations of memantine were incubated with 6-OHDA-lesioned PC12 cells, and Nur77/Nurr1 and their related signaling molecules were examined by Western blot and immunocytochemistry. Nur77-deficient PC12 cells were used to verify the influences of Nur77 on neurodegeneration and memantine-mediated neuroprotection. We found that memantine reversed Nur77 upregulation and restored Nurr1 downregulation in 6-OHDA-lesioned PC12 cells. 6-OHDA incubation caused Nur77 translocation from the nucleus to cytosol and induced co-localization of Cyt c/HSP60/Nur77 in the cytosol. Memantine strongly reduced the sub-cellular translocations of Nur77/Cyt c/HSP60 under 6-OHDA-induced oxidative condition. Knockdown of Nur77 enhanced the viability of PC12 cells exposed to 6-OHDA, while memantine-induced neuroprotection was much less in the cells with Nur77 knockdown than in those without it. We conclude that Nur77 plays a crucial role in modulating mitochondrial impairment and contributes to neurodegeneration under the experimental PD condition. Memantine effectively suppresses such Nur77-mediated neurodegeneration and promotes survival signaling through post-translational modification of Nurr1. Nur77 and Nurr1 present a contra-directionally coupling interaction in memantine-mediated neuroprotection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disorder following Alzheimer’s disease (AD) and is mainly characterized by a disturbance of the central dopaminergic system by dopaminergic neurodegeneration. Although the etiology of PD is far from clear, the disease is generally recognized to be associated with chronic neuroinflammatory responses and neuronal apoptosis [1, 2]. Increasing evidence suggests that oxidative- and/or inflammatory-related mediators, such as tumor necrosis factor-α (TNF-α), glutamate, and interleukin-1 (IL-1) contribute to the pathogenesis of PD and modulate the progression [3–5]. Among these inflammatory mediators, a family of orphan nuclear receptors, nuclear receptor subfamily 4 (Nur), has also been suggested to play an important role [6–8].
The family members of Nur are differentially involved, as transcriptional factors, in neuroinflammation, neuronal development, and maintenance [7, 9, 10]. In humans, three members have been identified: nerve growth factor IB (Nur77 or NR4A1 ), nuclear receptor related 1 (Nurr1 or NR4A2 ), and neuron-derived orphan receptor 1 (Nor-1 or NR4A3 ). Among the three members, Nurr1 was the first member discovered to be associated with familial parkinsonism, and it functions mainly in transcriptional activation to regulate a battery of genes expressed in dopaminergic (DA) neurons [10], thereby preventing the loss of dopaminergic neurons partially via limiting the production of neurotoxic mediators and modulating neurotrophic factor signaling [11, 12]. Different from Nurr1, Nur77 is less investigated in terms of its role in PD. Nur77 usually acts as a transcription factor to potently modulate the inflammatory responses in macrophages, T cells, vascular inflammation, and infarcted myocardium [13–18]. In macrophages, Nur77 activated the NF-kappa B signaling pathway and potentiated the induction of pro-inflammatory gene expression [19]. Also, Nur77 enhances mouse resistance to lipopolysaccharide (LPS)-induced sepsis by inhibiting NF-κB activity and suppressing aberrant cytokine production [17]. In addition, recent studies have indicated that Nur77 is implicated in mitochondrial dysfunction and subsequent initiation of apoptosis via its intracellular translocation from the nucleus to mitochondria [20, 21]. Cheng et al. found that the translocation of Nur77 from the nucleus to the mitochondria in cardiomyocytes results in the loss of mitochondrial integrity, subsequently leading to the apoptosis in response to ischemia/reperfusion injury [13]. Nur77 may translocate from nucleus into the mitochondria through interaction with the mitochondrial outer membrane protein Nix and thus cause excessive mitochondria clearance. It subsequently activates the mitochondrial signaling pathway and integrates a nuclear receptor with autophagy for irreversible cell death [22]. Kiss et al. demonstrated that by targeting mitochondria, Nur77 may convert Bcl-2, an anti-apoptotic protein, into a proapoptotic molecule, and induce STAT1, thereby enhancing Bim expression and subsequently activating the mitochondrial pathway for apoptosis [23]. It has been reported that JNK-mediated phosphorylation of Nur77 positively regulates its translocation to the mitochondria and negatively regulates the stability of Nur77. Moreover, cyclic AMP/PKA signaling switches the functions of Nur77 from degradation to triggering apoptosis [24]. However, the precise impact and molecular mechanisms underlying the actions of Nur77 in the transcriptional activation and repression of inflammatory responses and apoptosis in PD etiology are unknown.
Memantine (1-amino-3,5-dimethyladamantane) is known as an uncompetitive N-methyl-d-aspartate receptor (NMDAR) antagonist and has been clinically approved to ameliorate the cognitive impairment experienced in moderate to severe Alzheimer’s disease (AD). Recently, several lines of evidence have demonstrated its potential in neuroprotective applications against neurological diseases, such as ischemic stroke, Parkinson’s disease dementia (PDD), and dementia with Lewy bodies (DLB) [25–27]. The possible mechanisms involved in these applications include its NMDAR antagonist activity against NMDA receptor-mediated excitotoxicity, its inhibition of the activity of the cellular histone deacetylase (HDAC), and its inhibition of the activation of microglia, as well as anti-inflammation [27, 28]. However, the precise mechanisms underlying its neuroprotection, especially in PD condition, are far from being understood.
Given the key roles of Nurr1 in the regulation of NMDAR-mediated neuroprotection [29], and the crucial roles of Nur77 in apoptosis, inflammation, and mitochondrial dysfunction, as the downstream of NMDA-mediated signaling, we sought to determine whether Nur77/Nurr1 are differentially involved in memantine-mediated neuroprotection in the in vitro PD model, 6-OHDA-lesioned pheochromocytoma (PC12) cells. We mainly investigated (1) the regulatory association between Nur77 and Nurr1 and effects of memantine on Nur77/Nurr1, and (2) the involvement of cytochrome c (Cyt C) and heat shock protein 60 (HSP60) in the regulation of Nur77/Nurr1 in the PD model.
Materials and Methods
Cell Culture and Treatments
The PC12 cell culture was prepared as described in our previous study and other studies [30]. Briefly, PC12 cells were routinely maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % fetal bovine serum, 5 % horse serum, 100 U/ml benzyl penicillin, and 100 mg/l streptomycin (Gibco Life Technologies, Rockville, MD, USA). For all experiments, the cells were seeded in 96-well plates or 6-well plates at a density of 1.0 × 105 cells/ml for 24 h. For the memantine toxicity measurement experiment, PC12 cells were treated with either conditioned media alone, 6-OHDA (100 μM), or 6-OHDA (100 μM) + the following concentrations of memantine in conditioned media for 24 h: 1, 5, 10, and 20 μM (Sigma-Aldrich, St. Louis, MO, USA). After the 24-h incubation, the Cell Counting Kit-8 (CCK8) assay was used to examine the optimal concentration of memantine for providing neuroprotection to 6-OHDA-lesioned PC12 cells. Then, four experimental groups were treated with either media (DMEM), memantine (10 μM) only, 6-OHDA (100 μM), or 6-OHDA (100 μM) + memantine (10 μM). After 24 h, PC12 cell death was quantified via the CCK8 and the apoptotic cells.
Assays of CCK8, LDH, Glutamate, IL-6, TNF-α, and Measurement of Apoptotic Cells
PC12 cell viability was measured by the CCK8 assay (Dojindo Molecular Technologies, Rockville, MD, USA) according to the manufacturer’s instructions. Briefly, 10 μl of the CCK8 kit reagent was added to the cells treated with 6-OHDA or 6-OHDA + memantine in 96-well plates and incubated at 37 °C for 1 h. The cell viability was assessed at an absorbance of 570 nm with the enzyme-linked immunosorbent assay (ELISA) plate reader. Each treatment group was replicated in three wells. All results were normalized to optical density (OD) values measured from an identically conditioned well without cells [31]. The results for the CCK8 values are expressed as a percentage of the control group, which was set as 100 %. To further investigate the apoptosis, PC12 cells were seeded at a density of 1 × 105 cells/well in 24-well plates. After incubation with 6-OHDA (100 μM) or 6-OHDA (100 μM) + memantine (10 μM) for 24 h, PC12 cell apoptosis was evaluated by flow cytometry (Bender MedSystems, Burlingame, CA, USA).
Cell viability was also measured by determining the activity of the LDH released into the medium. After the 6-OHDA or 6-OHDA + memantine treatments, released LDH was measured, and the cells were lysed to obtain total LDH. Total and released LDH activity was measured following the specifications of the in vitro toxicology assay kit LDH-based Tox-7 (Sigma-Aldrich, St. Louis, MO, USA), and released LDH was normalized to total LDH. The data are represented as the percentage of LDH in the 6-OHDA group, which was designated as 100 %. The concentrations of glutamate, IL-6, and TNF-α in the medium were measured using the Glutamate Assay Protocol (BioVision, CA, USA) and IL-6 and TNF-α ELISA kits at an absorbance of 450 nm with an ELISA plate reader.
Lentiviral Vector Construction/Infection and Short Hairpin Ribonucleic Acid (shRNA) Interference
Lentiviral vectors were used for the knockdown of Nur77 as previously described [32]. Briefly, 293T cells were cultured in DMEM [25 mM N-2-hydroxyethyl piperazine-N0-2-ethanesulfonic acid (HEPES); Gibco] containing 10 % fetal bovine serum (FBS), 5 % horse serum (HoS), penicillin (50 IU/ml), streptomycin (50 mg/ml), and l-glutamine (2 mM; Gibco) and transfected with pMDL-pRRE, pVSV-G, pRSV-REV, and Nur77 using the calcium phosphate method. Viral particles contained in the supernatant were harvested at days 1 and 2 after transfection. After ultracentrifugation, viral titers were determined as previously described [33]. Briefly, serial dilutions of the concentrated virus were used for the transduction of 293T cells. Twenty-four hours after the transduction, total genomic deoxyribonucleic acid (DNA) was isolated using a DNeasy blood and tissue kit (Qiagen) according to the manufacturer’s instructions. The number of inserted vector copies was determined by reverse transcription-polymerase chain reaction (RT-PCR) using the vector DNA for the standard curve: forward primer, 5′ CTGCAGCAGCAGAACAATTTG 3′; reverse primer, 5′ CCCCAGACTGTGAGTTGCAA 3′.
Transduction of PC12 Cells and the Selection of Transduced Cells
PC12 cells were seeded on a six-well plate at a density of 1.0 × 105 cells/ml and allowed to adhere for 24 h. For transduction, the medium was replaced with DMEM + 10 μg/ml polybrene containing viral particles [multiplicity of infection (MOI) 50 for knockdown]. After overnight exposure to the virus, the medium was replaced with DMEM + 10 % FBS + 5 % HoS, and the cells were cultured for another 24 h. Selection with puromycin (2 μg/ml final concentration) was then carried out for 3–4 days, by which time transfection was evident. Depending on the transfection efficiency, most transfections did not establish puromycin-resistant colonies, but the retrovirus-producing cell lines were used for generating viral vectors. The stable colonies were amplified after 10–14 days of selection with puromycin. The transduction efficiency was evaluated by real-time quantitative (RQ)-PCR.
Immunofluorescence and Confocal Microscopy
For immunofluorescence analysis, the procedures were performed with modifications as described in previous studies [34–36]. Briefly, 1 × 105 cells/ml from four experimental groups were plated on confocal Petri dishes in serum-containing media for 24 h. The cells were then incubated in conditioned media alone, memantine (10 μM) alone, 6-OHDA (100 μM) alone, or 6-OHDA (100 μM) + memantine (10 μM) before staining for immunofluorescence. After three washes in phosphate buffered saline (PBS), fixation and permeabilization of the cells was conducted with 4 % paraformaldehyde in PBS for 15 min at room temperature, and the cells were rinsed with PBS and then incubated with PBS-T (0.3 % Triton X-100 in PBS) for 15 min. After three more washes with PBS, the cells were incubated with blocking buffer [PBS, 3 % bovine serum albumin (BSA)] for 60 min at room temperature. The following primary antibodies were used: rabbit anti-Nurr1 (Sigma; 1:200), goat anti-Nur77 (Sigma; 1:200), mouse anti-Cyt c (Abcam; 1:250), and rabbit anti-HSP60 (Cell Signaling; 1:800). The specimens were incubated with the primary antibodies in PBS containing 3 % BSA overnight at 4 °C. For immunofluorescence, on the following day, after additional rinsing with PBS, the samples were incubated with fluorescent-labeled secondary antibodies (Alexa 488-, Alexa 568-, Alexa 555-, or Alexa 647-labeled IgG; Invitrogen) in PBS containing 3 % BSA for 60 min at room temperature. After rinsing with PBS, 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (300 nmol/l; Invitrogen, Carlsbad, CA, USA) was used for counterstaining for 3–5 min. The confocal analysis was performed using a Zeiss LSM 710/Meta Station (Carl Zeiss, Jena, Germany) equipped with a digital camera (Hamamatsu, Hamamatsu, Japan) and operated by QED imaging software.
Protein Extraction, Subcellular Fractionation, and Western Blot Analysis
After 6-OHDA or 6-OHDA + memantine treatment, the cells were harvested using cell scrapers, washed in ice-cold PBS (0.0067 M), and lysed with two different ice-cold lysis buffers [30, 37]. The buffer for total cellular extracts contained 10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.1 mM ethylene diamine tetraacetic acid (EDTA), 0.1 mM phenylmethanesulfonyl fluoride, 1 mM dithiothreitol, 0.2 % NP-40, 1 μg/ml leupeptin, 1 μg/ml pepstatin, and 2 μg/ml aprotinin was used. The same buffer, without dithiothreitol and with addition of 10 mM NaF, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, and 1 mM Na3VO4, was used for the evaluation of protein phosphorylation. The supernatants were collected for protein determination by bicinchoninic acid (BCA) assay (Pierce, Inc., Rockford, IL, USA), and the proteins were run in NuPage Bis-Tris 10 % gels (Invitrogen, Carlsbad, CA, USA) and transferred to polyvinylidene fluoride (PVDF) membranes (Amersham Bioscience, Ltd., Buckinghamshire, UK). The membranes were blocked in 5 % skim milk, 0.05 % Tween 20, and Tris-buffered saline (TBS) for 1 h. The PVDF membranes were incubated in the following primary antibodies overnight at 4 °C: rabbit anti-Nurr1 (Sigma; 1:500), rabbit anti-Nur77 (Proteintech; 1:500), rabbit anti-tyrosine hydroxylase (TH) (Santa Cruz Biotechnology; 1:500), rabbit anti-dopamine transporter (DAT) (Santa; 1:500), rabbit anti-brain-derived neurotrophic factor (BDNF) (Santa; 1:500), rabbit anti-phosphatidylinositol 3 kinase (PI3K)/p-PI3K (Cell Signaling; 1:500), rabbit anti-AKT/p-AKT (Cell Signaling; 1:500), mouse anti-Cyt c (Abcam; 1:1000), rabbit anti-Lamin B1 (Cell Signaling; 1:1000), or rabbit anti-β-actin (Cell Signaling; 1:1000). The next day, horseradish peroxidase-conjugated secondary antibodies (Cell Signaling, Danvers, MA, USA) were applied. Peroxidase-conjugated streptavidin and substrate were used for detection. Negative controls were prepared by omitting the primary antibodies. For inhibition of the extracellular signal-regulated protein kinases (ERK), c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinases (MAPKs), their inhibitors, i.e., PD98059 (50 μM), SP600125 (10 μM), and SB202190 (100 μM), were respectively pre-incubated for 1 h before 6-OHDA addition. For the protein extractions prepared from the cytosolic and nuclear fractions, a method was used that was described by Garcia-Yagüe [38]. Briefly, the cells were washed three times with cold PBS and harvested by centrifugation at 1100 rpm for 10 min. The cell pellet was resuspended in 3 pellet volumes of cold buffer A [20 mM HEPES, pH 7.0, 0.15 mM ethylene diamine tetraacetic acid (EDTA), 0.015 mM ethylene glycol tetraacetic acid (EGTA), 10 mM potassium chloride (KCl), 1 % Nonidet P-40, 1 mM phenylmethylsulfonyl fluoride, 20 mM sodium fluoride (NaF), 1 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 1 μg/ml leupeptin] and incubated on ice for 30 min. Then, the homogenate was centrifuged at 500×g for 5 min. The supernatants were taken as the cytosolic fraction. The nuclear pellet was resuspended in 5 volumes of cold buffer B [10 mM HEPES, pH 8.0, 0.1 mM EDTA, 0.1 mM sodium chloride (NaCl), 25 % glycerol, 1 mM phenylmethylsulfonyl fluoride, 20 mM NaF, 1 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 1 μg/ml leupeptin]. After centrifugation under the same conditions indicated above, the nuclei were resuspended in loading buffer containing 0.5 % sodium dodecyl sulfate (SDS). The cytosolic and nuclear fractions were resolved using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted with the indicated antibodies. The images were analyzed using NIH Image J software.
Statistical Analysis
The data are expressed as the mean ± standard error of the mean (SEM). The data related to the CCK8, IL-6, TNF-α, glutamate, LDH, and flow cytometry analyses, and the different protein quantifications by Western blot were analyzed using one-way analysis of variance (ANOVA) followed by Bonferroni’s comparison post hoc analysis (SPSS 15.0 program, Chicago, IL, USA). Differences with p values of less than 0.05 were regarded as statistically significant.
Results
Memantine Rescued PC12 Cells from 6-OHDA Injury
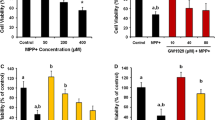
The CCK8 value in the 24 h 6-OHDA-incubated group was significantly reduced to 53 % of the control group (***p < 0.001, 6-OHDA vs. controls, n = 5 independent experiments, Fig. 1a); memantine dose-dependently and significantly attenuated this reduction (Fig. 1a). According to previous studies, 24 h 6-OHDA (100 μM) incubation produces a valid PD in vitro model (Supplementary Fig. S1). At a concentration of 10 μM, memantine improved the viability of PC12 cells more efficiently than other concentrations. Therefore, we chose to use this concentration in the subsequent experiments. Indeed, previous reports also showed that memantine were neuroprotective when the concentrations were ≧5 μM [39, 40]. We detected the apoptosis in 6-OHDA and 6-OHDA plus memantine-treated groups by flow cytometry. Memantine significantly attenuated the 6-OHDA-lesioned cell death of PC12 cells (Fig. 1b–f). Consistently, memantine significantly attenuated the 6-OHDA-induced decrease in the expression of TH/DAT (TH—10.5 % of the control in the 6-OHDA-treated group, 90 % in the group of 6-OHDA plus memantine, n = 5, ***p < 0.001; DAT—16.8 % of the control in the 6-OHDA-treated group, 79.1 % in the group of 6-OHDA plus memantine, n = 5, ***p < 0.001) (Fig. 7b–d). We also measured the excitotoxicity mediators LDH and glutamate and pro-inflammatory cytokines IL-6 and TNF-α in 6-OHDA-treated PC12 cells with or without memantine incubation (Fig. 2). Our result showed that in 6-OHDA-incubated PC12 cells, LDH and glutamate increased 1.69- and 2.22-fold, respectively, compared with controls (n = 5, ***p < 0.001; Fig. 2c, d), but memantine incubation abolished this elevation even though it did not restore the mediators to normal levels (n = 5, ## p < 0.01; Fig. 2c, d). In addition, IL-6 and TNF-α levels increased 2.79- and 3.17-fold, respectively, in 6-OHDA-lesioned PC12 cells compared with untreated cells (n = 5, ***p < 0.001; Fig. 2a, b); memantine incubation profoundly eliminated this upregulation (n = 5, ## p < 0.01; Fig. 2a, b). Taken together, these results strongly indicate that memantine displays significant neuroprotection in 6-OHDA-lesioned PC12 cells via the anti-oxidative stress and anti-inflammatory responses.

Memantine protects PC12 cells from 6-OHDA injury. a PC12 cells were incubated with 100 μM 6-OHDA alone or in combination with different concentrations (1, 5, 10, 20 μM) of memantine for 24 h. The viability of the PC12 cells was measured via the CCK8 assay. The results are expressed as a relative ratio of the blank and are expressed as the mean ± SEM of 5 independent experiments. ***p < 0.001, one-way ANOVA followed by Bonferroni’s comparison post hoc analysis (Bonferroni’s t test) vs. the blank group; ## p < 0.01, ### p < 0.001, Bonferroni’s t test vs. the 6-OHDA-treated culture. b–f Memantine protected PC12 cells from 6-OHDA-induced apoptosis. PC12 cells were exposed to 100 μM 6-OHDA with/without 10 μM memantine for 24 h, and apoptosis was quantified by flow cytometry with double staining Annexin V-FITC and PI for the b blank group, c memantine-treated group, d 6-OHDA-treated group, and e 6-OHDA + memantine group. f The bar chart shows the apoptotic rate of PC12 cells for (b)–(e). The results are expressed as the mean ± SEM of 5 independent experiments. ***p < 0.001, one-way ANOVA followed by Bonferroni’s comparison post hoc analysis (Bonferroni’s t test) vs. the blank group; ### p < 0.001, Bonferroni’s t test vs. the 6-OHDA-treated group

Memantine attenuates the 6-OHDA-mediated elevation in LDH, glutamate, TNF-α, and IL-6. PC12 cells were incubated with 100 μM 6-OHDA with/without 10 μM memantine for 24 h, and the expressions of a IL-6, b TNF-α, c glutamate, and d LDH were measured using an IL-6 and TNF-α ELISA kit, the glutamate assay protocol, and an LDH cytotoxicity detection kit, respectively. The results are the mean ± SEM of 5 independent experiments. ***p < 0.001, one-way ANOVA followed by Bonferroni’s comparison post hoc analysis (Bonferroni’s t test) vs. the blank group; ## p < 0.01 and ### p < 0.001, Bonferroni’s t test vs. the 6-OHDA-treated group
Memantine Restored the Levels of Nur77/Nurr1 in 6-OHDA-Lesioned PC12 Cells
Nurr1 and Nur77 are believed to play differential roles in neuronal survival and inflammatory responses [6, 41]. Our results showed that 6-OHDA (100 μM) time-dependently increased the expression of Nur77 but decreased the total protein level of Nurr1 (Fig. 3a, b). Because the 24-h incubation with 6-OHDA produced the most obvious changes in the expression of Nur77 and Nurr1, we accordingly chose this incubation time for further experiments. We analyzed the total protein expression with different concentrations of memantine (0, 1, 5, 10, 20 μM) following 24 h 6-OHDA incubation. We found that only 10 μM memantine profoundly attenuated the magnitude of the alteration in Nur77 expression following 24-h 6-OHDA incubation (Fig. 3c, d). Interestingly, the 6-OHDA-induced downregulation of Nurr1 was obviously restored by memantine incubation (1, 5, 10, and 20 μM concentrations), with the 10 μM concentration showing the most robust restoration and reaching the basal level.

Memantine conversely regulates Nur77 and Nurr1 expression induced in the 6-OHDA-lesioned PC12 cells. a 6-OHDA induces a time-dependent change in the expressions of Nur77 and Nurr1 in PC12 cells. Western blot analysis shows Nur77, Nurr1, and β-actin protein levels in PC12 cells after treatment with 6-OHDA at 100 μM for various time points. b The diagram shows the relative quantitation of Nur77 and Nurr1 protein levels compared with that of β-actin in (a). The data are expressed as the relative ratios of the 0 h group, which were set to 1.0, and are expressed as the mean ± SEM of 5 independent experiments. *p < 0.05 and ***p < 0.001, Bonferroni’s t test vs. the 0 h group. c Memantine significantly reversed the upregulation of Nur77 expression and downregulation of Nurr1 expression in 6-OHDA-lesioned PC12 cells. Western blot analysis shows Nur77, Nurr1, and β-actin protein levels in 6-OHDA-lesioned PC12 cells accompanied by different concentrations of memantine treatment. d The diagram shows the relative quantitation of the Nur77 and Nurr1 protein levels compared with that of β-actin. The data are expressed as the relative ratios of the blank group, which were set to 1.0, and are the mean ± SEM of 5 independent experiments. **p < 0.01, ***p < 0.001, Bonferroni’s t test vs. the blank group; ## p < 0.01, ## p < 0.001, Bonferroni’s t test vs. the 6-OHDA group
To determine the expression and localization of Nurr1 and Nur77 in 6-OHDA-lesioned PC12 cells with/without memantine treatment, immunocytochemistry and Western blot were examined. We found that under normal conditions, the majority of Nurr1 was predominantly localized in the nucleus, and Nur77 was partially localized in the cytosol but also could be found in the nucleus of PC12 cells (Fig. 4a). Memantine treatment alone did not change their expression or localization in PC12 cells. However, 6-OHDA incubation clearly increased the density of cytosolic Nur77 (Fig. 4a [j]) and decreased the density of nuclear Nurr1 protein (Fig. 4a [k]). Memantine treatment restored the density of cytosolic Nur77 and nuclear Nurr1 to the normal control levels (Fig. 4a [n], [o]). Following 6-OHDA incubation, the Western blot showed that the expression of cytosolic Nur77 was increased 2.58-fold (compared with untreated control, n = 5 experiments, p < 0.001), whereas this increase was attenuated to 1.36-fold of the control after memantine treatment (Fig. 4b, d). By contrast, in 6-OHDA-lesioned PC12 cells, the levels of nuclear Nur77 and Nurr1 were significantly decreased to 37 and 20 % of the normal controls, respectively, and were reversed to 65 and 77 %, respectively, after memantine treatment (Fig. 4c–e). Interestingly, we observed that the expression of cytosolic Nurr1 was at very low level (nearly undetectable) when compared with nuclear Nurr1 (Fig. 4b). This Western blot result is partially consistent with Renaud’s study [42] and further verified our immunocytochemistry findings. These observations further demonstrated that memantine could regulate the contra-directional interaction between Nur77 and Nurr1 in DA neurons.

Memantine protects PC12 cells from 6-OHDA lesion via regulating the expression and subcellular localization of Nur77 and Nurr1. a After incubation with 100 μM 6-OHDA with/without 10 μM memantine for 24 h, PC12 cells were immunostained for Nur77 (b, f, j, n), Nurr1 (c, g, k, o), and the nucleus (a, e, i, m) using confocal microscopy; the merged images (d, h, l, p) are shown. An upregulation of cytosolic Nur77 was caused by 6-OHDA (j), with a translocation of Nur77 from the nucleus to the cytosol, shown by the brighter green fluorescence located in the cytosol and the lighter green fluorescence located in the nucleus (j; arrows); 6-OHDA induced a downregulation of Nurr1 predominantly in the nucleus (k; arrows), with a subsequent decreased co-localization of Nur77 and Nurr1 in the nucleus (i). The addition of memantine prevents the sub-cellular translocation of Nur77 (n; arrows) and the decrease in Nurr1 (o; arrows) and reversed them to the normal levels to some extent, with a restoration of the co-localization of Nur77 and Nurr1 in the nucleus (p) (scale bar = 20 μm). b, c Subcellular fractionations were performed in PC12 cells exposed to 6-OHDA (100 μM) with/without memantine (10 μM) for 24 h. The lysates from the cytosol (b) and nucleus (c) were probed for Nur77 and Nurr1 and normalized to β-actin (cytosolic marker; internal control) and Lamin B1 (nuclear marker; internal control). d, e The bar charts display the relative quantitation of Nur77 and Nurr1 protein levels compared with that of β-actin or Lamin B1, respectively. The values are set up as relative ratios of the blank group, which were set to 1.0, and expressed as the mean ± SEM of 5 independent experiments. ***p < 0.001, Bonferroni’s t test vs. the blank group; ### p < 0.001, Bonferroni’s t test vs. the 6-OHDA group
It has been well documented that ERK/JNK and MAPK p38 signaling pathways are associated with inflammation and apoptosis and cellular survival, and more importantly, recent studies have suggested it is also correlated with Nur77/Nurr1 [43]. Our observations led us to explore a link between Nur77 and Nurr1 in the 6-OHDA-treated PC12 cells. 6-OHDA incubation increased the protein levels of Nur77 for 2.73-fold as compared with the controls (Fig. 5). However, PD98059 and SP600125 almost abolished the increased expression of Nur77 in 6-OHDA-treated PC12 cells (Fig. 5). In contrast, SB202190 had no appreciable effect on Nur77 expression (Fig. 5). On the other side, the decrease in Nurr1 protein (0.16-fold of the control level) induced by 6-OHDA was restored to 0.61- and 0.37-fold of the control level following PD98059 and SP600125 additions, respectively, while SB202190 did not display such effect.

Inhibitors of ERK and JNK prevent the 6-OHDA-induced changes of Nur77/Nurr1 expression in PC12 cells. a PC12 cells were treated with 6-OHDA (100 μM), with or without the inhibitors (PD98059, SP60015, and SB202190) for ERK, JNK, and p38 MAPK, respectively. Total cellular lysates were processed by Western blot for Nur77, Nurr1, and β-actin protein levels. b, c The bar charts show the relative quantitation of Nur77 and Nurr1 protein levels in (a) compared with that of β-actin, and only the inhibitors of ERK and JNK (PD98059 and SP60015, respectively) prevented the changes in Nur77 and Nurr1 induced by 6-OHDA. The data are expressed as the relative ratios of the blank group, which was set to 1.0, and expressed as the mean ± SEM of 5 independent experiments. ***p < 0.001, Bonferroni’s t test vs. the blank group; ### p < 0.001, Bonferroni’s t test vs. the 6-OHDA group
Considering that Nur77 is closely correlated to mitochondrial functions, we measured mitochondrial dysfunction by detecting Cyt c and HSP60 expression via immunocytochemistry. Memantine incubation alone did not change the expression and localization of Cyt c in PC12 cells (Fig. 6a [c, g]). However, 6-OHDA incubation obviously increased the density of cytosolic Cyt c (Fig. 6a [k], g). Following memantine treatment, the increased density of cytosolic Cyt c was significantly attenuated and restored to the normal control levels (Fig. 6a [o], g), which is consistent with the changes in Nur77. Strikingly, we observed a remarkable accumulation and translocation of Nur77 from the cytosol to the nucleus, and Nur77 was co-localized with Cyt c in the cytosol following 6-OHDA incubation (Fig. 6a [l], b). The presence of Nur77 in the nuclear fraction was significantly reduced with concomitant increases in the cytosolic fraction, and the co-localization of Cyt c and Nur77 was also significantly attenuated by memantine treatment (arrows, Fig. 6a [p], b, c). This finding strongly demonstrates that memantine not only downregulated the increased expressions of Nur77 and Cyt c in the cytosol but also prevented Nur77 translocation and co-localization with Cyt c in 6-OHDA-lesioned PC12 cells. Interestingly and strikingly, we found that the change in HSP60 was consistent with those in Cyt c and Nur77 (Fig. 6d, e, f, h). The presence of Nur77 in the nuclear fraction was significantly reduced with concomitant increases in the cytosolic fraction, and the co-localization of HSP60 and Nur77 was also significantly attenuated by memantine treatment (arrows, Fig. 6d [p], e, f).

Memantine represses the 6-OHDA-induced Nur77 translocation and co-localization with Cyt c/HSP60. After incubation with 100 μM 6-OHDA with/without 10 μM memantine for 24 h, PC12 cells were immunostained for the nucleus (a, e, i, m), Nur77 (b, f, j, n), and Cyt c /HSP60 (c, g, k, o). a An upregulated cytosolic and downregulated nuclear Nur77 expression was induced by 6-OHDA (j; arrows), with a translocation of Nur77 from nucleus to cytosol; 6-OHDA also caused increased and more diffused Cyt c expression present in the cytosol (k; arrows) and the co-localization of Nur77/Cyt c (l; arrows). Additional memantine incubation abolished the upregulation of cytosolic Nur77 (n) and Cyt c (o) expression, and Nur77 subcellular translocation (p; arrows) (scale bar = 20 μm). b, c The individual pictures for A l and p are shown magnified. Higher-magnification images of framed areas are located in the upper right corners. The co-localization of Nur77 and Cyt c appeared yellow in cytosol (b), while additional memantine treatment partially abolished this co-localization and reversed diffused Cyt c back to original scattered state (c) (scale bar = 10 μm). d Treatment with 6-OHDA induced increasing cytosolic but decreasing nuclear expression of Nur77 (j; arrows), with an upregulating cytosolic HSP60 expression (k; arrows) and their co-localization (l; arrows). Additional memantine treatment attenuated the increase in cytosolic Nur77 (n) and HSP60 (o) levels, and inhibited Nur77 subcellular translocation and its co-localization with HSP60 (p; arrows) (scale bar = 20 μm). e, f The individual pictures for D l and p are shown. Higher-magnification images of the framed areas are located in the upper right corner (e) or in the lower left corner (f). The co-localization of Nur77 and HSP60 appeared yellow (e), while additional memantine treatment partially attenuated this co-localization (f) (scale bar = 10 μm). g, h The bar charts show the relative quantitation of mean fluorescent intensity (MFI) of Nur77/Cyt c and Nur77/HSP60 in A and D. The data are expressed as the mean ± SEM of 5 independent experiments. ***p < 0.001, Bonferroni’s t test vs. the blank group; ### p < 0.001, Bonferroni’s t test vs. the 6-OHDA group
Knockdown of Nur77 Enhanced Cell Viability and Increased the Levels of Nurr1 and TH/DAT, but Reduced Cyt c Expression Following Memantine Treatment
Is the regulation of Nur77 translocation and expression necessary for the neuroprotective action of memantine in 6-OHDA-lesioned PC12 cells? We knocked down Nur77 expression using a lentiviral vector and compared the cell viability and Nurr1, TH, and DAT expression following 6-OHDA incubation with/without memantine treatment in these mutant PC12 cell cultures (Nur77−/− cells). The CCK8 value in the 6-OHDA-incubated Nur77−/− cells was increased 21 % in comparison with control shRNA-infected cells (Nur77+/+ cells) (## p < 0.01, Nur77−/− vs. Nur77+/+, n = 5 independent experiments; Fig. 7a). Following 6-OHDA incubation, memantine (10 μM) co-application resulted in a 20 % decrease in the CCK8 value in Nur77−/− cells in comparison with the same treatment in Nur77+/+ cells (&p < 0.05, n = 5; Fig. 7a) and a slight 15 % increase in comparison with the lentivirally delivered group without memantine treatment ($ p < 0.05, Nur77−/− with memantine vs. without memantine, n = 5; Fig. 7a). Similar results were also obtained for the expressions of Nurr1, TH, and DAT. The expression of Nurr1, TH, and DAT in 6-OHDA-incubated PC12 cells with Nur77−/− was significantly increased 2.84-fold, 4.2-fold, and 2.0-fold, respectively, in comparison with control shRNA-infected cells (## p < 0.01, 6-OHDA with Nur77−/− vs. Nur77+/+, n = 5; Fig. 7b–e). However, in 6-OHDA-lesioned PC12 cells, the lentivirally delivered shNur77 with memantine (10 μM) treatment resulted in a slight 16, 17, and 16 % decrease in Nurr1, TH, and DAT expression, respectively, in comparison with the non-lentivirally delivered group with memantine (&p < 0.05, memantine with Nur77−/− vs. Nur77+/+, n = 5; Fig. 7b–e) and a 28, 25, and 80 % increase, respectively, in comparison with the lentivirally delivered group without memantine treatment ($ p < 0.05, Nur77−/− with memantine vs. without memantine, n = 5; Fig. 7b–e).

Lentiviral knockdown of Nur77 has a neuroprotective effect in 6-OHDA-lesioned PC12 cells treated with memantine. PC12 cells were subjected to incubation with conditioned media, 6-OHDA (100 μM), 6-OHDA (100 μM) + negative virus, 6-OHDA (100 μM) + Nur77−/−, 6-OHDA (100 μM) + memantine (10 μM), 6-OHDA (100 μM) + memantine (10 μM) + Nur77−/− for 24 h. The cell viability (a); protein levels of TH, DAT, Nurr1, and Cyt c (b–f); and the immunofluorescent analysis of Cyt c and HSP60 expression (g) were measured via the CCK8 assay, Western blot, and confocal microscopy, respectively. a The data for cell viability are expressed as the relative ratios of the blank group, which was set to 1.0, and are expressed as the mean ± SEM of 5 independent experiments. ***p < 0.001, Bonferroni’s t test vs. the blank group; ## p < 0.01, Bonferroni’s t test, 6-OHDA + Nur77−/− vs. 6-OHDA + negative virus; &p < 0.05, Bonferroni’s t test, 6-OHDA + memantine + Nur77−/− vs. 6-OHDA + memantine; $ p < 0.05, Bonferroni’s t test, 6-OHDA + memantine + Nur77−/− vs. 6-OHDA + Nur77−/−. b – f The bar charts (c – f) show the relative quantitation of the specific protein levels of (b) compared with that of β-actin. The data are expressed as the relative ratios of the blank group, which was set to 1.0, and are expressed as the mean ± SEM of 5 independent experiments. *p < 0.05, ***p < 0.001 Bonferroni’s t test vs. blank group; ## p < 0.01, ### p < 0.001, Bonferroni’s t test, 6-OHDA + Nur77−/− vs. 6-OHDA + negative virus; &p < 0.05, Bonferroni’s t test, 6-OHDA + memantine + Nur77−/− vs. 6-OHDA + memantine; $ p < 0.05, Bonferroni’s t test, 6-OHDA + memantine + Nur77−/− vs. 6-OHDA + Nur77−/−. g PC12 cells were immunostained for HSP60 (b, f, j, n), Cyt c (c, g, k, o), and the nucleus (a, e, i, m) (scale bar = 20 μm)
Interestingly, in 6-OHDA-lesioned PC12 cells, the expression of Cyt c was inversely related to that of Nurr1, TH, and DAT. The expression of Cyt c with Nur77−/− was significantly decreased 44 % in comparison with control shRNA-infected cells (## p < 0.01, 6-OHDA with Nur77−/− vs. Nur77+/+, n = 5; Fig. 7b, f). However, in 6-OHDA-lesioned PC12 cells, the lentivirally delivered shNur77 with memantine (10 μM) treatment resulted in a slight 22 % increase in Cyt c in comparison with the non-lentivirally delivered group with memantine (&p < 0.05, memantine with Nur77−/− vs. Nur77+/+, n = 5; Fig. 7b, f) and a 30 % decrease in comparison with the lentivirally delivered group without memantine treatment ($ p < 0.05, Nur77−/− with memantine vs. without memantine, n = 5; Fig. 7b, f). Immunocytochemistry via confocal microscopy was further utilized to investigate the changes in Cyt c and HSP60 in the Nur77 knockdown condition. Incubation of the Nur77 knockdown with 6-OHDA clearly decreased the densities of Cyt c and HSP60 in comparison with the Nur77+/+ group (Fig. 7g [f, g, h, b, c, d]). In the 6-OHDA-lesioned PC12 cells, memantine treatment with Nur77+/+ significantly decreased the densities of Cyt c and HSP60 in comparison with the Nur77−/− group only (Fig. 7g [j, k, l, f, g, h]). Strikingly, we observed that memantine treatment slightly decreased the densities of Cyt c and HSP60 in the 6-OHDA-lesioned PC12 cells with the Nur77 knockdown in comparison with the Nur77−/− group (Fig. 7g [n, o, p, f, g, h]), whereas memantine treatment with Nur77−/− significantly increased the densities of Cyt c and HSP60 compared with memantine treatment with the Nur77+/+ group (Fig. 7g [n, o, p, j, k, l]). This finding strongly demonstrates that memantine has effects on Cyt c and HSP60 partially via the modulation of Nur77.
Discussion
We investigated the effects of memantine on the in vitro PD model and gained four principal findings in this study: (1) the contra-directional coupling of Nur77/Nurr1, i.e., the upregulation of Nur77 and downregulation of Nurr1, contributes to the pathogenesis of PD; (2) memantine reverses the upregulation of Nur77 and downregulation of Nurr1; (3) Nur77 knockdown attenuates 6-OHDA-induced cell damage; and (4) memantine partially inhibits the translocation of Nur77 to the cytosol and decreases Cyt c and HSP60 release from mitochondria. Our data suggest that memantine-mediated neuroprotection is, at least partially, dependent on contra-directional coupling interaction between Nur77 and Nurr1 in cellular reactions against oxidative stress.
An important observation in this study was that memantine reduced dopaminergic neuronal death in 6-OHDA-incubated PC12 cells. The 6-OHDA-lesioned PC12 cells have been widely used as an in vitro PD model since the pathological and/or biochemical characteristics of PD can be mimicked in these cells with 6-OHDA injury [44–48]. They can be used to define important cellular actors of cell death presumably critical for the dopaminergic degeneration [44–48]. In the current study, the 6-OHDA-incubated PC12 cells exhibited a major decrease in cell viability, downregulated TH and DAT, and increased apoptosis. However, memantine significantly restored the reduced cell viability, attenuated the increased apoptosis, and significantly restored the reduction in the levels of TH and DAT in 6-OHDA-lesioned PC12 cells. Our results also showed that inflammatory mediators (i.e., IL-6 and TNF-α) and oxidative predictors (i.e., glutamate and LDH release) were significantly increased in 6-OHDA-induced PC12 cells; however, these elevations were clearly prevented after memantine incubation (Fig. 2a–d). These findings strongly demonstrate that memantine provides pronounced neuroprotection in this in vitro model of PD.
The disruption or mutation of the Nurr1 gene is well documented to contribute to the cellular pathology of PD and to increase the vulnerability of DA neurons to neurotoxicity [6, 9, 49]. However, another member of the family of nuclear receptors, Nur77, is less studied in the pathological and physiological aspects of DA neurons. Because we noticed that memantine obviously prevented DA neuronal death, we asked whether memantine could have similar or different effects on the regulation of Nur77 and Nurr1 expression and looked for correlations between DA neuronal death and Nur77/Nurr1 following memantine treatment. This finding (Fig. 3a, b) suggests that the total decreased cellular level of Nurr1 following the 24-h 6-OHDA incubation indicates obvious DA neuronal death, which is consistent with other studies [50–53]. Strikingly, we noticed a contrasting trend, a time-dependent increase in the total cellular levels of Nur77, while Nurr1 was profoundly decreased in PD patients and closely correlated with the severity of PD progression [51, 52, 54, 55]. Therefore, we propose that in the pathogenesis of PD, the increased Nur77 is concurrently contributing to DA neuronal death as a detrimental factor, while Nurr1 acts as a crucial transcriptional mediator for neuronal survival and neurogenesis.
The current results showed that, following memantine incubation, the decrease in the total level of Nurr1 was reversed, while the increased level of Nur77 was attenuated in a dose-dependent manner, with 10 μM memantine resulting in the most significant effect. This finding (Fig. 3) strongly implies that memantine may concurrently modulate signal transductions that involve both Nur77 and Nurr1. More interestingly, the differential regulation of Nur77 and Nurr1 by memantine strongly demonstrates that Nur77 and Nurr1 deserve crucial attention as possible targets of memantine and as mediators of toxicity. However, whether the observed transcriptional regulation of Nur77/Nurrl following memantine treatment is co-localized in PC12 cells and how they change remains unknown. Therefore, we further examined the exact mechanisms of the effects of memantine on Nur77 and Nurr1. Our immunocytochemistry data revealed changes similar to the Western blot analysis for Nur77 and Nurr1, indicating that memantine provides neuroprotection in 6-OHDA-mediated PC12, at least partially, by regulating transcriptional Nur77 and Nurr1 expressions. We noted that Nurr1 was co-localized with the majority of Nur77 in PC12 cells, with the former mainly in the nucleus and the latter in both the nucleus and cytosol under normal conditions. Following 6-OHDA incubation, however, Nur77 and Nurr1 displayed opposite changes, i.e., upregulation of the former and downregulation of latter, while memantine reversed such opposite changes. This finding strengthened our belief that under oxidative stress, the expression of Nur77 and Nurr1 is regulated in opposite directions; however, memantine modulates the contra-directional regulation of Nur77 and Nurr1.
Furthermore, our Western blotting data show that in 6-OHDA-lesioned PC12 cells, the total protein of Nurr1 largely decreased, and Nurr1 decreased significantly in the nuclear fraction and was almost undetectable in the cytosol. This finding (Fig. 4b–e) is consistent with our immunocytochemistry result. It seems that after 6-OHDA incubation, Nurr1 translocates from the nucleus into the cytosol but degrades very quickly. Indeed, previous evidence also supports this point of view [56, 57]. Apparently, degradation of Nurr1 in the cytosol leads to the functional deletion of Nurr1 as a nuclear transcription factor, while memantine is able to rescue dopaminergic neurons from death by, at least partially, attenuating the reduction in nuclear Nurr1 at subcellular levels. In addition, we observed that, surprisingly, the upregulation and downregulation of Nur77 in the cytosol and nucleus, respectively, reveals the translocation of Nur77 from the nucleus to the cytosol, which was also directly observed in the immunocytochemistry results (Fig. 4a). Considering that the total cellular expression level of Nur77 was upregulated in 6-OHDA-lesioned PC12 cells, we propose that in response to 6-OHDA-induced inflammation, total Nur77 expression is probably upregulated and accompanied by partial nuclear Nur77 translocation to the cytosol. Besides, memantine significantly reversed the changes in Nur77, strongly implying that memantine may prevent Nur77 upregulation and subcellular translocation in response to inflammatory or oxidative stress stimulation.
Our data raise several conundrums. The first is regarding the biological significance of the memantine-mediated reversal of the translocation of Nur77 from the nucleus to the cytosol induced by 6-OHDA. It has been well documented that the translocation of Nur77 from the nucleus is closely correlated to mitochondrial dysfunction and subsequent apoptosis [13], and this led us to further investigate mitochondria-related mediators, including Cyt c and HSP60. This finding indicates that the translocation of Nur77 from the nucleus to the cytosol is accompanied by the Cyt c release response to oxidative stress. The second conundrum is that memantine treatment not only attenuated both the increased cytosolic Cyt c release and cytosolic Nur77 accumulation but also profoundly prevented their 6-OHDA-induced co-localization in the cytoplasm. This finding strongly demonstrates that memantine not only attenuates the increased expression of cytosolic Nur77 and prevents the translocation of Nur77 but also modifies mitochondrial dysfunction under oxidative stress. This hypothesis was further strengthened by a similar observation showing a consistent result for HSP60 expression (Fig. 6d–f).
To further determine whether memantine affects 6-OHDA-lesioned PC12 cells via Nur77 and how memantine influences Cyt c and HSP60, we knocked down Nur77 and examined cell viability and TH, DAT, Nurr1, and Cyt c/HSP60 expression levels. Our results revealed the third conundrum that the CCK8 value in 6-OHDA-incubated PC12 with Nur77−/− was significantly increased in comparison with control shRNA-infected cells, strongly demonstrating that Nur77 has a harmful inflammatory influence here. In 6-OHDA-induced PC12 cells, the finding that lentivirally delivered shNur77 with memantine treatment resulted in a significant decrease in the CCK8 value in comparison with non-lentivirally delivered group with memantine and a slight increase in comparison with the lentivirally delivered group without memantine treatment strongly implies that memantine effects are partially via the modulation of a Nur77-mediated pathway. This CCK8 result is consistent with results regarding the expressions of DAT, TH, and Nurr1 in Nur77 knockdown PC12 cells with/without memantine treatment, further indicating that the modulation of Nur77 may directly regulate DA neuronal death. These findings further verified the third conundrum that Nur77 may be a molecular pharmacological target by memantine in 6-OHDA-lesioned PC12 cells. Interestingly, in Nur77 knockdown PC12 cells, the levels of Cyt c and HSP60 in different experimental groups contrasted with those of CCK8, TH, DAT, and Nurr1 (Fig. 7). Because increased Cyt c and HSP60 represent the mitochondrial dysfunction response to oxidative stress, the changes in Cyt c and HSP60 observed in Nur77 knockdown 6-OHDA-lesioned PC12 cells with memantine treatment further demonstrated that memantine affects Cyt c and HSP60 partially via the modulation of Nur77. Taken together, this finding strongly indicates that the prevention of Nur77 translocation and co-localization with Cyt c/HSP60 by memantine represents a novel approach and target to inhibit DA neuronal death.
Because Nurr1 is correlated with the differentiation of DA neurons [6, 12] and Nur77 is involved in inflammation and apoptosis [13, 14], we also investigated their effector signaling pathways (Fig. 5, Supplementary Fig. S2). The results (Supplementary Fig. S2) strongly indicate that memantine has an effect in mediating differentiation- and neurogenesis-related pathways. Interestingly, Chen et al. found that AKT could substantially disrupt the interaction of cytosolic Nur77 with Bcl-2 via the phosphorylation of Nur77, subsequently preventing Cyt c release and apoptosis [58]. Therefore, Cyt c release and apoptosis after 6-OHDA lesioning may be partially attributed to the downregulation of PI3K/AKT signaling, whereas memantine restored the PI3K/AKT levels (Supplementary Fig. S2). Because AKT is thought to be one of the downstream effectors of Nurr1 [59], we note that AKT may act as a mediator of the coupling between Nur77 and Nurr1, while this finding indicates that memantine may have effects on pro-differentiation/neurogenesis and anti-inflammation/apoptosis via the regulation of the contra-directional coupling between Nur77 and Nurr1. Our results showed that the ERK/JNK inhibitors PD98059 and SP600125 had opposite effects on Nurr1 and Nur77 levels under oxidative stress, strongly indicating that the downregulation of Nurr1 and the upregulation of Nur77 levels were, at least partially, attributable to ERK/JNK signaling pathways in the in vitro PD model.
In summary, we have demonstrated an increase in the cytosolic Nur77 and its translocation from the nucleus to cytosol together with the reduction of nuclear Nurr1 following the oxidative stress and memantine-induced reversal of such pathophysiological changes. The current study indicates that memantine may modulate Nur77 and Nurr1 in a contra-directionally coupling manner in oxidative stress. This finding strongly demonstrates that memantine not only influences Nurr1 by post-translational modification but also regulates Nur77 and the mitochondrial dysfunction response to oxidative stress. The memantine-induced regulation of the rapid induction and temporal modulation of Nur77 and Nurr1 transcripts in oxidative and inflammatory stress fits well with the properties expected of transcriptional activity. This is a novel mechanism underlying memantine-mediated neuroprotection, in addition to its roles in the antagonism of NMDA receptors and anti-inflammation [28, 60]. This study provides a clue for developing an alternative approach for the treatment of neurodegenerative diseases by memantine-induced reversal of Nur77 upregulation and Nurr1 downregulation. A better understanding of the roles and relationships between memantine, Nur77, and Nurr1 may open new perspectives for the role of memantine in the modulation of neurodegenerative disorders such as PD.
Abbreviations
- AD:
-
Alzheimer’s disease
- AKT:
-
Also known as protein kinase B
- Annexin V-FITC:
-
Annexin V-fluorescein isothiocyanate
- ANOVA:
-
Analysis of variance
- BCA:
-
Bicinchoninic acid
- BDNF:
-
Brain-derived neurotrophic factor
- BSA:
-
Bovine serum albumin
- CCK8:
-
Cell Counting Kit-8
- Cyt c:
-
Cytochrome c
- DA:
-
Dopamine
- DAT:
-
Dopamine transporter
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- DNA:
-
Deoxyribonucleic acid
- EDTA:
-
Ethylene diamine tetraacetic acid
- EGTA:
-
Ethylene glycol tetraacetic acid
- ELISA:
-
Enzyme-linked immunosorbent assay
- ERK:
-
Extracellular regulated protein kinases
- FBS:
-
Fetal bovine serum
- HDAC:
-
Histone deacetylase
- HEPES:
-
N-2-hydroxyethyl piperazine-N0-2-ethanesulfonic acid
- HSP60:
-
Heat shock protein 60
- IL-1:
-
Interleukin-1
- IL-6:
-
Interleukin-6
- JNK:
-
c-Jun N-terminal kinase
- KCL:
-
Potassium chloride
- LBD:
-
Dementia with Lewy bodies
- LDH:
-
Lactate dehydrogenase
- MAPK:
-
Mitogen-activated protein kinases
- NaF:
-
Sodium fluoride
- NaCl:
-
Sodium chloride
- NMDA:
-
N-Methyl-d-aspartate
- NMDAR:
-
N-Methyl-d-aspartate receptor
- NR4A1:
-
Nuclear receptor subfamily 4, group A, member 1
- Nur77:
- Nurr1:
- OD:
-
Optical density
- PBS:
-
Phosphate buffered saline
- PC12 cells:
-
Pheochromocytoma 12 cells
- PD:
-
Parkinson’s disease
- PDD:
-
Parkinson’s disease dementia
- PI:
-
Propidium iodide
- PI3K:
-
Phosphatidylinositol 3 kinase
- PVDF:
-
Polyvinylidene fluoride
- RNA:
-
Ribonucleic acid
- RPMI:
-
Roswell Park Memorial Institute
- RQ-PCR:
-
Real-time quantitative-polymerase chain reaction
- RT-PCR:
-
Reverse transcription-polymerase chain reaction
- SDS:
-
Sodium dodecyl sulfate
- SDS-PAGE:
-
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- SEM:
-
Standard error of the mean
- SPSS:
-
Statistical Product and Service Solutions
- TBS:
-
Tris-buffered saline
- TH:
-
Tyrosine hydroxylase
- TNF-α:
-
Tumor necrosis factor-α
- 6-OHDA:
-
6-Hydroxydopamine
References
Wang G, Pan J, Chen SD (2012) Kinases and kinase signaling pathways: potential therapeutic targets in Parkinson’s disease. Prog Neurobiol 98(2):207–221
Gemma C (2010) Neuroimmunomodulation and aging. Aging Dis 1(3):169–172
Long-Smith CM, Sullivan AM, Nolan YM (2009) The influence of microglia on the pathogenesis of Parkinson’s disease. Prog Neurobiol 89(3):277–287
Spulber S, Schultzberg M (2010) Connection between inflammatory processes and transmitter function—modulatory effects of interleukin-1. Prog Neurobiol 90(2):256–262
Labandeira-Garcia JL, Rodriguez-Pallares J, Villar-Cheda B, Rodríguez-Perez AI, Garrido-Gil P, Guerra MJ (2011) Aging, angiotensin system and dopaminergic degeneration in the substantia nigra. Aging Dis 2(3):257–274
Decressac M, Volakakis N, Björklund A, Perlmann T (2013) NURR1 in Parkinson disease—from pathogenesis to therapeutic potential. Nat Rev Neurol 9(11):629–636
Lévesque D, Rouillard C (2007) Nur77 and retinoid X receptors: crucial factors in dopamine-related neuroadaptation. Trends Neurosci 30(1):22–30
Nolan YM, Sullivan AM, Toulouse A (2013) Parkinson’s disease in the nuclear age of neuroinflammation. Trends Mol Med 19(3):187–196
Jankovic J, Chen S, Le WD (2005) The role of Nurr1 in the development of dopaminergic neurons and Parkinson’s disease. Prog Neurobiol 77(1-2):128–138
Le WD, Xu P, Jankovic J, Jiang H, Appel SH, Smith RG, Vassilatis DK (2003) Mutations in NR4A2 associated with familial Parkinson disease. Nat Genet 33(1):85–89
Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, Gage FH, Glass CK (2009) A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 137(1):47–59
Decressac M, Kadkhodaei B, Mattsson B, Laguna A, Perlmann T, Björklund A (2012) α-Synuclein-induced down-regulation of Nurr1 disrupts GDNF signaling in nigral dopamine neurons. Sci Transl Med 4(163):163ra156
Cheng Z, Völkers M, Din S, Avitabile D, Khan M, Gude N, Mohsin S, Bo T, Truffa S, Alvarez R, Mason M, Fischer KM, Konstandin MH, Zhang XK, Heller Brown J, Sussman MA (2011) Mitochondrial translocation of Nur77 mediates cardiomyocyte apoptosis. Eur Heart J 32(17):2179–2188
Wang A, Rud J, Olson CMJ, Anguita J, Osborne BA (2009) Phosphorylation of Nur77 by the MEK-ERK-RSK cascade induces mitochondrial translocation and apoptosis in T cells. J Immunol 183(5):3268–3277
Schott J, Reitter S, Philipp J, Haneke K, Schafer H, Stoecklin G (2014) Translational regulation of specific mRNAs controls feedback inhibition and survival during macrophage activation. PLoS Genet 10(6), e1004368
Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG, Iwamoto Y, Liao R, Zirlik A, Scherer-Crosbie M, Hedrick CC, Libby P, Nahrendorf M, Weissleder R, Swirski FK (2014) Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res 114(10):1611–1622
Li L, Liu Y, Chen HZ, Li FW, Wu JF, Zhang HK, He JP, Xing YZ, Chen Y, Wang WJ, Tian XY, Li AZ, Zhang Q, Huang PQ, Han J, Lin T, Wu Q (2015) Impeding the interaction between Nur77 and p38 reduces LPS-induced inflammation. Nat Chem Biol 11(5):339–346
Papac-Milicevic N, Breuss JM, Zaujec J, Ryban L, Plyushch T, Wagner GA, Fenzl S, Dremsek P, Cabaravdic M, Steiner M, Glass CK, Binder CJ, Uhrin P, Binder BR (2012) The interferon stimulated gene 12 inactivates vasculoprotective functions of NR4A nuclear receptors. Circ Res 110(8):e50–63
Pei L, Castrillo A, Tontonoz P (2006) Regulation of macrophage inflammatory gene expression by the orphan nuclear receptor Nur77. Mol Endocrinol 20(4):786–794
Mohan HM, Aherne CM, Rogers AC, Baird AW, Winter DC, Murphy EP (2012) Molecular pathways: the role of NR4A orphan nuclear receptors in cancer. Clin Cancer Res 18(12):3223–3228
Lindenboim L, Borner C, Stein R (2011) Nuclear proteins acting on mitochondria. Biochim Biophys Acta 1813(4):584–596
Wang WJ, Wang Y, Chen HZ, Xing YZ, Li FW, Zhang Q, Zhou B, Zhang HK, Zhang J, Bian XL, Li L, Liu Y, Zhao BX, Chen Y, Wu R, Li AZ, Yao LM, Chen P, Zhang Y, Tian XY, Beermann F, Wu M, Han J, Huang PQ, Lin T, Wu Q (2014) Orphan nuclear receptor TR3 acts in autophagic cell death via mitochondrial signaling pathway. Nat Chem Biol 10(2):133–140
Kiss B, Toth K, Sarang Z, Garabuczi E, Szondy Z (2015) Retinoids induce Nur77-dependent apoptosis in mouse thymocytes. Biochim Biophys Acta 1853(3):660–670
Lin H, Lin Q, Liu M, Lin Y, Wang X, Chen H, Xia Z, Lu B, Ding F, Wu Q, Wang HR (2014) PKA/Smurf1 signaling-mediated stabilization of Nur77 is required for anticancer drug cisplatin-induced apoptosis. Oncogene 33(13):1629–1639
Aarsland D, Ballard C, Walker Z, Bostrom F, Alves G, Kossakowski K, Leroi I, Pozo-Rodriguez F, Minthon L, Londos E (2009) Memantine in patients with Parkinson’s disease dementia or dementia with Lewy bodies: a double-blind, placebo-controlled, multicentre trial. Lancet Neurol 8(7):613–618
Emre M, Tsolaki M, Bonuccelli U, Destée A, Tolosa E, Kutzelnigg A, Ceballos-Baumann A, Zdravkovic S, Bladström A, Jones R, Investigators S (2010) Memantine for patients with Parkinson’s disease dementia or dementia with Lewy bodies: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 9(10):969–977
Montagne A, Hébert M, Jullienne A, Lesept F, Le Béhot A, Louessard M, Gauberti M, Orset C, Ali C, Agin V, Maubert E, Vivien D (2012) Memantine improves safety of thrombolysis for stroke. Stroke 43(10):2774–2781
Wu HM, Tzeng NS, Qian L, Wei SJ, Hu X, Chen SH, Rawls SM, Flood P, Hong JS, Lu RB (2009) Novel neuroprotective mechanisms of memantine: increase in neurotrophic factor release from astroglia and anti-inflammation by preventing microglial activation. Neuropsychopharmacology 34(10):2344–2357
Barneda-Zahonero B, Servitja JM, Badiola N, Miñano-Molina AJ, Fadó R, Saura CA, Rodríguez-Alvarez J (2012) Nurr1 protein is required for N-methyl-D-aspartic acid (NMDA) receptor-mediated neuronal survival. J Biol Chem 287(14):11351–11362
Xu YQ, Long L, Yan JQ, Wei L, Pan MQ, Gao HM, Zhou P, Liu M, Zhu CS, Tang BS, Wang Q (2013) Simvastatin induces neuroprotection in 6-OHDA-lesioned PC12 via the PI3K/AKT/caspase 3 pathway and anti-inflammatory responses. CNS Neurosci Ther 19(3):170–177
Du M, Wu M, Fu D, Yang S, Chen J, Wilson K, Lyons TJ (2013) Effects of modified LDL and HDL on retinal pigment epithelial cells: a role in diabetic retinopathy. Diabetologia 56(10):2318–2328
Maijenburg MW, Gilissen C, Melief SM, Kleijer M, Weijer K, Ten Brinke A, Roelofs H, Van Tiel CM, Veltman JA, de Vries CJ, van der Schoot CE, Voermans C (2012) Nuclear receptors Nur77 and Nurr1 modulate mesenchymal stromal cell migration. Stem Cells Dev 21(2):228–238
Sastry L, Johnson T, Hobson MJ, Smucker B, Cornetta K (2002) Titering lentiviral vectors: comparison of DNA, RNA and marker expression methods. Gene Ther 9(17):1155–1162
Kaufman AM, Milnerwood AJ, Sepers MD, Coquinco A, She K, Wang L, Lee H, Craig AM, Cynader M, Raymond LA (2012) Opposing roles of synaptic and extrasynaptic NMDA receptor signaling in cocultured striatal and cortical neurons. J Neurosci 32(12):3992–4003
Chung S, Moon JI, Leung A, Aldrich D, Lukianov S, Kitayama Y, Park S, Li Y, Bolshakov VY, Lamonerie T, Kim KS (2011) ES cell-derived renewable and functional midbrain dopaminergic progenitors. Proc Natl Acad Sci U S A 108(23):9703–9708
Lee KW, Ma L, Yan X, Liu B, Zhang XK, Cohen P (2005) Rapid apoptosis induction by IGFBP-3 involves an insulin-like growth factor-independent nucleomitochondrial translocation of RXRalpha/Nur77. J Biol Chem 280(17):16942–16948
Rodriguez-Blanco J, Martín V, Herrera F, García-Santos G, Antolín I, Rodriguez C (2008) Intracellular signaling pathways involved in post-mitotic dopaminergic PC12 cell death induced by 6-hydroxydopamine. J Neurochem 107(1):127–140
García-Yagüe ÁJ, Rada P, Rojo AI, Lastres-Becker I, Cuadrado A (2013) Nuclear import and export signals control the subcellular localization of Nurr1 protein in response to oxidative stress. J Biol Chem 288(8):5506–5517
Wroge CM, Hogins J, Eisenman L, Mennerick S (2012) Synaptic NMDA receptors mediate hypoxic excitotoxic death. J Neurosci 32(19):6732–6742
Volbracht C, van Beek J, Zhu C, Blomgren K, Leist M (2006) Neuroprotective properties of memantine in different in vitro and in vivo models of excitotoxicity. Eur J Neurosci 23(10):2611–2622
Hamers AA, Hanna RN, Nowyhed H, Hedrick CC, de Vries CJ (2013) NR4A nuclear receptors in immunity and atherosclerosis. Curr Opin Lipidol 24(5):381–385
Renaud J, Chiasson K, Bournival J, Rouillard C, Martinoli MG (2014) 17β-estradiol delays 6-OHDA-induced apoptosis by acting on Nur77 translocation from the nucleus to the cytoplasm. Neurotox Res 25(1):124–134
Jacobs CM, Boldingh KA, Slagsvold HH, Thoresen GH, Paulsen RE (2004) ERK2 prohibits apoptosis-induced subcellular translocation of orphan nuclear receptor NGFI-B/TR3. J Biol Chem 279:50097–50101
Zhu G, Chen G, Shi L, Feng J, Wang Y, Ye C, Feng W, Niu J, Huang Z (2015) PEGylated rhFGF-2 conveys long-term neuroprotection and improves neuronal function in a rat model of Parkinson’s disease. Mol Neurobiol 51(1):32–42
Basile JR, Binmadi NO, Zhou H, Yang YH, Paoli A, Proia P (2013) Supraphysiological doses of performance enhancing anabolic-androgenic steroids exert direct toxic effects on neuron-like cells. Front Cell Neurosci 7:69
Zhao J, Cheng Y, Yang C, Lau S, Lao L, Shuai B, Cai J, Rong J (2015) Botanical drug puerarin attenuates 6-hydroxydopamine (6-OHDA)-Induced neurotoxicity via upregulating mitochondrial enzyme arginase-2. Mol Neurobiol. In press
Tsai KL, Cheng YY, Leu HB, Lee YY, Chen TJ, Liu DH, Kao CL (2015) Investigating the role of Sirt1-modulated oxidative stress in relation to benign paroxysmal positional vertigo and Parkinson’s disease. Neurobiol Aging 36(9):2607–2616
Huleatt PB, Khoo ML, Chua YY, Tan TW, Liew RS, Balogh B, Deme R, Goloncser F et al (2015) Novel arylalkenylpropargylamines as neuroprotective, potent, and selective monoamine oxidase B inhibitors for the treatment of Parkinson’s disease. J Med Chem 58 (3):1400–1419
Jiang C, Wan X, He Y, Pan T, Jankovic J, Le W (2005) Age-dependent dopaminergic dysfunction in Nurr1 knockout mice. Exp Neurol 191(1):154–162
Besong-Agbo D, Wolf E, Jessen F, Oechsner M, Hametner E, Poewe W, Reindl M, Oertel WH, Noelker C, Bacher M, Dodel R (2013) Naturally occurring α-synuclein autoantibody levels are lower in patients with Parkinson disease. Neurology 80(2):169–175
Liu H, Wei L, Tao Q, Deng H, Ming M, Xu P, Le W (2012) Decreased NURR1 and PITX3 gene expression in Chinese patients with Parkinson’s disease. Eur J Neurol 19(6):870–875
Mollenhauer B, Locascio JJ, Schulz-Schaeffer W, Sixel-Döring F, Trenkwalder C, Schlossmacher MG (2011) α-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: a cohort study. Lancet Neurol 10(3):230–240
Shi M, Liu C, Cook TJ, Bullock KM, Zhao Y, Ginghina C, Li Y, Aro P, Dator R, He C, Hipp MJ, Zabetian CP, Peskind ER, Hu SC, Quinn JF, Galasko DR, Banks WA, Zhang J (2014) Plasma exosomal α-synuclein is likely CNS-derived and increased in Parkinson’s disease. Acta Neuropathol 128(5):639–650
Wang Y, Shi M, Chung KA, Zabetian CP, Leverenz JB, Berg D, Srulijes K, Trojanowski JQ, Lee VM, Siderowf AD, Hurtig H, Litvan I, Schiess MC, Peskind ER, Masuda M, Hasegawa M, Lin X, Pan C, Galasko D, Goldstein DS, Jensen PH, Yang H, Cain KC, Zhang J (2012) Phosphorylated α-synuclein in Parkinson’s disease. Sci Transl Med 4(121):121ra120
Shi M, Bradner J, Hancock AM, Chung KA, Quinn JF, Peskind ER, Galasko D, Jankovic J, Zabetian CP, Kim HM, Leverenz JB, Montine TJ, Ginghina C, Kang UJ, Cain KC, Wang Y, Aasly J, Goldstein D, Zhang J (2011) Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann Neurol 69(3):570–580
Jo AY, Kim MY, Lee HS, Rhee YH, Lee JE, Baek KH, Park CH, Koh HC, Shin I, Lee YS, Lee SH (2009) Generation of dopamine neurons with improved cell survival and phenotype maintenance using a degradation-resistant nurr1 mutant. Stem Cells 27(9):2238–2246
Lin X, Parisiadou L, Sgobio C, Liu G, Yu J, Sun L, Shim H, Gu XL, Luo J, Long CX, Ding J, Mateo Y, Sullivan PH, Wu LG, Goldstein DS, Lovinger D, Cai H (2012) Conditional expression of Parkinson’s disease-related mutant α-synuclein in the midbrain dopaminergic neurons causes progressive neurodegeneration and degradation of transcription factor nuclear receptor related 1. J Neurosci 32(27):9248–9264
Chen HZ, Zhao BX, Zhao WX, Li L, Zhang B, Wu Q (2008) Akt phosphorylates the TR3 orphan receptor and blocks its targeting to the mitochondria. Carcinogenesis 29(11):2078–2088
Volpicelli F, Caiazzo M, Greco D, Consales C, Leone L, Perrone-Capano C, Colucci D’Amato L, di Porzio U (2007) Bdnf gene is a downstream target of Nurr1 transcription factor in rat midbrain neurons in vitro. J Neurochem 102(2):441–453
Rosi S, Ramirez-Amaya V, Vazdarjanova A, Esparza EE, Larkin PB, Fike JR, Wenk GL, Barnes CA (2009) Accuracy of hippocampal network activity is disrupted by neuroinflammation: rescue by memantine. Brain 132(Pt 9):2464–2477
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant no. 81271427, 81471291), 973 Project (2011CB510000), and Scientific Research Foundation of Guangzhou (grant no. 2014 J4100210), National Natural Science Foundations of Guangdong of China (2014A020212068) to Q.W. Y.X. was partially supported by NIH AT004422 and Vivian L Smith Neurologic Foundation.
Author contributions
X.W., HG, J.Z., Y.X., X.L., and Q.W. conceived and designed the experiments; X.W., H.G., J.Z., Y.X., X.L., J.L., and Q.W. performed the experiments; X.W., J.Z., Y.X., D.C., and Q.W. analyzed the data; Q.W., X.C., L.M., Z.Z., and B.T. contributed reagents/materials/analysis tools; X.C., K.J., Y.X., and Q.W. wrote the paper.
Additional information
Competing financial interests: The authors declare no competing financial interests.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Xiaobo Wei, Huimin Gao and Jing Zou contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 1482 kb)
Rights and permissions
About this article

Cite this article
Wei, X., Gao, H., Zou, J. et al. Contra-directional Coupling of Nur77 and Nurr1 in Neurodegeneration: A Novel Mechanism for Memantine-Induced Anti-inflammation and Anti-mitochondrial Impairment. Mol Neurobiol 53, 5876–5892 (2016). https://doi.org/10.1007/s12035-015-9477-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9477-7