
Abstract
Cocaine abuse has been shown to accelerate the progression of human immunodeficiency virus (HIV)-1-associated neurological disorders (HANDs) partially through increasing neuroinflammatory response mediated by activated astrocytes; however, the detailed molecular mechanism of cocaine-mediated astrocyte activation is unclear. In the current study, we demonstrated increased astrogliosis in the cortical regions of brains from HIV+ cocaine abusers compared with the HIV+ group without cocaine abuse. We next sought to explore whether cocaine exposure could result in increased expression of glial fibrillary acidic protein (GFAP), a filament protein critical for astrocyte activation. Exposure of cocaine to astrocytes resulted in rapid translocation of sigma receptor to the plasma membrane with subsequent activation of downstream signaling pathways. Using a pharmacological approach, we provide evidence that cocaine-mediated upregulation of GFAP expression involved activation of mitogen-activated protein kinase (MAPK) signaling with subsequent downstream activation of the early growth response gene 1 (Egr-1). Egr-1 activation, in turn, caused transcriptional regulation of GFAP. Corroboration of these findings in vivo demonstrated increased expression of GFAP in the cortical region of mice treated with cocaine compared with the saline injected controls. A thorough understanding of how cocaine mediates astrogliosis could have implications for the development of therapeutic interventions aimed at HIV-infected cocaine abusers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Although the advent of combined anti-retroviral therapy has changed the phenotype of HIV infection from a death sentence to a manageable condition [1], as individuals on therapy continue to live longer, complications of the CNS such as HIV-associated neurological disorders (HAND) are on a rise [2, 3]. Adding complexity to this is the use of illicit drugs which further increase the prevalence and severity of HAND [4, 5]. Cocaine, often abused by HIV-infected patients, has been suggested to hasten as well as worsen disease pathogenesis [6]. While the effects of HIV viral proteins have been well studied [7, 8], how drugs of abuse such as cocaine can cause glial activation and concomitant neuronal toxicity remains less clear. In the current study, we sought to examine the molecular mechanisms of cocaine-mediated induction of GFAP expression in astrocyte cultures and in mice injected with cocaine. The basis for this notions stems from a previous study demonstrating that acute cocaine administration in a mouse model triggered induction of GFAP with increased astrocytic proliferation and changes in cell morphology [9].
A characteristic feature of astrocytic activation is increased expression of GFAP, an intermediary filament protein, that is specific for mature astrocytes, and is likely involved in regulating the shape and movement of astrocytes [10]. While the information on cocaine-mediated regulation of GFAP is extant [11–13], there is paucity of information on the mechanism(s) involved in this process. Originally proposed to be a subtype of opioid receptor, the sigma-1 receptor (σ-1R) is now recognized as the non-opioid receptor family that binds to diverse class of psychotropic drugs including cocaine [14]. Expression of σ-1R is strongly upregulated in the brain after cocaine administration [15, 16]. σ-1R has also been closely linked to HIV-related pathogenesis [17, 18]. Activation of this receptor in microglia has been shown to increase HIV expression in microglia [18]. Our previous studies also demonstrated that cocaine-mediated activation of σ-1R modulates the expression of monocyte chemotactic protein 1 (MCP-1) in microglia, a pivotal chemokine critical for migration of HIV-infected mononuclear phagocytes across the blood-brain barrier [19].
Acute cocaine administration has been shown to elicit rapid and transient induction of several immediate-early genes in the brain, including the early growth response-1 (Egr-1) gene [20, 21], which belongs to a zinc finger family of transcription factors. It has been suggested that these proteins function as molecular switches to coordinate changes in gene expression and regulate the cellular responses. In the present study, we explored the intracellular mechanism(s) by which cocaine induced the expression of GFAP. Such an understanding will have clinical relevance in developing therapeutics aimed at targeting astrogliosis in cocaine abusers with or without HIV infection.
Materials and Method
Animals
Eighteen 2-month-old male C57BL/6N mice (Charles River, Sulzfeld, Germany) were purchased from Charles River Laboratories, Inc. (Wilmington, MA). All the animals were housed under conditions of constant temperature and humidity on a 12-h light, 12-h dark cycle, with lights on at 0700 h. Food and water were available ad libitum. Animals were deeply anesthetized by overdose of isoflurane followed by pneumothorax prior to perfusion. All animal procedures were performed according to the protocols approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center.
Cell Cultures
Primary mice astrocyte cells were obtained from 1-to 3-day-old C57BL/6N newborn pups. After digestion and dissociation of the dissected brain cortices in Hank’s buffered salt solution supplemented with trypsin (1 mg/ml), cells were pelleted, resuspended and plated, and maintained in CO2 incubator at 37 °C for 7 days. Cells were then shaken gently to remove loosely attached cells, and the attached cells that remained and that were primarily astrocytes were allowed to grow for 3–4 days, followed by passaging and seeding them on culture dishes. Seven days post-plating, cultures consisted of 97 % astrocytes as assessed by immunostaining using the GFAP antibody. Mouse primary astrocytes were cultured in DMEM medium (Invitrogen, Life Technologies, Carlsbad, CA) containing 10 % heat-inactivated fetal bovine serum, 2 mM glutamine, 1 mM pyruvate, penicillin (100 units/ml), and streptomycin (100 μg/ml). Human astrocytic cell line A172 (ATCC no. CRL-1620; American Type Culture Collection) was cultured as described previously [22].
Semi-Quantitative and Real-Time PCR
Total RNA was extracted with Trizol reagent (Invitrogen, CA) according to the manufacturer’s instructions and our previous reports [19]. One microgram of RNA was used for complementary DNA (cDNA) synthesis according to manufacturer’s instructions (Thermo Scientific, MA). The resulting cDNA was appropriately diluted and amplified using Titanium TaqDNA polymerase and human σ-1R primers (forward 5′-TATCGCAGTGCTGATCCA-′, reverse 5′-TACTCCACCATCCACGTGTT-3′). PCR products were examined by running on a 1.5 % agarose gel. Real-time RT2 qPCR primer sets for human GFAP primers (forward 5′-GGCGCTCAATGCTGGCTTCA-3′, reverse 5′-TCTGCCTCCAGCCTCAGGTT-3′) were obtained from SA Biosciences (Frederick, MD). Quantitative analyses of messenger RNA (mRNA) were conducted using ABI 7500 Fast Real-Time PCR system (Applied Biosystems, CA). Data were normalized using the cycle threshold (Ct) values for 18 s in each sample. To calculate relative amounts of mRNA, the average Ct values were subtracted from 18 s values for each target gene to provide changes in Ct value. Fold change in expression was calculated as log2 relative units.
Short Interfering (si) RNA Transfection
Astrocytes were transfected with 20 nmol/l short interfering RNAs (siRNAs) specific for either σ-1R or Egr-1 obtained from Thermo Scientific Dharmacon RNAi Technologies (Accell SMART Pool). The knockdown efficiency of siRNAs was determined 2 days post-transfection by Western blotting.
Transfection with Plasmid Constructs
Astrocytes were transfected with plasmid constructs containing either wild-type (WT) or dominant negative [23] forms of Egr-1 using Lipo 2000 (Invitrogen, CA) according to the manufacturer’s protocol. Knockdown efficiencies were examined by Western blotting.
Western Blotting
Treated cells were lysed using the Mammalian Cell Lysis Kit (Sigma, MO). Equal amounts of the proteins were electrophoresed in a sodium dodecyl sulfate-polyacrylamide gel (12 %) under reducing conditions followed by transfer to PVDF membranes. The blots were blocked with 5 % non-fat dry milk in phosphate-buffered saline. Western blots were then probed with antibodies recognizing the phosphorylated and total forms of extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), p38 (1:800, Cell Signaling, MA), GFAP (1:10,000, Santa Cruz, CA), Egr-1 (1:800, Santa Cruz, CA), σ-1R (1:800, Cell Signaling, MA), and β-actin (1:4000, Sigma). Cells were then incubated with goat-anti-rabbit secondary Ab (1:5000; Invitrogen, CA). Signals were detected by chemiluminescence (Pierce, IL). All experiments were repeated at least three times individually, and representative blots are presented in the figures.
Lipid Raft Isolation and Analysis
Lipid raft were isolated from confluent A172 cells treated with cocaine according to the previous report [24]. Briefly, lysates were mixed with 1 ml of 85 % (w/v) sucrose, were overlaid with 35 and 5 % sucrose (5 ml each), and were centrifuged for 24 h at 39,000 rpm (Beckman SW 4 Rotor) and 4 °C. Ten 1 ml fractions were collected from the top to bottom. The concentration of cholesterol and protein in each fraction was analyzed with lipid raft maker GM-1 antibody (1:1000, Abcam, CA) and a BCA protein assay kit (Pierce, IL).
Immunocytochemistry
Prior to treatment, cells were seeded on coverslips to perform immunocytochemical analysis of Egr-1 or GFAP. Cells were fixed with 4 % paraformaldehyde for 15 min at room temperature following treatment. The cells were then permeabilized with PBS containing 0.3 % Triton X-100 and incubated with 10 % normal goat serum PBS blocking solution for 1 h. Following blocking, cells were incubated at 4 °C overnight with anti-Egr-1 (1:200, Santa Cruz, CA) rabbit polyclonal antibody or anti-GFAP (1:200, Santa Cruz, CA) mouse polyclonal antibody. Following extensive washing, goat anti-rabbit and anti-mouse Alexa Fluor 488-conjugated secondary antibodies were used for signal detection. For negative controls, cells were treated as described above but without the primary antibody. Cells were mounted with Prolong Gold containing DAPI to stain nuclei. Images were captured with an 40× objective.
Immunohistochemistry
Animals were divided into three groups (n = 6). These groups were treated with (1) saline, (2) cocaine, or (3) BD1047 and cocaine. Cocaine was injected intraperitoneally at a dose of 20 mg/kg once daily. In the BD1047 plus cocaine group, BD1047 was first injected for 2 days at the concentration of 3.5 μg/kg, followed by cocaine injection for subsequent 7 days. Animals were sacrificed 1 h after the final dose of cocaine for isolation of brain tissue for sectioning. Mice were perfused transcardially using chilled 4 % paraformaldehyde. Free-floating sections encompassing the entire brain were sectioned at 40 μm using a cryostat. For GFAP and Egr-1 co-immunostaining, tissue sections were incubated with anti-Egr-1 (1:200, Santa Cruz, CA), rabbit anti-mouse and anti-GFAP (1:200, Sigma, MO) rabbit anti-mouse antibodies overnight at 4 °C. Fluorescent-tagged goat anti-mouse (594 nm, red), goat anti-rabbit (488 nm, green) secondary antibodies were used to probe GFAP and Egr-1 expression, respectively. Immunostained floating tissue samples were gelatin mounted and examined for co-localization of GFAP and Egr-1 protein. Images were captured at wavelengths encompassing the emission spectra of the probes with a 40× objective.
For immunohistochemistry, sections (5 μm thick) from paraffin-embedded human cortex (National NeuroAIDS Tissue Consortium) (Table 1) were chosen followed by deparaffinization and rehydration. Immunostaining with GFAP antibody involved boiling the tissue sections in Tris/EDTA buffer (pH 9) for about 20 min for efficient antigen retrieval. Sections were blocked with 10 % normal goat serum (NGS) for 1 h at RT and then incubated in primary anti-GFAP (1:50, Sigma) antibody at 4 °C overnight. The sections were then incubated with biotin-labeled secondary antibody (Southern Biotechnology Associates) at a 1:500 dilution for 2 h at RT followed by incubation with streptavidin-conjugated horseradish peroxidase (HRP; Southern Biotechnology Associates) at a 1:500 dilution for 1 h at RT. HRP-labeled areas were detected using a solution containing 0.3 mg/ml of 3-3′-diaminobenzidine (DAB) in 100 mM Tris, pH 7.4, and 0.02 % hydrogen peroxide (H2O2). Sections were examined under bright-field light microscopy, and images were captured with a 40× objective.
Statistical Analysis
All the data are expressed as the mean ± SEM. Statistical significance was evaluated with Student t test for between two groups or ANOVA followed by the Newman-Keuls’ test for multiple groups. p < 0.05 was considered as statistically significant difference.
Results
Upregulation of GFAP Expression in Astrocytes from HIV-Infected Drug (Cocaine) Abusers
To establish the clinical relevance of GFAP expression in the context of HIV-1 infection with drug (cocaine) abuse, we examined GFAP expression levels in the sections of postmortem brain tissues from HIV−, HIV+/no cocaine, and HIV+/cocaine drug abusers (Fig. 1a). Because of the inherent difficulty in finding tissues from unidrug abusers, we resorted to samples from polydrug abusers that included a history of cocaine abuse. As shown in Fig. 1b, there was a significant increase in the expression of GFAP positive cells in the cortical sections of brains from HIV+ positive patients compared with the uninfected controls due to the global activation of astrocytes in the HIV-infected brain. Interestingly, HIV+/cocaine abusers exhibited an even more increased activation of astrocytes exemplified by hypertrophic astrocytes, compared with the HIV+ individuals.

Induction of GFAP expression in astrocytes from HIV-infected drug (cocaine) abusers. a Representative images after immunostaining for GFAP showed the increased expression of GFAP positive cells in the cortical sections of brains from HIV+ cocaine abuser patients compared with the HIV+ sections with the latter group demonstrating increased GFAP expression compared to the uninfected controls. Scale bar 20 μm. b Cell counting using ImageJ indicates that drug (cocaine) abuse resulted in a significant increase of GFAP positive cells per brain section. n = 3 sections, threshold = 50, *p < 0.05 vs HIV−/cocaine− group, # p < 0.05 vs HIV+/cocaine− group
Cocaine-Mediated Upregulation of GFAP mRNA and Protein in Astrocytes
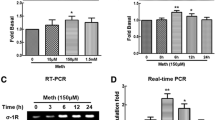
In the present study, as an initial investigation to examine the potential modulation of GFAP expression following cocaine treatment, human astrocyte A172 cells were first serum-starved overnight followed by exposure to varying concentrations of cocaine (1–100 μM) for 3 h. Messenger RNA levels of GFAP were determined by real-time PCR. The rationale for using the chosen concentrations of cocaine is based on the premise that in human volunteers, cocaine concentrations in the plasma following intranasal administration of 1.5 mg/kg cocaine usually range from 0.4 to 1.6 μM [25], while plasma cocaine concentrations in tolerant abusers reach levels of up to 13 μM [26]. Additionally, cocaine concentrations of up to 100 μM and higher have been reported in postmortem brains of chronic human cocaine users following acute intoxication [27]. As shown in Fig. 2a, GFAP mRNA was maximally upregulated in A172 cells exposed to 10 μM cocaine. A concentration of 10 μM cocaine was thus used for all the subsequent experiments.

Cocaine induced upregulation of GFAP expression in human A172 cells and mice primary astrocytes. a Cocaine-mediated induction of GFAP mRNA expression by real-time RT-PCR. Cells were incubated with various concentrations of cocaine (1–100 μM) for 12 h and processed for RNA isolation and real-time PCR. b Time course of cocaine-mediated induction of GFAP expression in A172 cells. c Time course of cocaine-mediated induction of GFAP expression in mouse primary astrocytes. d Representative image of GFAP staining in mouse primary astrocytes. Scale bar 20 μm. All data are presented as mean ± SEM of four individual experiments. *p < 0.05, **p < 0.01 vs control group
Having determined that cocaine mediated increased expression of GFAP mRNA, we next examined whether the mRNA upregulation translated into increased protein expression. A172 cells were treated with cocaine for various time points (1 to 24 h), followed by cell lysis and subjected to immunoblotting. As shown in Fig. 2b, cocaine time-dependently upregulated GFAP protein expression with maximal expression at 12 h post-cocaine treatment in A172 cells. To further confirm the result in mouse primary astrocytes, cells were serum-starved overnight followed by treatment with 10 μM cocaine for 1 to 24 h. As shown in Fig. 2c, cocaine exposure resulted in induction of GFAP in mouse primary astrocytes. Confirmation of this finding by immunostaining also revealed increased GFAP expression with a characteristic alteration in the morphology of primary astrocytes following 10 μM cocaine treatment for 6 h (Fig. 2d). As is evident in the representative image, cocaine-treated astrocytes displayed more hypertrophic looking cells with larger cell bodies and increased cellular size. Cumulatively, these data clearly demonstrated cocaine-mediated induction of GFAP both at the mRNA and protein levels in astrocytes.
Cocaine-Induced Activation of MAPK (ERK, JNK, p38) Pathways
To better understand how cocaine modulated GFAP expression in astrocytes, we next sought to elucidate the signaling pathways involved in this process. Since mitogen-activated protein kinase (MAPK) pathways play critical roles in cocaine-mediated signaling [28, 29], the next logical step was to examine the role of these pathways in cocaine-mediated induction of GFAP. As shown in Fig. 3a, treatment of A172 cells with 10 μM cocaine resulted in time-dependent increase in phosphorylation of ERK, JNK, p38 kinases. Further, using a pharmacological approach, we were able to confirm activation of the respective signaling pathways following cocaine treatment. Pretreatment of cells with either the MEK (U0126), JNK (SP600125), or p38 (SB203580) inhibitor(s) followed by treatment with 10 μM cocaine for 30 min resulted in amelioration of cocaine-mediated induction of ERK, JNK, and p38 (Fig. 3b–d), respectively.

MAPK pathways are activated during cocaine exposure. a Treatment of A172 cells with cocaine resulted in increased phosphorylation of ERK, JNK, and p38 kinases. b Pretreatment of cells with ERK pathway inhibitor U0126 (5 μM) abrogated cocaine-mediated phosphorylation of ERK. c Pretreatment of cells with p38 pathway inhibitor SB203680 (10 μM) decreased the phosphorylation of p38. d Pretreatment of cells with JNK pathway inhibitor SP600125 (10 μM) abrogated cocaine-mediated phosphorylation of JNK. All data are presented as mean ± SEM of four individual experiments. **p < 0.01 vs control group; # p < 0.05 vs cocaine-treated group
Involvement of Egr-1 in Cocaine-Mediated Upregulation of GFAP in Astrocytes
Since acute cocaine administration has been shown to elicit rapid and transient induction of several immediate-early genes in the brain, including Egr-1, c-fos, and junB [30] and Egr-1 has been suggested to function as a molecular switch that coordinates changes in GFAP expression [7], we sought to examine whether cocaine-mediated induction of GFAP expression involved activation of Egr-1 in astrocytes. As shown in Fig. 4a, b, cocaine upregulated expression of Egr-1 in a time-dependent manner with peak expression elicited between 1 and 3 h and a decline thereafter, both in A172 cells and in mouse primary astrocytes. This was further validated by immunostaining A172 cells for Egr-1 expression. As shown in Fig. 4c, cells treated with 10 μM cocaine for 15 min demonstrated increased immunostaining for Egr-1 compared with untreated cells.

Involvement of Egr-1 in cocaine-mediated upregulation of GFAP. Cocaine induced time-dependent activation of Egr-1 expression in lysates from a A172 cells and b mouse primary astrocytes. c Representative images of Egr-1 staining in mouse primary astrocytes. Scale bars 20 μm. d Western blot demonstrating the knockdown efficiency of Egr-1 in A172 cells. e Egr-1 siRNA but not non-specific siRNA inhibited cocaine-mediated induction of GFAP. f DN-Egr-1 but not WT-Egr-1 inhibited cocaine-mediated induction of GFAP. All data are presented as mean ± SEM of three individual experiments. *p < 0.05, **p < 0.01 vs control group
We next examined the role of Egr-1 in cocaine-mediated induction of GFAP using the gene silencing approach. Briefly, A172 cells were transfected with either the siRNA for Egr-1 or a scrambled construct and assessed for Egr-1 knockdown. As shown in Fig. 3d, transfection of A172 with Egr-1 siRNA resulted in efficient knockdown of Egr-1 protein as evidenced by Western blotting. Intriguingly, Egr-1 siRNA significantly abrogated cocaine-mediated upregulation of GFAP expression (Fig. 4e). To further validate the involvement of the Egr-1 in cocaine-induced regulation of GFAP, cells were transfected with either WT or DN constructs of Egr-1, followed by exposure with 10 μM cocaine for 12 h. Cocaine-mediated induction of GFAP was attenuated in cells transfected with the DN-Egr-1 construct but not in cells transfected with the WT-Egr-1 construct (Fig. 4f). Together, these data clearly underscore the role of Egr-1 in cocaine-mediated induction of GFAP.
MAPK (ERK, JNK, p38) Pathways are Involved in Cocaine-Mediated Egr-1 and GFAP Expression in Astrocytes
Having determined the activation of ERK, JNK, and p38 MAPKs in cocaine-treated A172 cells, the next logical step then was to examine whether there existed a link that could tie together the activation of ERK1/2, JNK, and p38 MAPKs with Egr-1. Similar to the studies on signaling molecules described above, A172 cells were pretreated with inhibitors specific for the respective signaling pathways prior to stimulation with cocaine and assessed for expression of GFAP. As shown in Fig. 5a, all three MAPK pathways were demonstrated to be involved in cocaine-induced activation of Egr-1. Next, we wanted to address the functional role of MAPKs in cocaine-mediated induction of GFAP expression. Briefly, A172 cells were pretreated with the respective MAPK inhibitors followed by exposure to cocaine and assessed for expression of GFAP by immunoblotting. As shown in Fig. 5b, all the three pharmacological inhibitors abrogated cocaine-mediated induction of GFAP expression. Collectively, these findings thus confirmed the involvement of ERK, p38 as well as the JNK MAPK cascade in cocaine-mediated induction of GFAP in A172 cells.

ERK1/2, JNK, and p38 pathways are involved in cocaine-mediated upregulation of GFAP expression in astrocytes. a Pharmacological inhibition of ERK1/2 and JNK pathways by MEK (U0126—5 μM), JNK (SP600125—10 μM), and p38 (SB203580—10 μM) inhibitors resulted in amelioration of cocaine-mediated induction of Egr-1. b Pretreatment of cells with MEK (U0126—5 μM), JNK (SP600125—10 μM), and p38 (SB203580—10 μM) inhibitors resulted in amelioration of cocaine-mediated induction of GFAP. All data are presented as mean ± SEM of three individual experiments. **p < 0.01, ***p < 0.001 vs control group; # p < 0.05 vs cocaine-treated group
Cocaine-Mediated Sctivation of Sigma-1 Receptor (σ-1R) and its Translocation to the Lipid Rafts
Several studies have demonstrated the translocation of ER residing σ-1R following stimulation with ligands such as cocaine and (+) pentazocin [31]. It has been shown that upon ligand binding, σ-1Rs translocate from the mitochondrial ER membrane (MAM) to other areas of the cell including the extended ER reticular network, the plasmalemmal area, or the plasma membrane lipid raft domains where they interact and regulate the function of a variety of ion channels, receptors, or kinases [19]. To examine the role of σ-1R in cocaine-mediated induction of GFAP, we first examined whether A172 cells did indeed express σ-1R. As shown in Fig. 5a, these cells expressed both the σ-1R RNA (Fig. 6a) and protein (Fig. 6b) as demonstrated by RT-PCR and immunoblotting, respectively. To assess the translocation of σ-1R following treatment with cocaine, stimulated A172 cells were lysed and fractionated by ultracentrifugation for isolation of the lipid-rich domains of the plasma membrane. As shown in Fig. 6c, there was a significant increase in σ-1R levels in the lipid raft domain of cocaine-treated cells compared with the untreated cells. Lipid raft domain was characterized by the GM-1 marker. Further validation of this phenomenon was confirmed by immunofluorescence. Briefly, A172 cells were treated with cocaine and immunostained using antibodies specific for GM-1 and σ-1R. The overlay images indicated that cocaine treatment resulted in translocation of σ-1R to the lipid raft with its colocalization with the GM-1-rich microdomains. Untreated cells exhibited a diffuse pattern of σ-1R staining in the cytoplasm (Fig. 6d).

Cocaine exposure results in translocation of σ-1R to lipid-rich domains within the astrocytes. a RT-PCR and Western blotting demonstrating the expression of σ-1R in both human and primary mice astrocytes. b Detergent-soluble and detergent-resistant membrane fractions were separated using density gradient ultracentrifugation. Western blot analysis of the lysates from two fractions demonstrated translocation of the σ-1R to the lipid raft fractions (GM-1-rich region) compared with lysates from untreated cultures. c Representative image of immunofluorescence staining demonstrating increased colocalization of the σ-1R with the lipid raft GM-1-rich microdomains. Untreated cells exhibited a diffuse pattern of σ-1R staining in the cytoplasm. Scale bar 20 μm
Engagement of σ-1R is Critical for Cocaine-Mediated Induction of GFAP Expression
Having determined that cocaine activated σ-1R and triggered its translocation to the lipid raft microdomains, we next sought to assess the role of σ-1R in cocaine-mediated induction of GFAP in astrocytes. To examine the involvement of σ-1R in this process, we used σ-1R antagonist BD1047 (10 mM) to block its function. As shown in Fig. 6, cocaine-mediated induction of Egr-1 (Fig. 7a) and GFAP (Fig. 7b) was significantly attenuated in cells pretreated with the known σ-1R antagonist BD1047. To further validate the role of σ-1R in cocaine-mediated regulation of GFAP expression, we used the knockdown approach by transfecting A172 cells with either the σ-1R or the scrambled siRNA construct. As shown in Fig. 7c, d, σ-1R siRNA transfected cells exhibited significant abrogation of cocaine-mediated induction of Egr-1 as well as GFAP, thereby underscoring the role of σ-1R in this process.

σ-1R is critical for cocaine-induced GFAP expression. Pretreatment of A172 with the σ-1R antagonist BD1047 inhibited cocaine-mediated induction of Egr-1 expression (a) and GFAP expression (b). σ-1R siRNA but not non-specific siRNA inhibited cocaine-mediated induction of Egr-1 (c) and GFAP (d) expression. All the data are presented as mean ± SEM of four individual experiments. *p < 0.05, **p < 0.01 vs control group; # p < 0.05, ## p < 0.01 vs cocaine-treated group
Cocaine-Mediated Induction of GFAP In Vivo Involves Activation of σ-1R
To validate the role of cocaine/σ-1R axis in cocaine-mediated induction of GFAP in vivo, we resorted to treating mice with cocaine in the presence or absence of the σ-1R antagonist BD1047. Briefly, mice were divided into three groups—(a) saline; (b) cocaine; and (c) BD1047 and cocaine treated. For the BD1047-treated group, mice were pretreated for 2 days with IP injection of BD1047 (3.5 μg/kg) prior to cocaine administration, which was given daily at a concentration of 20 mg/kg for 7 days. As expected from our cell culture studies, cocaine administration in the cortex resulted in increased expression of both Egr-1 and GFAP compared with saline-injected mice in the same region, as demonstrated by immunohistochemistry, and this effect was ameliorated in mice pretreated with BD1047 (Fig. 8a). To further confirm our findings, cortical regions were isolated from the three groups of mice and homogenates from these regions were assessed for expression of both Egr-1 and GFAP by Western blot. As shown in Fig. 8b, cocaine-administered mice demonstrated increased expression of both Egr-1 and GFAP in brain tissues compared with the saline group. Mice pretreated with BD1047 demonstrated attenuated expression of both Egr-1 and GFAP compared with the cocaine-treated animals.

Cocaine-mediated activation of Egr-1 expression and GFAP upregulation involves σ-1R activation in vivo. a Cocaine administration resulted in increased expression of GFAP and Egr-1 (middle panels) compared with saline-injected animals (top panels). Pretreatment of the mice with σ-1R antagonist BD1047 followed by cocaine administration (bottom panels) failed to upregulate both GFAP and Egr-1 expressions. Scale bar 20 μm. b Western blotting demonstrating that cocaine administration to mice resulted in increased expression of Egr-1 and GFAP in brain homogenates and that this effect of cocaine was ameliorated in mice pretreated with the σ-1R antagonist BD1047. All the data are presented as mean ± SEM of four individual experiments. *p < 0.05, **p < 0.01 vs control group; # p < 0.05 vs cocaine-treated group
Discussion
Although antiviral therapies have had a profound impact on controlling systemic HIV-1 load, the poor penetrative property of these anti-retrovirus drugs coupled with increased longevity of infected individuals makes the brain a sanctuary for virus-induced toxicity, thereby leading to increased prevalence of HAND [2]. Adding fuel to this problem is the increased use of illicit drug abuse that is also a common problem in HIV-1-infected individuals [32]. Intriguingly, increased use of the psychostimulant cocaine in HIV-1-infected individuals has been linked to increased progression of HAND [4, 6]. One of the hallmark features of HAND is reactive astrocytosis, exemplified by enhanced GFAP expression, cellular hypertrophy, and astrocyte dysfunction [1]. While the role of HIV proteins such as Tat and GP120 in mediating astrocytosis is well characterized, very few studies have explored the detailed mechanism(s) by which cocaine triggers astrocyte activation exemplified by increased expression of GFAP expression and leading to enhanced cognitive decline and disease pathogenesis in cocaine abusers.
Astrocyte activation is a finely tuned process of progressive change encompassing changes in gene expression and cellular morphology that are subtly regulated by complex intercellular and intracellular signaling pathways [33]. The extent of various molecular changes involved in this process is not well characterized. Upregulation of the intermediate filament proteins, in particular GFAP, by reactive astrocytes is perhaps the best-known hallmark feature of reactive astrogliosis [33, 34]. Since cocaine abuse has been associated with glial activation, we undertook the present study to explore the molecular pathways involved in cocaine-mediated activation of astrocytes. Herein, we report that cocaine exposure of both human astrocyte cell line and primary mouse astrocytes resulted in both transcriptional and translational induction of GFAP. These findings are in agreement with previous in vivo report indicating the induction of GFAP expression following acute cocaine injection in mice [9]. Intriguingly, using human brain cortical sections from HIV+/cocaine drug abusers, we found prominent upregulation of GFAP compared with either the HIV+ or the control-uninfected groups. These findings add further validity to previous reports [22, 35] indicating cocaine-mediated exacerbation of astrocyte activation and the ensuing neuroinflammatory responses in the context of HIV infection.
σ-1R are unique drug-binding proteins that are present in the CNS, as well as in the periphery [36]. Cocaine is known to exhibit moderate affinity for σ-1R which are expressed in most neuronal cells [14]. The role of σ-1R in cocaine-induced immune alteration and HIV expression has been the focus of many studies, for example, findings from Gekker et al. demonstrated that σ-1R antagonist BD1047 blocked cocaine-stimulated increase of HIV-1 expression in microglial cells [18]. Moreover, σ-1R activation via alteration of lipid components is known to cause a remodeling of plasma membrane lipid rafts, thereby facilitating signal transduction triggered by growth factors and other mediators [19, 31]. In agreement with these findings, our data also demonstrated cytoplasmic localization of σ-1R in untreated cells, with a translocation of the receptor to the lipid raft domains of the plasma membrane following cocaine exposure. Our data implicated that in addition to inflicting addictive properties [15, 37], activation of σ-1R in the brain may also be involved in the development of cocaine-induced activation of astrocytes and that, this in part, could be a possible mechanism of increased neuropathogenesis observed in HIV-infected cocaine abusers.
Downstream of σ-1R activation, we also demonstrated cocaine-mediated activation of various signaling pathways, which ultimately led to increased expression of GFAP in astrocytes, and further that, this effect of cocaine was significantly abrogated in cells pretreated with either BD1037 or transfected with σ-1R siRNA. Cocaine-mediated activation of MAPK signal transduction has been documented in reactive astrocytes in diverse animal models of CNS pathogenesis [28, 38]. A recent study also demonstrated a functional link between JNK activation and GFAP accumulation in astrocytes [39]. Consistent with these findings, using both pharmacologic and genetic approaches, we also demonstrated activation of ERK1/2, JNK, and p38 MAPK pathways in astrocytes exposed to cocaine.
The zinc finger family of transcription factors, of which Egr-1 is a member, undergo a rapid and transient activation following growth factor(s) and/or environmental stimuli including hypoxia, physical force, or vascular injury [23]. Previous studies have suggested a correlation between increased activation of Egr-1 to increased expression of pivotal regulators of inflammation such as cytokines (IL-1β), chemokines (MIP-2, MCP-1) as well as GFAP expression [40, 41]. Our findings have demonstrated a time-dependent upregulation of Egr-1 expression in astrocytes treated with cocaine. Further validation of the role of Egr-1 involved both the pharmacological and genetic approaches. These findings suggested that cocaine-mediated activation of ERK and JNK MAPK pathways were upstream of Egr-1. Interestingly, our findings are in agreement with a report from Fan et al. demonstrating that HIV Tat-mediated astrogliosis involves activation of Egr-1 and transcriptional upregulation of GFAP [7]. It is likely that Egr-1 is one of the common signaling molecules critical for both Tat and cocaine-mediated activation of astrocytes.
In summary, our findings have demonstrated a detailed molecular pathway of cocaine-mediated induction of GFAP in astrocytes both in vitro and in vivo. Our findings suggest that in astrocytes, ligation of σ-1R following exposure of cocaine triggers its translocation to the plasma membrane, leading to activation of the MAPK pathway cascade with the downstream activation of the transcription factor Egr-1, which, in turn, transcriptionally upregulates GFAP expression. Our finding of increased astrogliosis in brain sections of patients with HIV and cocaine abuse compared with the HIV-infected group further corroborates the role of cocaine in activating cellular pathways leading to exacerbated pathology of HAND. These findings are in agreement with those reporting the role of HIV protein Tat in upregulating GFAP in astrocyte [7]. It is intriguing that drugs of abuse such as cocaine can hijack the host signaling response similar to the virus/viral proteins. These findings have clinical ramifications for the development of therapeutic strategies aimed at dampening astrogliosis as a treatment modality for cocaine abusers with or without HIV infection.
Reference
Sacktor N, Lyles RH, Skolasky R, Kleeberger C, Selnes OA, Miller EN, Becker JT, Cohen B, McArthur JC (2001) HIV-associated neurologic disease incidence changes: Multicenter AIDS Cohort Study, 1990-1998. Neurology 56(2):257–260
Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE (2005) Influence of HAART on HIV-related CNS disease and neuroinflammation. J Neuropathol Exp Neurol 64(6):529–536
Albright AV, Soldan SS, Gonzalez-Scarano F (2003) Pathogenesis of human immunodeficiency virus-induced neurological disease. J Neurovirol 9(2):222–227. doi:10.1080/13550280390194073
Purohit V, Rapaka R, Shurtleff D (2011) Drugs of abuse, dopamine, and HIV-associated neurocognitive disorders/HIV-associated dementia. Mol Neurobiol 44(1):102–110. doi:10.1007/s12035-011-8195-z
Grassi MP, Perin C, Clerici F, Zocchetti C, Borella M, Cargnel A, Mangoni A (1997) Effects of HIV seropositivity and drug abuse on cognitive function. Eur Neurol 37(1):48–52
Goodkin K, Shapshak P, Metsch LR, McCoy CB, Crandall KA, Kumar M, Fujimura RK, McCoy V, Zhang BT, Reyblat S, Xin KQ, Kumar AM (1998) Cocaine abuse and HIV-1 infection: epidemiology and neuropathogenesis. J Neuroimmunol 83(1–2):88–101
Fan Y, Zou W, Green LA, Kim BO, He JJ (2011) Activation of Egr-1 expression in astrocytes by HIV-1 Tat: new insights into astrocyte-mediated Tat neurotoxicity. J Neuroimmune Pharmacol 6(1):121–129. doi:10.1007/s11481-010-9217-8
Stanley LC, Mrak RE, Woody RC, Perrot LJ, Zhang S, Marshak DR, Nelson SJ, Griffin WS (1994) Glial cytokines as neuropathogenic factors in HIV infection: pathogenic similarities to Alzheimer's disease. J Neuropathol Exp Neurol 53(3):231–238
Fattore L, Puddu MC, Picciau S, Cappai A, Fratta W, Serra GP, Spiga S (2002) Astroglial in vivo response to cocaine in mouse dentate gyrus: a quantitative and qualitative analysis by confocal microscopy. Neuroscience 110(1):1–6
Eddleston M, Mucke L (1993) Molecular profile of reactive astrocytes—implications for their role in neurologic disease. Neuroscience 54(1):15–36
Haile CN, Hiroi N, Nestler EJ, Kosten TA (2001) Differential behavioral responses to cocaine are associated with dynamics of mesolimbic dopamine proteins in Lewis and Fischer 344 rats. Synapse 41(3):179–190. doi:10.1002/syn.1073
Bowers MS, Kalivas PW (2003) Forebrain astroglial plasticity is induced following withdrawal from repeated cocaine administration. Eur J Neurosci 17(6):1273–1278
Hemby SE (2006) Assessment of genome and proteome profiles in cocaine abuse. Prog Brain Res 158:173–195. doi:10.1016/S0079-6123(06)58009-4
Sharkey J, Glen KA, Wolfe S, Kuhar MJ (1988) Cocaine binding at sigma receptors. Eur J Pharmacol 149(1–2):171–174
Liu Y, Chen GD, Lerner MR, Brackett DJ, Matsumoto RR (2005) Cocaine up-regulates Fra-2 and sigma-1 receptor gene and protein expression in brain regions involved in addiction and reward. J Pharmacol Exp Ther 314(2):770–779. doi:10.1124/jpet.105.084525
Romieu P, Phan VL, Martin-Fardon R, Maurice T (2002) Involvement of the sigma(1) receptor in cocaine-induced conditioned place preference: possible dependence on dopamine uptake blockade. Neuropsychopharmacology 26(4):444–455. doi:10.1016/S0893-133X(01)00391-8
Roth MD, Whittaker KM, Choi R, Tashkin DP, Baldwin GC (2005) Cocaine and sigma-1 receptors modulate HIV infection, chemokine receptors, and the HPA axis in the huPBL-SCID model. J Leukoc Biol 78(6):1198–1203. doi:10.1189/jlb.0405219
Gekker G, Hu S, Sheng WS, Rock RB, Lokensgard JR, Peterson PK (2006) Cocaine-induced HIV-1 expression in microglia involves sigma-1 receptors and transforming growth factor-beta1. Int Immunopharmacol 6(6):1029–1033. doi:10.1016/j.intimp.2005.12.005
Yao H, Yang Y, Kim KJ, Bethel-Brown C, Gong N, Funa K, Gendelman HE, Su TP, Wang JQ, Buch S (2010) Molecular mechanisms involving sigma receptor-mediated induction of MCP-1: implication for increased monocyte transmigration. Blood 115(23):4951–4962. doi:10.1182/blood-2010-01-266221
Jouvert P, Revel MO, Lazaris A, Aunis D, Langley K, Zwiller J (2004) Activation of the cGMP pathway in dopaminergic structures reduces cocaine-induced EGR-1 expression and locomotor activity. J Neurosci 24(47):10716–10725. doi:10.1523/JNEUROSCI. 1398-04.2004
Hope B, Kosofsky B, Hyman SE, Nestler EJ (1992) Regulation of immediate early gene expression and AP-1 binding in the rat nucleus accumbens by chronic cocaine. Proc Natl Acad Sci U S A 89(13):5764–5768
Yang Y, Yao H, Lu Y, Wang C, Buch S (2010) Cocaine potentiates astrocyte toxicity mediated by human immunodeficiency virus (HIV-1) protein gp120. PLoS One 5(10):e13427. doi:10.1371/journal.pone.0013427
Khachigian LM, Lindner V, Williams AJ, Collins T (1996) Egr-1-induced endothelial gene expression: a common theme in vascular injury. Science 271(5254):1427–1431
Cayrol R, Wosik K, Berard JL, Dodelet-Devillers A, Ifergan I, Kebir H, Haqqani AS, Kreymborg K, Krug S, Moumdjian R, Bouthillier A, Becher B, Arbour N, David S, Stanimirovic D, Prat A (2008) Activated leukocyte cell adhesion molecule promotes leukocyte trafficking into the central nervous system. Nat Immunol 9(2):137–145. doi:10.1038/ni1551
Van Dyke C, Barash PG, Jatlow P, Byck R (1976) Cocaine: plasma concentrations after intranasal application in man. Science 191(4229):859–861
Stephens BG, Jentzen JM, Karch S, Mash DC, Wetli CV (2004) Criteria for the interpretation of cocaine levels in human biological samples and their relation to the cause of death. Am J Forensic Med Pathol 25(1):1–10
Kalasinsky KS, Bosy TZ, Schmunk GA, Ang L, Adams V, Gore SB, Smialek J, Furukawa Y, Guttman M, Kish SJ (2000) Regional distribution of cocaine in postmortem brain of chronic human cocaine users. J Forensic Sci 45(5):1041–1048
Fan L, Sawbridge D, George V, Teng L, Bailey A, Kitchen I, Li JM (2009) Chronic cocaine-induced cardiac oxidative stress and mitogen-activated protein kinase activation: the role of Nox2 oxidase. J Pharmacol Exp Ther 328(1):99–106. doi:10.1124/jpet.108.145201
Li G, Xiao Y, Zhang L (2005) Cocaine induces apoptosis in fetal rat myocardial cells through the p38 mitogen-activated protein kinase and mitochondrial/cytochrome c pathways. J Pharmacol Exp Ther 312(1):112–119. doi:10.1124/jpet.104.073494
Drago J, Gerfen CR, Westphal H, Steiner H (1996) D1 dopamine receptor-deficient mouse: cocaine-induced regulation of immediate-early gene and substance P expression in the striatum. Neuroscience 74(3):813–823
Hayashi T, Su TP (2003) Intracellular dynamics of sigma-1 receptors (sigma(1) binding sites) in NG108-15 cells. J Pharmacol Exp Ther 306(2):726–733. doi:10.1124/jpet.103.051292
Larrat EP, Zierler S (1993) Entangled epidemics: cocaine use and HIV disease. J Psychoactive Drugs 25(3):207–221
Sofroniew MV (2009) Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci 32(12):638–647. doi:10.1016/j.tins.2009.08.002
Laping NJ, Teter B, Nichols NR, Rozovsky I, Finch CE (1994) Glial fibrillary acidic protein: regulation by hormones, cytokines, and growth factors. Brain Pathol 4(3):259–275
Yao H, Allen JE, Zhu X, Callen S, Buch S (2009) Cocaine and human immunodeficiency virus type 1 gp120 mediate neurotoxicity through overlapping signaling pathways. J Neurovirol 15(2):164–175. doi:10.1080/13550280902755375
Walker JM, Bowen WD, Walker FO, Matsumoto RR, De Costa B, Rice KC (1990) Sigma receptors: biology and function. Pharmacol Rev 42(4):355–402
Sabino V, Cottone P, Blasio A, Iyer MR, Steardo L, Rice KC, Conti B, Koob GF, Zorrilla EP (2011) Activation of sigma-receptors induces binge-like drinking in Sardinian alcohol-preferring rats. Neuropsychopharmacology 36(6):1207–1218. doi:10.1038/npp.2011.5
Barber SA, Uhrlaub JL, DeWitt JB, Tarwater PM, Zink MC (2004) Dysregulation of mitogen-activated protein kinase signaling pathways in simian immunodeficiency virus encephalitis. Am J Pathol 164(2):355–362. doi:10.1016/S0002-9440(10)63125-2
Gadea A, Schinelli S, Gallo V (2008) Endothelin-1 regulates astrocyte proliferation and reactive gliosis via a JNK/c-Jun signaling pathway. J Neurosci 28(10):2394–2408. doi:10.1523/JNEUROSCI. 5652-07.2008
Beck H, Semisch M, Culmsee C, Plesnila N, Hatzopoulos AK (2008) Egr-1 regulates expression of the glial scar component phosphacan in astrocytes after experimental stroke. Am J Pathol 173(1):77–92. doi:10.2353/ajpath.2008.070648
Gashler A, Sukhatme VP (1995) Early growth response protein 1 (Egr-1): prototype of a zinc-finger family of transcription factors. Prog Nucleic Acid Res Mol Biol 50:191–224
Acknowledgments
This work was supported by grants DA020392, DA023397, and DA024442 from the National Institutes of Health.
Conflict of Interest
The authors declare no competing financial interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yang, L., Yao, H., Chen, X. et al. Role of Sigma Receptor in Cocaine-Mediated Induction of Glial Fibrillary Acidic Protein: Implications for HAND. Mol Neurobiol 53, 1329–1342 (2016). https://doi.org/10.1007/s12035-015-9094-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9094-5