
Abstract
Heterogeneous nuclear ribonucleoprotein A2/B1 (hnRNP A2/B1) plays an important role in influence of pre-messenger RNA (pre-mRNA) processing and mRNA metabolism and transportation in cells. Increasing evidence indicates that hnRNP A2/B1 played an important role in development and progression of various human cancers. Forty cases of normal and human glioma tissue samples were analyzed using immunohistochemistry to reveal the expression of hnRNP A2/B1 protein in the samples. Then, knockdown of hnRNP A2/B1 expression induced by RNA interference (RNAi) method was used to analyze the role of hnRNP A2/B1 in glioblastoma cell viability, adhesion, migration, invasion, and chemoresistance for temozolomide (TMZ). The data showed that hnRNP A2/B1 protein was overexpressed in glioma tissue specimens and associated with advanced glioma grades. Knockdown of hnRNP A2/B1 could reduce glioblastoma cell viability, adhesion, migration, invasion, and chemoresistance for TMZ capacity, but induced tumor cells to apoptosis and reactive oxygen species (ROS) generation in glioma U251 and SHG44 cells. Molecularly, hnRNP A2/B1 knockdown reduced expression of phospho-STAT3 and MMP-2. Detection of hnRNP A2/B1 expression may be useful as a biomarker for prediction of glioma progression and knockdown of hnRNP A2/B1 expression as a novel strategy in future control of glioblastoma in clinic.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Brain tumors are the most common malignancy and one of the leading cancer-related deaths in children and young adults, as showed by the American Brain Tumor Association (ABTA). Brain tumors can be benign or malignant and occur in different parts of the brain, which significantly affect quality of life because of their invasive and infiltrative character in the limited space of the intracranial cavity. Glioma, accounting for almost half of all primary brain tumors, is a lethal intracranial tumor arisen from glial cells. Gliomas are classified by cell type, by grade, and by location and further categorized according to their pathology grade [1, 2]. Despite recent advancement in aggressive surgery, radiation, and chemotherapies, the life expectancy of patients with glioblastoma multiform (GBM), the most frequently diagnosed malignant glioma (World Health Organization [WHO] grade IV), is approximately 14 months under the current standard of care [3]. Their diffuse invasion is regarded as one of the main barriers for treatment failure and tumor recurrence [4, 5].
To date, the major treatment strategies for glioblastoma typically consist of a gross total resection of the lesion, when feasible, followed by focal radiotherapy and systematical chemotherapy. Such therapies have been developed to improve surgical resection of completely surgical lesion removal, optimize radiotherapy with the beam to suit the irregular profiles, and use of temozolomide chemotherapy instead of carmustine for decrease in toxicity [6]. However, due to specific glioblastoma characteristics, these treatment advancements remain insufficient to reduce the morbidity and mortality of glioma. Thus, novel approaches are needed to improve the clinical outcome by increasing our understanding of the basic molecular pathogenesis of this lethal disease, which may include a more comprehensive view and study of the altered genes and pathways in glioma cells, the underlying molecular mechanisms responsible for alterations of the hallmark pathobiological features of this disease, the identification and evaluation of novel biomarkers for early detection and prediction of disease progression for glioma patients, and the novel therapeutic strategies for targeting keystone genetic lesions and their pathways in control of gliomas [2, 7].
Toward this end, heterogeneous nuclear ribonucleoproteins (hnRNPs) are a family of RNA-binding proteins that consist of approximately 20 major hnRNP proteins. They were involved in various aspects of messenger RNA (mRNA) key biogenesis [8, 9]. As a key member of the hnRNP family, hnRNP A2/B1 is a complex formed by protein B1 and A2 at a rational proportion [10]. hnRNP A/B regulates the splicing and transportation of mRNA in cells and plays a role in regulation of cell growth and carcinogenesis [11]. Previous studies have showed that hnRNP A2/B1 were overexpressed in lung cancer, which may be as a biomarker for early detection of lung cancer [12], while up-regulation of hnRNP A2/B1 protein may be a candidate gene for screening of early pancreatic cancer and gastric adenocarcinoma [13]. Knockdown of hnRNPA1 and A2/B1 expression induced apoptosis of breast cancer cells [14]. hnRNP A2/B1 may play an important role in regulation of hypoxia, the common feature inherent to the biologic processes of embryogenesis and carcinogenesis [15]. hnRNP A2/B1 overexpression may probably save as a biomarker for prediction of survival of glioblastoma patients [16]. Thus, in this study, we first detected hnRNP A2/B1 expression in glioma tissue specimens and then knocked down its expression in two glioma cell lines to assess the changed cell phenotypes and events.
Materials and Methods
Human Tissue Samples
This study was approved by the medical review board of Chongqing Medical University (Chongqing, China). The data on clinical characteristics of patients were presented in Table 1. Thirty-two cases of human brain tissue samples were obtained from glioma patients who underwent surgical resection of glioma lesions and signed an informed consent form in the Department of Neurosurgery of The Second Affiliated Hospital of Chongqing Medical University. Eight cases of normal human brain tissues were also taken from the area of the peripheral tissues away from the tumor lesions or from nonneoplastic brain resected due to hemorrhage. Tissues were then snap-frozen in liquid nitrogen immediately and stored in liquid nitrogen until use.
Immunohistochemistry
The tissue sections were subjected to the standard SP immunohistochemistry procedures using the Histostain™-Plus kit (#SP-9002 from Zymed, CA, USA) according to the manufacturer’s instructions. The color development was followed as previously described [17], and the sections were then covered with cover slips in a neutral resin. The stained sections were then reviewed and scored by two pathologists. Images were acquired with a digital camera attached to a microscope (DM6000 B, Leica Microsystems Ltd. Germany), and the photographs were analyzed using the Image-Pro Plus 6.0 computer program provided by the Leica. In this research, all the lighting conditions were held constant across all imaging studies and the individual collecting and analyzing the photomicrographs was blinded to treatment condition.
Cell Lines and Culture
Human malignant glioma U251 and SHG44 cell lines were obtained from KeyGen Biotech (glioblastoma multiforme, WHO grade IV; NanJing KeyGen Biotech Co., Ltd., Nanjing, China.). They were cultured in the recommended medium supplemented with 10 % fetal bovine serum (FBS) in a 5 % CO2 incubator at 37 °C. The medium was replaced every 2–3 days with a complete medium, and subcultures were performed when cells in monolayer reached confluence. Cell morphology was examined using an inverted microscope (NIKON TE2000-U, Tokyo, Japan). All culture media and supplements above were obtained from Hyclone (Thermo Fisher Scientific (China) Co., Ltd., Beijing, China). Normal human astrocytes (NHAs) were maintained in astrocyte growth medium (AGM). The NHAs and AGM were obtained from Lonza Group Ltd. (Basel, Switzerland).
Immunofluorescence
Cells were seeded on cover slips precoated with polylysine in a 6-well plate. The cells were fixed in 4 % paraformaldehyde prepared in PBS for 30 min, permeabilized in 0.5 % Triton X-100 prepared in PBS for 5 min and incubated in blocking buffer (5 % goat serum prepared in PBS) at 37 °C for 30 min. Then, they were incubated in blocking buffer containing mouse anti-hnRNP A2/B1 antibody (Santa Cruz, CA, USA; 1:100) at 4 °C overnight followed by incubation in blocking buffer containing Cy3-conjugated goat anti-mouse antibody (Beyotime Institute of Biotechnology; 1:100), FITC-conjugated goat anti-mouse antibody (Beyotime Institute of Biotechnology; 1:100), and DAPI (Santa Cruz, CA, USA; 50 ng/ml) at 37 °C for 1 h. All the wash buffer used before every step was PBS. Then all the results were detected by a fluorescence microscope (Nikon TE2000-U, Tokyo, Japan).
Stable Gene Transfection
To knockdown of hnRNP A2/B1 expression, tumor cells were grown overnight and then transfected with an hnRNP A2/B1 small hairpin RNA (shRNA) vector using a FuGENE® HD transfection reagent (Hoffmann-La Roche Ltd., Germany) according to the manufacturer’s instructions. Forty-eight hours after that, the cells were maintained in a complete medium containing puromycin dihydrochloride from Sigma-Aldrich Co. (St Louis, MO, USA; 2 μg/ml for U251 cell lines and 4 μg/ml for SHG44) for 2 weeks and then subcloned the individual puromycin-resistant cells to 96-well plates and expanded the cells into puromycin-resistant sublines. The stable sublines transfected with hnRNP A2/B1-specific pLKO.1-shRNA vector (H1 sense primer: 5′-CCGGCAGAAATACCATACCATCAATCTCGAGATTGATGGTATGGTATTTCTGTTTTTG-3′, H1 antisense primer: 5′-AATTCAAAAACAGAAATACCATACCATCAATCTCGAGATTGATGGTATGGTATTTCTG-3′; H2 sense primer: 5′-CCGGAGAAGCTGTTTGTTGGCGGAACTCGAGTTCCGCCAACAAACAGCTTCTTTTTTG-3′, H2 antisense primer: 5′-AATTCAAAAAAGAAGCTGTTTGTTGGCGGAACTCGAGTTCCGCCAACAAACAGCTTCT-3′; H3 sense primer: 5′-CCGGTGACAACTATGGAGGAGGAAACTCGAGTTTCCTCCTCCATAGTTGTCATTTTTG-3′, H3 antisense primer: 5′-AATTCAAAAATGACAACTATGGAGGAGGAAACTCGAGTTTCCTCCTCCATAGTTGTCA-3′) were identified as U251-H1, U251-H2, U251-H3, SHG44-H1, SHG44-H2, and SHG44-H3, respectively.
RNA Isolation and Quantitative Reverse Transcription-PCR
Total RNA was isolated from cell lines using a TRIzol reagent (TaKaRa, Dalian, China) according to the instructions. The concentration of these RNA samples was then measured using a spectrophotometer at 260 and 280 nm (A260/280), and the RNA samples were reverse-transcribed into cDNA using a PrimeScript™ RT reagent Kit (TaKaRa). qPCR amplification was performed in a Roche-480 with a SYBR Green RT-PCR kit (TaKaRa), and qPCR conditions were an initial 95 °C for 3 min and then 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The primers for hnRNP A2B1 were 5′-AGCGACTGAGTCCGCGATGGA-3′ and 5′-GCAGGATCCCTCATTACCACACAGT-3′ and primers for 18 s RNA as an internal control. The experiments were performed in triplicate and repeated at least twice. The amount of target was calculated by the 2-ΔΔCT method using and Light Cycle 480 SW1.5 software (Roche).
Protein Extraction and Western Blot
The tissue and cultured cells were lysed in a RIPA Lysis Buffer (P0013B, Beyotime Institute of Biotechnology, Beijing, China) containing phenylmethanesulfonyl fluoride (PMSF, ST506, Beyotime Institute of Biotechnology) according to the manufacturer’s instructions. The protein samples were quantified using a Pierce® BCA Protein Assay Kit (Thermo Scientific, USA). The equal amount protein samples (50 μg) were separated by SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes, then probed with individual antibodies according to the instruction book. After that, the protein bands were revealed by a developing reagent (P0019, Beyotime Institute of Biotechnology) and photographed by a chemiluminescence imaging system (ChemiDoc XRS+, Bio-Rad, USA). The amount of protein in each band was then quantified using the Quantity One 4.6 computer software. The results were normalized as a ratio of hnRNP A2/B1 to internal reference using the Gaussian Model Trace.
The primary antibodies used were rabbit monoclonal anti-STAT3 (Epitomics, Inc. CA, USA; at a dilution of 1:1000), rabbit anti-phospho-STAT3 (Epitomics; 1:1000), mouse anti-hnRNP A2/B1 (Santa Cruz, CA, USA; 1:200), mouse anti-MMP-2 (Santa Cruz Biotechnology; 1:200), mouse anti-MMP-9 (Santa Cruz Biotechnology; 1:200), rabbit anti-β-actin (Epitomics; 1:1000), and rabbit anti-GAPDH (Epitomics; 1:1000). The secondary antibodies used were goat anti-rabbit IgG (MultiSciences, 1:10,000) and goat anti-mouse IgG (MultiSciences, 1:10,000). The wash buffers were PBS and PBS-T (Tween-20, 1:2000).
Cell Viability Assay
The cell viability was determined by the water-soluble tetrazolium-1 (WST-1, Hoffmann-La Roche Ltd., Germany). In brief, cells were seeded at a density of 1 × 104 cells/well and cultured in 96-well plates containing 0.2 ml culture medium and 10 % FBS for up to 96 h. At the experiments, WST (10 μl/well) was added and incubated for 4 h. The cell viability was then determined by measuring the absorbance at 450 nm in a Tecan F039300 Sunrise Microplate Reader (Tecan (Shanghai) Trading Co., Ltd. Shanghai, China). Cell growth curves were drawn from the mean value of three wells with repeated experiments. And for anticancer agent sensitivity assay, cells (1 × 104 cells/well) were plated in 96-well plates and cultured overnight. After 24-h treatment with an anticancer agent TMZ (Sigma), cell viability was determined as described above.
Tumor Cell Colony Formation Assay
For colony formation assays, cells (500/well) were seeded in triplicate on 6-well plates containing culture medium (2 ml/well) supplemented with 10 % FBS and puromycin dihydrochloride (2 μg/ml for stable transfected U251 cell lines and 4 μg/ml for stable transfected SHG44 cell lines). After 30 days, the colonies were washed with phosphate-buffered saline (PBS) for three times, fixed with 4 % paraformaldehyde for half an hour, stained with crystal violet for half an hour, and then washed by distilled water. Cell colonies with more than 50 cells in size were counted.
For colony formation assay in soft agar, the 6-well plates were first poured with 5 % base agar (1 ml/well) and solidified for 2 h. Next, 1 ml of 0.3 % agar containing with a complete medium and stable cell sublines (500/ml) was layered onto the surface of the bottom agar. After incubated for 30 days with refed medium every 3 days, the cell colonies (>50 μm) were photographed and counted. The experiments were repeated twice.
Flow Cytometric Assay to Detect Cell Cycle and Apoptosis and Reactive Oxygen Species
The stable sublines were seeded and grown in a 6-well plate. When the cell reached approximately 85 % confluency, the cells were harvested and then washed twice with PBS, fixed in precooled 70 % ethanol overnight at 4 °C. In the next day, the cells were washed twice in PBS, and suspended in 500 μl PBS containing RNase A (50 μg/ml) and propidium iodide (PI; 50 μg/ml) and incubated for 30 min at 37 °C water bath. The cell cycle distribution was analyzed by using a FCM (FACSVantage SE, Becton Dickinson, USA).
To determine cell apoptosis, the same-treated cells were harvested and incubated with reagents from the Annexin V-FITC apoptosis kit (BioVision, Inc., CA, USA) according to the manufacturer’s protocol.
To quantitate reactive oxygen species (ROS) generation, cells were incubated in the dark with 10 μM DCFH-DA (Cellular Reactive Oxygen Species Detection Assay Kit, Applygen Technologies Inc., Beijing, China) for 60 min. After washing three times with PBS, cells were suspended and examined with flow cytometry.
All of the experiments were assayed in triplicate and repeated at least twice.
Tumor Cell Adhesion Assay
The 96-wells plates were first coated with 100 μl Matrigel and kept in a CO2 incubator for 1 h. Cells (2 × 104/well) were then seeded in these 96-well plates and incubated for 2 h at 37 °C in a CO2 incubator with 5 % CO2. After that, the unattached cells were washed away three times with PBS and WST (10 μl/well) was added into the cell culture and incubated for 4 h, and the absorbance was measured at 450 nm with a microplate reader.
Transwell Tumor Cell Invasion and Migration Assay
Transwell chambers (with 8 μm pore membranes, BD Biosciences, CA, USA) were used to analyze tumor cell invasion capacity in 24-wells plates. The chambers were coated with Matrigel (BD Biosciences, CA, USA) and reconstituted with serum-free medium for 24 h before the experiment. The stabled sublines containing 200 μl serum-free medium were plated in the upper chamber and the 600 μl medium supplemented with 10 % FBS was added to the lower chamber. Sixteen hours after incubation, cells on the top chambers of the Transwell were removed; cells on the lower surface of the membranes were fixed with 4 % paraformaldehyde for half an hour, stained with crystal violet for half an hour, and then washed by distilled water. The invaded cells were photographed under a light inverted microscope (Nikon TE2000-U, Tokyo, Japan). Thirty-three percent acetic acid was added to the 24-wells plate to destain the dyed invasive cells, and the quantitative value was measured by the absorbance at 570 nm on a microplate reader.
The membranes without Matrigel coating were used as tumor cell migration assay, and the cells were incubated for 12 h.
Statistical Analysis
The two-tailed unpaired Student’s t test was used to evaluate differences between two groups. For experiments involving multiple comparisons, one-way ANOVA and Tukey’s post hoc test were used to evaluate differences. The SPSS software was used in the statistical analysis. Data were considered to be statistically significant if p < 0.05 or p < 0.01 as appropriate.
Results
Overexpression of hnRNP A2/B1 Protein in Human Glioma
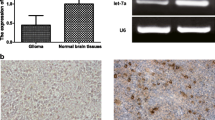
In this study, we first assessed the expression of hnRNP A2/B1 protein in human glioma tissue samples. As shown in Fig. 1a–f, expression of hnRNP A2/B1 protein in grade II–IV of glioma tissues was much higher than that of the normal brain tissues. Expression of hnRNP A2/B1 protein was associated with advanced tumor grade of gliomas. Next, we determined hnRNP A2/B1 expression in two glioma U251 and SHG44 cell lines and found that hnRNP A2/B1 was highly expressed in these two cell lines (Fig. 1g). And it is found that hnRNP A2/B1 hnRNP A2/B1 were located in the cellular nucleus from the immunofluorescence images (Fig. 1h). Thus, we stably transfected hnRNP A2/B1 shRNA into these two glioma U251 and SHG44 cell lines.

Immunohistochemistry detection of hnRNP A2/B1 expression in glioma tissue specimens. a hnRNP A2/B1 expression in normal brain samples (×400); b–e hnRNP A2/B1 expression in grade 1–4 clinic glioma samples (×400); f summarized data of all 40 cases in triplicate were analyzed in this study. g HnRNP A2/B1 protein expression in NHAs, glioma U251, and SHG44 cell lines. h Immunofluorescent staining of glioma cells for hnRNP A2/B1 (red in U251 and green in SHG44) and nuclei (blue). Error bars represent the standard error of the mean of quadruplicates. Compared with normal brain samples, *p < 0.01
RNAi on U251 and SHG44 Cells
Compared to the hnRNP A2/B1 expressions in the U251 and SHG44 cells, respectively, the hnRNP A2/B1 mRNA and protein expressions were significantly reduced in the stably hnRNP A2/B1 shRNA transfected glioma U251 and SHG44 cell sublines (U251-H1, U251-H2, U251-H3, SHG44-H1, and SHG44-H2) (Fig. 2). Because of plasmids transfection efficiency were different, the U251-H1 and SHG44-H1 transfected cells (hnRNP A2/B1 mRNA expression decreased >80 % and protein expression decreased >40 %) were finally selected as the target cells to research the effect of hnRNP A2/B1 knockdown on glioblastoma cells. The U251-H3 and SHG44-H3 transfected cells (hnRNP A2/B1 mRNA expression decreased <25 %, the protein expression decreased 13 % in U251-H3 cells, and the protein expression had no significant changes in SHG44-H3 cells) were excluded from this research. The U251-H2 and SHG44-H2 transfected cells (hnRNP A2/B1 mRNA expression decreased 50–65 % and protein expression decreased approximately 15–25 %) were used as the supplementary research cells of U251-H1 and SHG44-H1, respectively. There were no significant differences between U251 and U251-NC or SHG44 and SHG44-NC. The empty vector control cells U251-NC and SHG44-NC were used as the negative control groups.

Knockdown of hnRNP A2/B1 expression in glioma U251 and SHG44 cell lines. a qRT-PCR. The stable hnRNP A2/B1 knockdown U251 and SHG44 sublines were grown in cell culture dishes and subjected to qRT-PCR of hnRNP A2/B1 mRNA expression. Results are representative of five independent experiments. Compared with untransfected cell groups, *p < 0.01. b Western blot. The same cells were subjected to Western blot. Results are representative of three independent experiments. Error bars represent the standard error of the mean of quadruplicates. Compared with untransfected cell groups, *p < 0.05
Effects of hnRNP A2/B1 Knockdown on Regulation of Glioma Cell Viability, Colony Formation
Next, we assessed the effects of hnRNP A2/B1 knockdown on regulation of tumor cell viability and found that knockdown of hnRNP A2/B1 expression inhibited tumor cell viability. Specifically, compared to U251-NC and SHG44-NC cells, the growth rate of U251-H1 was reduced to 46 % (p < 0.05, Fig. 3) and SHG44-H1 to 38 % (p < 0.05, Fig. 3a) after 96-h incubation. Cell cycle analysis showed that hnRNP A2/B1 knockdown obviously increased percentage of tumor cells at G2/M phase (12.27 ± 1.397 % for U251-NC, 28.03 ± 8.794 % for U251-H1, 24.26 ± 7.244 % for SHG44-NC, and 63.71 ± 6.958 % for SHG44-H1; Fig. 3b). Flow cytometric apoptosis assay showed that apoptosis of U251-H1 (9.312 ± 2.705 %) and SHG44-H1 (9.67 ± 3.716 %) significantly increased compared to U251-NC (0.904 ± 0.3679 %) and SHG44-NC (0.942 ± 0.3710 %; Fig. 3c). Furthermore, the ROS generation was increased most significantly in U251-H1 and SHG44-H1 (Fig. 3d).

Effects of hnRNP A2/B1 knockdown on regulation of glioma cell subline viability. Cell cycle distribution and apoptosis. a Cell viability WST assay. The stable hnRNP A2/B1 knockdown U251 and SHG44 sublines were grown in cell culture dishes and subjected to WST assay. b Flow cytometric cell cycle distribution assay. c Flow cytometric apoptosis assay. d flow cytometric ROS generation assay. Results are representative of five independent experiments, respectively. Error bars represent the standard error of the mean of quadruplicates. Compared with the negative control groups, *p < 0.05
After that, we performed anchorage-dependent and -independent colony formation assays. The results of plate anchorage-dependent colony formation showed that there were much less colonies in U251-H1 and SHG44-H1 than in relevant negative contrast cells (Fig. 4a). The size of colonies was also smaller. Soft agar assay data showed that hnRNP A2/B1 knockdown exhibited a significant decrease in the number and size of colonies compared to control U251-NC and SHG44-NC cells (Fig. 4b).

Effects of hnRNP A2/B1 knockdown on regulation of glioma cell subline colony formation. a Anchorage-dependent colony formation assay. b Anchorage-independent soft agar colony formation assay. Results are representative of three independent experiments in duplicate. Error bars represent the standard error of the mean of quadruplicates. Compared with the negative control groups, *p < 0.01
Effects of hnRNP A2/B1 Knockdown on Regulation of Glioma Cell Adhesion, Migration, and Invasion Capacity
We next determined the effects of HnRNP A2/B1 knockdown on regulation of glioma cell adhesion, migration, and invasion capacity. From the quantitative data shown in Fig. 5a, we can conclude that U251-H1 and SHG44-H1 cell adhesive capacity was weaker than that of U251-NC and SHG44-NC cells. The number of cells that migrated across the polycarbonate membrane was also reduced after hnRNP A2/B1 knockdown (Fig. 5b), while tumor cell invasion was significantly inhibited in U251 and SHG44 infected cells (Fig. 5c).

Effects of hnRNP A2/B1 knockdown on regulation of glioma cell subline adhesion, migration, and invasion capacity. a Tumor cell adhesion assay. b Tumor cell migration assay. c Tumor cell invasion assay. Results are representative of five independent experiments. Error bars represent the standard error of the mean of quadruplicates. Compared with the negative control groups, *p < 0.01
Effects of hnRNP A2/B1 Knockdown on the TMZ Sensitivity of Glioma Cells
We investigated the cell survival rates of U251 and SHG44 cells after treatment with TMZ. The cell survival rates were evaluated at 24 h after treatment with 100 to 800 μmol/l TMZ. The survival rate of U251-NC was higher than the survival rate of U251-H1 cells after treatment with 100 to 400 μmol/l TMZ (Fig. 6a). We also evaluated apoptosis of hnRNP A2/B1 in U251 cells at 24 h after the treatment with 400 μmol/l TMZ and found that the decrease in hnRNP A2/B1 expression increased the number of apoptotic cells (Fig. 6b). Moreover, the survival rate of SHG44-H1 was lower than that of SHG44-NC cells after treatment with 100 to 400 μmol/l TMZ (Fig. 6c).

Effects of hnRNP A2/B1 knockdown on glioma cell chemoresistance. a Survival rate of U251-NC cells was higher than that of the U251-H1 cells after treatment with 100 to 400 μmol/l TMZ. b Apoptosis assay was done by flow cytometry using Annexin V and PI after 24 h the treatment of 400 μmol/l TMZ. U251-H1 cells increased the number of apoptotic cells compared with U251-NC cell. c Survival rate of SHG44-H1 cells was lower than that of SHG44-NC cells after treatment 100 to 400 μmol/l TMZ. Results are representative of five independent experiments
Effect of hnRNP A2/B1 Knockdown in Regulation of STAT3 and MMP Expression
To explore the molecular events after hnRNP A2/B1 knockdown, we determined the expression of cell growth and adhesion molecules STAT3 and matrix metalloproteinase (MMP). The data showed that phospho-STAT3 expression was significantly decreased in hnRNP A2/B1 knockdown cells, especially in U251-H1 and SHG44-H1 cells, whereas the total STAT3 did not have much changes in these cell lines (Fig. 7a). Expression of MMP-2 protein was decreased in hnRNP A2/B1 knocked down cells, especially in U251-H1 and SHG44-H1 cells, whereas MMP-9 expressed rarely in all these cell lines, and the results were not listed (Fig. 7b). Molecularly, hnRNP A2/B1 knockdown reduced expression of phospho-STAT3 and MMP-2, but had no effects on the total STAT3 and MMP-9.

Effects of hnRNP A2/B1 knockdown on regulation of STAT3 and MMP-2 expression in glioma cell sublines. a Western blot analysis of STAT3 expression in stable hnRNP A2/B1 knockdown U251 and SHG44 sublines. b Western blot analysis of MMP-2 expression in stable hnRNP A2/B1 knockdown U251 and SHG44 sublines. Results are representative of three independent experiments in duplicate. Error bars represent the standard error of the mean of quadruplicates. Compared with the negative control groups, *p < 0.05
Discussion
The hnRNP proteins A2 and B1 share many characteristics with the basic group of hnRNP protein family and have a similar overall structure consisting of two conserved amino terminal RNA-binding motifs (RBDs) linked to Gly-rich domains that contain an RGG box [18, 19] and a nuclear import/export signal termed M9 [20, 21]. A2 and B1 are produced by alternative splicing from a single copy gene and differ from each other only by a 12-amino-acid insertion in the N-terminal RBD of B1 [10]. hnRNP A2 and B1 proteins can form the core of the ribonucleoprotein complex and be named to hnRNP A2/B1 proteins [22]. They were among the most abundant pre-mRNA binding proteins of vertebrates in eukaryotic cells [23].
Recently, increasing evidence indicates that hnRNP A2/B1 played an important role in development and progression of various human cancers. For example, increased expression of hnRNP A2/B1 protein was observed in pancreatic cancer cells and lung cancer, which may be useful in detection of early stages of human cancers [12, 13]. In this current study, we found that hnRNP A2/B1 protein was overexpressed glioma tissue specimens and associated with advanced glioma grades. These data are consistent with the results published in a previous study [16], which demonstrated that hnRNP A2/B1 was upregulated in brain tumors, and expression and gene copy number of hnRNP A2/B1 were inversely correlated with survival of glioma patient, indicating that hnRNPA2B1 is a novel oncogene in glioblastoma and a potential new target for glioblastoma therapy.
Short hairpin RNA (shRNA) is one of the methods that can mediate the RNA interference (RNAi) effect. The knockdown approaches can be able to identify relevant biomolecular tumor network hubs as potential key targets [24, 25]. Thus, based on our ex vivo data on hnRNP A2/B1 overexpression in glioma tissue specimens, we use the shRNA approaches to knock down hnRNP A2/B1 expression in two glioma cell lines. Our data clearly demonstrated that knockdown of hnRNP A2/B1 mRNA and protein expressions inhibited U251 and SHG44 cell growth. More tumor cells were increased at G2/M phase of cell cycle, while apoptosis rate and ROS levels were also induced in hnRNP A2/B1-transfected glioma cell lines. Our current data support a previous study, in which hnRNP A2/B1 expression was required for glioblastoma cell transformation and tumorigenicity in vitro and in vivo [16].
Indeed, our current study revealed that hnRNP A2/B1 knockdown suppressed the anchor-dependent and -independent colony formation in glioma cells. All of our results supported the hypothesis that hnRNP A2/B1 expression is required for maintenance and proliferation of glioblastoma cells. Our further investigation showed that hnRNP A2/B1 knockdown inhibited cell adhesion, migration, and invasion capacity of glioma cells. These phenotypes are a multistep process of tumor metastasis in vivo. For example, tumor cell metastasis involves cell detachment from its original site, adhesion to extracellular matrix (ECM) proteins, proteolysis of the ECM, and migration through the disrupted matrix [26]. Thus, the cell adhesion to the ECM is critical for migration and invasion, especially in glioma cells [27]. Cell migration is the next step for glioma cell invasion, which is modulated sequentially by many factors, including actin-rich protrusions in the leading edge, cell adhesion, cell body translocation, and lagging edge detachment [28]. Our current data showed that hnRNP A2/B1 knockdown was able to suppress these steps, indicating that inhibition of hnRNP A2/B1 expression or activity could effectively inhibit glioma metastasis. Current standard-of-care therapy for GBM consists of surgical resection, followed by treatment with radiation therapy and the chemotherapeutic alkylating agent TMZ, the combination of which confers a median survival period of only 14.6 months [6]. Although most of the patients who benefit from TMZ have epigenetic silencing of the O6-methylguanine-DNA methyl-transferase (MGMT) DNA-repair gene by promoter methylation, tumors from approximately 55 % of GBM patients do not have methylation of the MGMT promoter and are relatively insensitive to TMZ [29]. Our current study finds that hnRNP A2/B1 suppression by shRNA increased cell death triggered by TMZ. Although further studies are needed to reveal the molecular mechanisms involved in these events, these results indicate that hnRNP A2/B1 knockdown increases the TMZ sensitivity of glioma cells.
The invasion of glioma cells into normal brain tissue is a key aspect of brain tumor pathology and the main cause of mortality in glioblastoma patients [29, 30]. Matrix metalloproteinases (MMPs), the zinc-dependent endopeptidases, can digest various components of the brain extracellular matrix and are essential mediators of glioblastoma tumor invasion [31]. Specifically, the main proteins MMP2 and MMP9 are crucial for glioma invasion, and their expression levels correlate directly with the pathologic grade of glioma [30, 32, 33]. In addition, our current study showed that hnRNP A2/B1 knockdown definitely inhibited high expression of MMP-2 protein in the two cell lines (MMP-9 rarely expressed in this study). These data was in accordance with other group’s [34], showing that expression of MMP-2 and, to a lesser extent, MMP-9 is associated with invasiveness, proliferation, and prognosis of astrocytoma cells. Thus, incorporation with previous studies of regulated cell phenotypes, hnRNP A2/B1, could mediate cell migration, invasion, and proliferation by regulation of MMP-2 protein expression in glioblastoma cells.
As an oncogenic transcription factor, STAT3 is a convergent point of many signaling pathways and has a major role in oncogenesis and tumor metastasis [35]. In contrast to normal cells, in which STAT3 tyrosine phosphorylation occurs transiently, it has been determined that STAT3 are persistently tyrosine phosphorylated (expression of pSTAT3, an indication for active STAT3 as a transcription factor) in most human malignancies [36, 37]. Numerous studies have revealed STAT3 to be required in many aspects of tumorigenesis and regulate the transcription of several genes that are involved in cell cycle progression, antiapoptosis, cell survival, and angiogenesis, such as cyclin D1, c-fos, c-Myc, pim-1, Bcl-XL, VEGF, and MMP-2 [38]. Based on these views, point efforts targeting it in anticancer drug development are proceeding [39, 40]. The presence of pSTAT3 in head and neck tumors can enhance tumor growth and invasion and may contribute to resistance to chemotherapy [37, 41]. STAT3 has also been shown to be constitutively activated in gliomas, and the finding that its constitutive activation as pSTAT3 is positively associated with glioma grade further suggests the potential pathologic significance of STAT3 activation in the aggressiveness of human gliomas [38, 42]. In this study, we found that the STAT3 activity was consistently activated in high expression of hnRNP A2/B1 protein cells. Knockdown of hnRNP A2/B1 expression was able to significantly decrease STAT3 protein phosphorylation, indicating that hnRNP A2/B1 protein most probably has an important role of activating STAT3 to promote cell proliferation, migration, and invasion in glioblastomas.
Not only that, Xie et al. [43] demonstrated that STAT3 regulates MMP-2 expression through direct interaction with the MMP-2 promoter via a STAT3-binding element or elements on the MMP-2 promoter. STAT3 signaling directly regulates MMP-2 expression, tumor invasion, and metastasis, and that STAT3 activation might be a crucial event in the development of metastasis [43].
Thus far, our current data and published data all support that hnRNP A2/B1 plays an important role in glioma development and progression, indicating that hnRNP A2/B1 is an oncogene in glioma [12, 16, 44]. A previous study demonstrated that oncogenic activity of hnRNP A2/B1 protein was to regulate several tumor suppressors and oncogenes [16]. In the present study, it is showed that knockdown of hnRNP A2/B1 could reduce glioblastoma cell viability, adhesion, migration, invasion, and chemoresistance for TMZ capacity, but induced tumor cells to apoptosis and ROS generation in highly invasive and metastatic glioblastoma cells. Blockade of hnRNP A2/B1 inhibited the activation of STAT3 and overexpression of MMP-2, and contributed to inhibit migration and invasion capacity and development and progression of glioblastoma cells. Other underlying molecular events by which hnRNP A2/B1 regulates glioblastoma cell phenotypes need to be investigated further. HnRNP A2/B1 was especially highly expressed in human glioma tissues and cell lines. Knockdown of hnRNP A2/B1 could reduce glioblastoma cell typical functions and phenotypes and regulate tumor-related signaling events. In conclusion, this study showed that hnRNP A2/B1 could be useful as a biomarker for prediction of glioma progression and target of hnRNP A2/B1 as a novel strategy for glioblastoma therapy.
References
Brat DJ, Prayson RA, Ryken TC, Olson JJ (2008) Diagnosis of malignant glioma: role of neuropathology. J Neurooncol 89(3):287–311
Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM et al (2007) Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev 21(21):2683–2710
Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY et al (2010) Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin 60(3):166–193
Giese A, Bjerkvig R, Berens ME, Westphal M (2003) Cost of migration: invasion of malignant gliomas and implications for treatment. J Clin Oncol 21(8):1624–1636
Prados MD, Levin V (2000) Biology and treatment of malignant glioma. Semin Oncol 27(3 Suppl 6):1–10
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B et al (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352(10):987–996
Ferla R, Haspinger E, Surmacz E (2012) Metformin inhibits leptin-induced growth and migration of glioblastoma cells. Oncol Lett 4(5):1077–1081
Dreyfuss G, Kim VN, Kataoka N (2002) Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol 3(3):195–205
Krecic AM, Swanson MS (1999) hnRNP complexes: composition, structure, and function. Curr Opin Cell Biol 11(3):363–371
Kozu T, Henrich B, Schafer KP (1995) Structure and expression of the gene (HNRPA2B1) encoding the human hnRNP protein A2/B1. Genomics 25(2):365–371
He Y, Brown MA, Rothnagel JA, Saunders NA, Smith R (2005) Roles of heterogeneous nuclear ribonucleoproteins A and B in cell proliferation. J Cell Sci 118(Pt 14):3173–3183
Fielding P, Turnbull L, Prime W, Walshaw M, Field JK (1999) Heterogeneous nuclear ribonucleoprotein A2/B1 up-regulation in bronchial lavage specimens: a clinical marker of early lung cancer detection. Clin Cancer Res 5(12):4048–4052
Yan-Sanders Y, Hammons GJ, Lyn-Cook BD (2002) Increased expression of heterogeneous nuclear ribonucleoprotein A2/B1 (hnRNP) in pancreatic tissue from smokers and pancreatic tumor cells. Cancer Lett 183(2):215–220
Patry C, Bouchard L, Labrecque P, Gendron D, Lemieux B et al (2003) Small interfering RNA-mediated reduction in heterogeneous nuclear ribonucleoparticule A1/A2 proteins induces apoptosis in human cancer cells but not in normal mortal cell lines. Cancer Res 63(22):7679–7688
Garayoa M, Man YG, Martinez A, Cuttitta F, Mulshine JL (2003) Downregulation of hnRNP A2/B1 expression in tumor cells under prolonged hypoxia. Am J Respir Cell Mol Biol 28(1):80–85
Golan-Gerstl R, Cohen M, Shilo A, Suh SS, Bakacs A et al (2011) Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res 71(13):4464–4472
Deng J, Huang Q, Wang F, Liu Y, Wang Z et al (2012) The role of caveolin-1 in blood–brain barrier disruption induced by focused ultrasound combined with microbubbles. J Mol Neurosci 46(3):677–687
Biamonti G, Ruggiu M, Saccone S, Della VG, Riva S (1994) Two homologous genes, originated by duplication, encode the human hnRNP proteins A2 and A1. Nucleic Acids Res 22(11):1996–2002
Burd CG, Swanson MS, Gorlach M, Dreyfuss G (1989) Primary structures of the heterogeneous nuclear ribonucleoprotein A2, B1, and C2 proteins: a diversity of RNA binding proteins is generated by small peptide inserts. Proc Natl Acad Sci U S A 86(24):9788–9792
Izaurralde E, Jarmolowski A, Beisel C, Mattaj IW, Dreyfuss G et al (1997) A role for the M9 transport signal of hnRNP A1 in mRNA nuclear export. J Cell Biol 137(1):27–35
Siomi H, Dreyfuss G (1995) A nuclear localization domain in the hnRNP A1 protein. J Cell Biol 129(3):551–560
He Y, Smith R (2009) Nuclear functions of heterogeneous nuclear ribonucleoproteins A/B. Cell Mol Life Sci 66(7):1239–1256
Kamma H, Horiguchi H, Wan L, Matsui M, Fujiwara M et al (1999) Molecular characterization of the hnRNP A2/B1 proteins: tissue-specific expression and novel isoforms. Exp Cell Res 246(2):399–411
Cooper TA, Wan L, Dreyfuss G (2009) RNA and disease. Cell 136(4):777–793
Rao DD, Vorhies JS, Senzer N, Nemunaitis J (2009) siRNA vs. shRNA: similarities and differences. Adv Drug Deliv Rev 61(9):746–759
Nakada M, Nakada S, Demuth T, Tran NL, Hoelzinger DB et al (2007) Molecular targets of glioma invasion. Cell Mol Life Sci 64(4):458–478
Ku BM, Lee YK, Ryu J, Jeong JY, Choi J et al (2011) CHI3L1 (YKL-40) is expressed in human gliomas and regulates the invasion, growth and survival of glioma cells. Int J Cancer 128(6):1316–1326
Sauvageot CM, Weatherbee JL, Kesari S, Winters SE, Barnes J et al (2009) Efficacy of the HSP90 inhibitor 17-AAG in human glioma cell lines and tumorigenic glioma stem cells. Neuro Oncol 11(2):109–121
Rahme GJ, Israel MA (2014) Id4 suppresses MMP2-mediated invasion of glioblastoma-derived cells by direct inactivation of Twist1 function. Oncogene.
Forsyth PA, Wong H, Laing TD et al (1999) Gelatinase-A (MMP-2), gelatinase-B (MMP-9) and membrane type matrix metalloproteinase-1 (MT1-MMP) are involved in different aspects of the pathophysiology of malignant gliomas. Br J Cancer 79(11–12):1828–1835
Rao JS (2003) Molecular mechanisms of glioma invasiveness: the role of proteases. Nat Rev Cancer 3(7):489–501
Choe G, Park JK, Jouben-Steele L et al (2002) Active matrix metalloproteinase 9 expression is associated with primary glioblastoma subtype. Clin Cancer Res 8(9):2894–2901
Kargiotis O, Chetty C, Gondi CS et al (2008) Adenovirus-mediated transfer of siRNA against MMP-2 mRNA results in impaired invasion and tumor-induced angiogenesis, induces apoptosis in vitro and inhibits tumor growth in vivo in glioblastoma. Oncogene 27(35):4830–4840
Wang M, Wang T, Liu S, Yoshida D, Teramoto A (2003) The expression of matrix metalloproteinase-2 and -9 in human gliomas of different pathological grades. Brain Tumor Pathol 20(2):65–72
Bromberg JF, Wrzeszczynska MH, Devgan G et al (1999) Stat3 as an oncogene. Cell 98(3):295–303
Bowman T, Garcia R, Turkson J, Jove R (2000) STATs in oncogenesis. Oncogene 19(21):2474–2488
Sansone P, Bromberg J (2012) Targeting the interleukin-6/Jak/stat pathway in human malignancies. J Clin Oncol 30(9):1005–1014
Lo HW, Cao X, Zhu H, Ali-Osman F (2008) Constitutively activated STAT3 frequently coexpresses with epidermal growth factor receptor in high-grade gliomas and targeting STAT3 sensitizes them to Iressa and alkylators. Clin Cancer Res 14(19):6042–6054
Turkson J, Ryan D, Kim JS et al (2001) Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J Biol Chem 276(48):45443–45455
Blaskovich MA, Sun J, Cantor A, Turkson J, Jove R, Sebti SM (2003) Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res 63(6):1270–1279
Lai SY, Childs EE, Xi S et al (2005) Erythropoietin-mediated activation of JAK-STAT signaling contributes to cellular invasion in head and neck squamous cell carcinoma. Oncogene 24(27):4442–4449
Rahaman SO, Vogelbaum MA, Haque SJ (2005) Aberrant Stat3 signaling by interleukin-4 in malignant glioma cells: involvement of IL-13Ralpha2. Cancer Res 65(7):2956–2963
Xie TX, Wei D, Liu M et al (2004) Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene 23(20):3550–3560
Zhou J, Nong L, Wloch M, Cantor A, Mulshine JL et al (2001) Expression of early lung cancer detection marker: hnRNP-A2/B1 and its relation to microsatellite alteration in non-small cell lung cancer. Lung Cancer 34(3):341–350
Grant Support
This work is supported by the National Natural Science Foundation of China (No. 30870723 and 81201066) and Chongqing Natural Science Foundation Project (No. CSTC2013jcyjA10054).
Conflict of Interest
No potential conflicts of interest were disclosed.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Deng, J., Chen, S., Wang, F. et al. Effects of hnRNP A2/B1 Knockdown on Inhibition of Glioblastoma Cell Invasion, Growth and Survival. Mol Neurobiol 53, 1132–1144 (2016). https://doi.org/10.1007/s12035-014-9080-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-014-9080-3