
Abstract
Trichostatin A (TSA), a pan-histone deacetylase inhibitor, exerts multiple neuroprotective properties. This study aims to examine whether TSA could enhance autophagy, thereby reduce neuronal apoptosis and ultimately attenuate early brain injury (EBI) following subarachnoid hemorrhage (SAH). SAH was performed through endovascular perforation method, and mortality, neurological score, and brain water content were evaluated at 24 h after surgery. Western blot were used for quantification of acetylated histone H3, LC3-II, LC3-I, Beclin-1, cytochrome c, Bax, and cleaved caspase-3 expression. Immunofluorescence was performed for colocalization of Beclin-1 and neuronal nuclei (NeuN). Apoptotic cell death of neurons was quantified with double staining of terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end-labeling (TUNEL) and NeuN. The autophagy inhibitor 3-methyladenine (3-MA) was used to manipulate the proposed pathway. Our results demonstrated that TSA reduced brain edema and alleviated neurological deficits at 24 h after SAH. TSA significantly increased acetylated histone H3, the LC3-II/LC3-I ratio, and Beclin-1 while decreased Bax and cleaved caspase-3 in the cortex. Beclin-1 and NeuN, TUNEL, and NeuN, respectively, were colocalized in cortical cells. Neuronal apoptosis in the ipsilateral basal cortex was significantly inhibited after TSA treatment. Conversely, 3-MA reversed the beneficial effects of TSA. These results proposed that TSA administration enhanced autophagy, which contributes to alleviation of neuronal apoptosis, improvement of neurological function, and attenuation of EBI following SAH.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Subarachnoid hemorrhage (SAH), which predominantly caused by rupture of intracranial aneurysm, is a disastrous cerebrovascular disease. Although account for only about 5 % of all stroke patients, it has placed a significant burden on society and economy, as it most often affects middle-aged patients, resulting in a combined mortality and disability rate higher than 50 % [1, 2]. Early brain injury (EBI), which defined as the events occurring within the first 72 h after the SAH onset, plays an important role in patient’s outcomes [1]. Apoptotic cell death of neuron is considered to be a significant contributor to SAH-induced EBI [3]. Neuronal apoptosis may be caused by increased intracranial pressure, cerebral ischemia, reperfusion, and the toxicity of blood breakdown components following SAH [2]. As a result, therapeutic strategies targeting apoptotic cascades of neurons may ameliorate secondary brain injury after SAH.
Autophagy, a conserved lysosomal degradation pathway, is essential for elimination of obsolete cellular proteins and damaged organelles to maintain cytoplasmic homeostasis [4]. The autophagic activity is significantly increased in neurons following SAH and lasts during the entire phase of EBI [5]. Autophagy appears to be a pro-survival mechanism, attempting to inhibit neuronal apoptosis in EBI after SAH [6–9]. However, the molecular mechanism of protective autophagy in SAH has not been completely clarified.
Acetylation/deacetylation of histones is a key post-translational modification mechanism which affects the accessibility of chromatin to the transcriptional machinery and gene expression [10]. Disruption of histones acetylation homeostasis has been recognized as a common neuropathological feature for central nervous system (CNS) disorders, including ischemic stroke, intracerebral hemorrhage, traumatic brain injury, and neurodegenerative diseases [11, 12]. As a classic histone deacetylase (HDAC) inhibitor, trichostatin A (TSA) provides a multitude of neuroprotective properties in the treatment of these disorders mentioned above, such as anti-excitotoxic [13], anti-inflammatory [14], anti-apoptotic [15], and neurotrophic effects [16], possibly via correction of hypoacetylation and transcriptional activation of disease-modifying genes (e.g., Hsp70) [17]. However, histones acetylation status in the brain following SAH has not been explored, and the therapeutic efficacy of HDAC inhibitors in SAH remains untested. On the other hand, increasing evidence suggests that HDAC inhibitors can stimulate autophagy process, depending on certain circumstance [18, 19]. For instance, Gammoh et al. demonstrated that the HDAC inhibitor suberoylanilide hydroxamic acid enhanced autophagy activity via inhibiting mTOR and upregulating light chain 3 (LC3) expression [20]. Thus, in the present study, we aim to examine the following two hypotheses: (1) post-insult treatment with TSA ameliorates EBI and improves neurological outcome after SAH via HDAC inhibition, and (2) TSA-enhanced protective autophagic pathway could attenuate neuronal apoptosis in the early stage of SAH.
Materials and Methods
Animals and SAH Model
All experimental procedures were conducted under the guideline of Institutional Animal Care and Use Committee (IACUC) of Zhejiang University. Rats get free access to food and water in a room with temperature maintained at 27 °C. Adult male Sprague–Dawley rats (230–270 g) were purchased from the Animal Center of Zhejiang Chinese Medical University (Hangzhou, China). We induced SAH model by endovascular perforation method as previously described [21]. Rats were anesthetized with pentobarbital (40 mg/kg, i.p.). The left external and internal carotid artery was exposed and a 4.0 monofilament nylon suture was inserted into the left internal carotid artery through the external carotid artery to perforate bifurcation of the anterior and middle cerebral artery. Sham-operated rats underwent identical procedures except the perforation. Rectal temperature was monitored at 37 °C with a heating pad during the surgery.
Experiment Protocol
Ninety-three rats were randomly assigned to four groups: sham group (n = 19), SAH + vehicle group (n = 26), SAH + TSA group (n = 23), and SAH + TSA + 3-methyladenine (3-MA) group (n = 25) (Fig. 1). Immediately after SAH induction, rats were administrated with vehicle (DMSO, 1 ml) or TSA (0.5 mg/kg, 1 ml, i.p.). 3-MA (300 nmol/rat, 3 μl) was intracerebroventricularly injected at 30 min before SAH onset. All drugs were purchased from Sigma-Aldrich (St. Louis, MO). Rats were sacrificed at 24 h after SAH. Five rats of each group were used for brain water content measurement, while another six rats of each group were used for immunofluorescence staining and terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end-labeling (TUNEL) staining; the rest of the rats were taken for Western blot.

Experimental design and animal group classification
Drug Administration
The intracerebroventricular infusion was performed according to our previous study [8]. Animals were anesthetized and positioned in a stereotaxic frame, the needle of a 10-μl syringe was inserted into the left lateral ventricle with the coordinates of 1.5 mm posterior, 0.8 mm lateral to bregma, and 3.5 mm below the horizontal plane of bregma. Then 3 μl 3-MA or vehicle was injected by a microinfusion pump at a speed of 1 μl/min 30 min before SAH onset. The syringe was left for another 30 s after injection, and the burr hole was plunged with bone wax. The drug dose selection was based on previous studies [6].
Evaluation of Mortality and Neurological Scores
Mortality was calculated at 24 h after SAH. Neurological scores were assessed according to the Garcia score system with minor modification by a partner blindly to the experiment [22]. Briefly, neurological deficits were determined from six aspects including spontaneous activity, symmetry in the movement of all limbs, forepaw outstretching, climbing, body proprioception, and response to vibrissae stimulation. The minimum score was 3 and maximum was 18. Lower score represents worse neurological function.
Assessment of SAH Grading
The evaluation of SAH severity was performed by a coworker who was blinded to the experiment according to a SAH grading system described before [23]. Subarachnoid blood clots scale in the six segments of basal cistern was assessed. Total score ranging from 0 to 18 was obtained.
Measurement of Brain Water Content
Brain edema was measured using the wet-weight/dry-weight method [24]. Rats were decapitated at 24 h after SAH injury. The brains were rapidly removed and divided into the right and left hemispheres and cerebellum. Then, the brain samples were weighed before (wet weight) and after (dry weight) drying in an oven at 100 °C for 72 h. The brain water content was calculated as (wet weight − dry weight) / wet weight × 100 % [25].
Western Blot
Rats were sacrificed at 24 h after SAH induction. Left cortical samples were obtained for Western blot [26]. Briefly, the samples were homogenized in lysis buffer and centrifuged at 10,000 rpm for 5 min at 4 °C; we collected the supernatants and determined protein concentrations with a BCA kit (Beyotime). Equal amounts (60 μg) of protein per sample were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and then transferred to polyvinylidene fluoride membranes. Afterward, membranes were blocked with blocking buffer (nonfat dry milk) for 2 h and incubated overnight at 4 °C with primary antibodies against Beclin 1 (1:1,500, Abcam), LC3 (1:500, Cell Signaling Technology), cleaved caspase-3 (1:1000, Abcam), Bax (1:400, Abcam), cytochrome c (1:500, Cell Signaling Technology), acetyl histone H3 (1:1000, Cell Signaling Technology), and β-actin (1:2000, Abcam). After incubation, the membranes were washed with TBST and incubated with horseradish-peroxidase conjugated secondary antibodies for 1 h at room temperature. The immunoreactive bands were visualized using an ECL Plus reagent kit (Beyotime) and quantified by the ImageJ software.
Double Immunofluorescence Staining
Immunofluorescent double staining of TUNEL and neuronal nuclei (NeuN) was performed to determine colocalization of apoptotic cells and neurons [27]. Briefly, brain samples were sectioned at 10-μm thickness. TUNEL staining was conducted according to the manufacturer’s instruction of In Situ Cell Death Dection Kit (Roche, Germany) and incubated with primary antibody against NeuN (1:200, Abcam) at 4 °C overnight. Afterward, the sections were incubated with rhodamine-conjugated secondary antibody (1:100, Jackson ImmunoResearch) and covered with 4′,6-diamidino-2-phenylindole (DAPI) (Beyotime). TUNEL-positive neurons were calculated as apoptosis index of neurons. Double immunofluorescence staining of Beclin 1 with NeuN was performed to clarify the colocalization of autophagy-positive cells and neurons. The primary antibodies used were against Beclin 1 (1:500, Abcam) and NeuN (1:200, Abcam), followed by incubation with corresponding secondary antibodies (1:100, Jackson ImmunoResearch). Subsequently, brain sections were covered with DAPI (Beyotime) and fluorescence signals were observed under a fluorescence microscope (Olympus, Japan).
Statistical Analysis
Data were presented as mean ± standard error. Multiple comparisons were analyzed using one-way analysis of variance (ANOVA) followed by Tukey’s test. Statistical analyses were performed with Mann–Whitney U test to compare the neurological score. Statistical significance was defined as p < 0.05.
Results
Mortality and SAH grade
Physiological variables (body weight, temperature) were monitored throughout the experiment, and there were no significant changes among the groups (data not shown). None of the sham group rats died. The mortality in the SAH + vehicle group was 30.8 % (8 of 26), 17.4 % (4 of 23) in the SAH + TSA group, and 24.0 % (6 of 25) in the SAH + TSA + 3-MA group (p > 0.05; Fig. 2b).

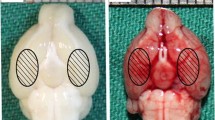
Representative pictures of brains from each group, and mortality, SAH grade, neurological score, and brain water content at 24 h post-SAH. a Typical brains from sham, SAH + vehicle, SAH + TSA, and SAH + TSA + 3-MA. Significant blood clots mainly distribute at the base of Willis circle and brain stem. The same part of basal cortical brain tissue was taken for tests (circled areas). b Mortality was 30.8, 17.4, and 24.0 % in the SAH + vehicle group, SAH + TSA group, and SAH + TSA + 3-MA group, respectively. c No obvious difference in severity of SAH bleeding among the SAH, SAH + TSA, and SAH + TSA + 3-MA groups (p > 0.05). d Neurobehavioral performance was impaired after SAH, and TSA alleviated neurologic dysfunction markedly, whereas 3-MA administration inhibited the beneficial effect of TSA (p < 0.05). e Brain water content increased significantly in the left and right hemispheres in SAH rats (p < 0.05), but not in the cerebellum (p > 0.05). TSA attenuated the left hemisphere’s brain edema significantly (p < 0.05), but not other brain regions (p > 0.05). However, pre-SAH injection of 3-MA increased brain water content compared to SAH + TSA group in the left hemisphere (p < 0.05). ##P < 0.01 versus sham group; **P < 0.01 versus SAH + vehicle group; $$P < 0.01 versus SAH + TSA group; $P < 0.05 versus SAH + TSA group
SAH-induced brain injury is tightly associated with the severity of bleeding. At 24 h post-SAH, the SAH scores were 0 in the sham group, 12.3 ± 1.6 in the SAH + vehicle group, 12.7 ± 1.8 in the SAH + TSA group, and 12.9 ± 1.1 in the SAH + TSA + 3-MA group. No significant difference of SAH severity was found among the three SAH groups. The blood distributed mainly around the Willis circle (p > 0.05; Fig. 2a, c).
TSA Attenuated Neurofunctional Deficits and Alleviated Brain Edema
The animals in the SAH + vehicle group demonstrated severe neurobehavioral dysfunction compared to the sham group (p < 0.01). After TSA treatment, there was a substantial improvement in neurological score at 24 h following SAH compared to the SAH + vehicle group (p < 0.01). Whereas administration of 3-MA, an autophagy inhibitor, inhibited the effect of TSA (p < 0.05; Fig. 2d).
As shown in Fig. 2e, brain water content increased markedly in the left hemisphere and right hemisphere in the SAH + vehicle group compared to the sham group (SAH + vehicle 79.62 ± 0.30 vs sham 78.74 ± 0.15; SAH + vehicle 79.22 ± 0.28 vs sham 78.64 ± 0.29, p < 0.01), administration of TSA significantly alleviated brain edema in the left hemisphere but not right hemisphere compared to the SAH + vehicle group (SAH + TSA 79.00 ± 0.22 vs SAH + vehicle 79.62 ± 0.30, p < 0.01), whereas 3-MA reduced the effect of TSA on brain edema in the left hemisphere (SAH + TSA + 3-MA 79.48 ± 0.22 vs SAH + TSA 79.00 ± 0.22, p < 0.01). However, there was no significant difference of brain edema in cerebellum among the groups (p > 0.05).
TSA Increased the Acetylation of Histone H3
To determine the effect of TSA on histone acetylation after SAH, we detected acetylated histones H3 by Western blot at 24 h subjected to SAH. It is appeared to be less histone H3 acetylation in the SAH + vehicle group compared to sham group (p < 0.05), while administration of TSA increased histone H3 acetylation at 24 h post-SAH (p < 0.01; Fig. 3a, b).

Effect of TSA on acetylated histones H3 expression in the left cerebral hemisphere at 24 h after SAH. a Representative picture of the acetylated histones H3 expression by Western blot. b Quantitative analysis of acetylated histones H3 level evaluated by Western blot. Acetylated histones H3 was significantly decreased in the SAH + vehicle group compared with sham group (p < 0.05). TSA can significantly enhanced acetylated histones H3 expression (p < 0.05). #P < 0.05 versus sham group; **P < 0.01 versus SAH + vehicle group
TSA Promoted Autophagy Activation and Increased Autophagy-Related Protein Expression
To explore the role of TSA on autophagy post-SAH, we evaluated the protein expression of Beclin 1 and LC3 by Western blot and colocalization of Beclin 1 and Neuron by double immunofluorescence staining at 24 h after SAH surgery. The expression of Beclin 1 increased significantly after SAH compared to the sham group (p < 0.05), while TSA treatment upregulated the expression of Beclin 1 compared to the SAH group, which was reversed by pre-SAH injection of 3-MA (p < 0.01; Fig. 4b, d). Similarly, the LC3 level was displayed as LC3-II/LC3-I ratio which enhanced after SAH onset compared to the sham group and promoted more by TSA treatment compared to the SAH + vehicle group (p < 0.01). However, 3-MA administration opposed the effect of TSA on LC3 expression (p < 0.05; Fig. 4a, c).

Effect of TSA on autophagy-related proteins (LC3 and Beclin 1) expression in the left cerebral hemisphere at 24 h after SAH. Representative Western blots of LC3 (a) and Beclin 1 (b) in the left cerebral hemisphere at 24 h after SAH. Similar trends were observed in the quantitative analysis of LC3 (c) and Beclin 1 (d) expression. Ratio of LC3II/LCI and Beclin 1 were increased substantially in the SAH + vehicle group compared to the sham group (p < 0.05), but increased more significantly in the SAH + TSA group (p < 0.05), and this increase was hindered by 3-MA administration (p < 0.05). #P < 0.05 versus sham group; ##P < 0.01 versus sham group; *P < 0.05 versus SAH + vehicle group; **P < 0.01 versus SAH + vehicle group; $P < 0.05 versus SAH + TSA group; $$P < 0.01 versus SAH + TSA group
With regard to Beclin 1 expression, the results from double immunofluorescence staining indicated that Beclin 1-positive cells were widespread within the left hemisphere cortex, and more intense staining was observed after TSA treatment, but immunoreactivity was less apparent after 3-MA administration (Fig. 5). On the other hand, Beclin 1-positive cells were mainly colocalized with neurons at 24 h after SAH (Fig. 5).

Immunofluorescent staining of NeuN (red), Beclin 1 (green), and DAPI (blue) in the left cerebral cortex at 24 h after SAH. Weak staining of Beclin 1 was observed in the sham group, while intense staining was detected in the SAH + vehicle group (p < 0.05). However, Beclin 1 immunoreactivity increased more after TSA treatment, and decreased significantly after 3-MA administration (p < 0.05). Merged images show that Beclin 1-positive cells mainly colocalized with neurons (arrows). Scale bar 20 μm (color figure online)
TSA Treatment Attenuated Neuronal Apoptosis and Downregulated the Apoptosis-Related Protein Expression
To confirm the role of TSA on neuronal apoptosis, protein levels of cleaved caspase-3, Bax, and cytochrome c were measured by Western blot, and colocalization of TUNEL and NeuN were performed by double-label staining. Western blot results demonstrated that upregulation of cleaved caspase-3 was observed in the SAH + vehicle group compared to the sham group, TSA treatment markedly abolished cleaved caspase-3 expression while injection of 3-MA significantly enhanced cleaved caspase-3 action (p < 0.05; Fig. 6a, d). Similar to the cleaved caspase-3, Bax and cytochrome c increased at 24 h after SAH, while compared to the sham group, and diminished substantially with TSA therapy. However, pre-SAH 3-MA administration inhibited the beneficial effect of TSA (p < 0.01; Fig. 6b, c, e, f).

Effect of TSA on apoptosis-related protein (cleaved caspase-3 and Bax) expression in the left cerebral hemisphere at 24 h after SAH. Typical Western blots of cleaved caspase-3 (a) and Bax (b) in the left cerebral hemisphere at 24 h after SAH. The significant increase of cleaved caspase-3 and Bax expression was reversed by TSA (p < 0.05), but 3-MA abolished the anti-apoptotic effect induced by TSA (p < 0.05). ##P < 0.01 versus sham group; *P < 0.05 versus SAH + vehicle group; $$P < 0.01 versus SAH + TSA group
Double-label staining of TUNEL and NeuN showed that TUNEL-positive cells mainly occurred in neurons. Barely detectable TUNEL-positive neurons were found in the cortex of left hemisphere in the sham group (p < 0.05; Fig. 7a, b). In the animals subjected to SAH, number of TUNEL and NeuN double-staining cells was much higher compared to the sham group (p < 0.01; Fig. 7a, b). However, TSA treatment attenuated neuronal apoptosis while 3-MA administration inhibited the favorable effect of TSA and aggravated neuronal apoptosis at 24 h post-SAH (p < 0.01; Fig. 7a, b).

Double-label staining of NeuN (red) and TUNEL (green) in the left cerebral cortex at 24 h after SAH. Nuclei were counterstained with DAPI (blue). a TUNEL-positive cells were barely detected in the sham group but widespread distributed in the left cerebral cortex after SAH. TSA decreased while 3-MA increased the amount of TUNEL-positive cells. Overlapped images show that TUNEL-positive cells mainly colocalized with neurons (arrows). Scale bar 20 μm. b Few apoptotic neurons were found in the sham group (p < 0.05), and SAH increased apoptotic neurons markedly at 24 h post-SAH (p < 0.05). TSA attenuates neuronal apoptosis remarkably, whereas 3-MA pre-SAH injection reversed the trend (p < 0.05). ##P < 0.01 versus sham group; **P < 0.01 versus SAH + vehicle group; $$P < 0.01 versus SAH + TSA group (color figure online)
Discussion
In this study, we showed that post-insult treatment with HDAC inhibitor TSA decreased brain edema and improved neurological outcomes at 24 h after SAH in a rat endovascular perforation model. The neuroprotective effect was associated with TSA-enhanced autophagy in neurons. Activation of autophagy alleviated neuronal apoptosis following SAH. These findings, for the first time, gave direct evidence of the beneficial effects of TSA on SAH-induced EBI.
Histone acetylation/deacetylation modifications directly influence chromatin structure and have an important role in the regulation of gene transcription. Two key enzymes, namely histone acetyltransferase (HAT) and HDAC, function antagonistically to maintain histone acetylation homeostasis, which is critical for cell survival. The pathological significance of disease-associated brain histone hypoacetylation has been observed in a variety of neurological disorders [28], in particular, in ischemic stroke [29], intracerebral hemorrhage [30], and traumatic brain injury [31]. HDAC inhibitors including TSA can restore adequate histone acetylation levels, which result in increased expression of the neuroprotective proteins (e.g., Hsp70 and Bcl-2) and attenuate brain damage and neurofunctional deficits in animals following acute brain injury [32]. In the present study, we found a lower level of acetylated histone H3 in the cerebral cortex at 24 h after SAH, suggesting that overdeacetylation may be involved in EBI. TSA, as a class I and II HDAC inhibitor, reversed SAH-induced histone H3 hypoacetylation, attenuated brain edema, and improved neurological function after SAH. Our results strengthened the neuroprotective potential of pharmacological strategies aimed at inhibiting HDAC in the setting of acute brain insult.
Neuronal apoptosis is a vital pathologic event in the pathogenesis of SAH-induced EBI [33]. The Bcl-2 family proteins, the adaptor protein such as apoptotic protease-activating factor 1 (Apaf-1), and the cysteine proteases play an important role in orchestrating apoptotic cell death of neuron [3]. As a widely expressed pro-apoptotic member of the Bcl-2 family, Bax can be activated following SAH [9], causing Bax-mediated channel formation and mitochondrial outer membranes permeabilization, which leads to the release of cytochrome c from mitochondria. In the cytosol, cytochrome c binds with Apaf-1 and procaspase-9 to assemble the apoptosome, which is essential for the activation of procaspase-9. Activated caspase-9 in turn cleaves and thereby activates caspase-3, contributing to apoptotic cell death [34]. In this study, we found that protein levels of cytochrome c, Bax, and cleaved caspase-3 were upregulated in the ipsilateral cortex of SAH rats, indicating that the mitochondrial apoptotic pathway may be activated after SAH. In addition, immunofluorescence staining demonstrated that apoptotic cell death of neurons was increased following SAH induction. Together, these results suggested that mitochondria-dependent proapoptotic pathway may involve in SAH-induced neuronal cell death.
Autophagy is an intracellular degradation mechanism which mediates the clearance of long-lived proteins and organelles [35]. Autophagy can be provoked in various pathological and physiological conditions, such as stress [36]. The activation of autophagy could be either deleterious or protective, depending on the specificity of the pathologic situation and stage [4]. Under the condition of SAH-induced EBI, autophagy was also activated and peaked at 24 h after hemorrhage [5]. Enhancement of autophagy by rapamycin decreased Bax-mediated cytochrome c release from the mitochondria to the cytosol, thereby prevented neurons from apoptosis and ameliorated EBI after SAH [8]. Conversely, 3-MA treatment inhibited autophagy, resulting in increased apoptotic cell death of neurons and exacerbated neurological deficits after SAH [37]. These data indicated that “self-eating” autophagy emerges to be beneficial and can inhibit “self-killing” apoptosis in EBI after experimental SAH [6]. Our study demonstrated that neuronal autophagy was stimulated at 24 h after SAH, revealed by elevated protein levels of LC3 and Beclin-1, increased LC3-II/LC3-I ratio, and coimmunofluorescence of Beclin-1 and NeuN in injured brain. The autophagic activation was further enhanced by TSA treatment, resulting in downregulation of Bax and cleaved caspase-3 expression, as well as decreased apoptotic cell death of neurons. 3-MA pre-treatment hindered TSA-induced autophagy activation, resulting in increased neuronal apoptosis after SAH. These findings suggest that induction of autophagy by TSA may prevent neurons from mitochondria-mediated apoptosis.
However, the mechanism of autophagy activation by TSA still remains unclear. As a histone deacetylase inhibitor, TSA could inhibit histone deacetylation, which resultantly reduce the binding force of histone with Beclin 1 gene sequences, and enhance some transcription factors binding to the Beclin 1 promoter, resultantly promote the Beclin 1 transcription and expression. And as we know, increased expression of Beclin 1 will contribute to the activation of autophagy. However, whether other pathway and mechanism involved in the TSA-induced autophagy activation is still unclear; therefore, in our future research, we will focus on exploring the underlying mechanism how TSA regulate autophagy.
Also, the underlying molecular mechanism of how did autophagy reduce neuronal apoptosis is not clarified in our study. We speculate that the potential underlying molecular mechanism may be that autophagy devours apoptosis-related factors such as damaged mitochondria and some pro-apoptotic proteins such as Bax and caspase-3, thus achieving the purpose of inhibiting apoptosis. In our research, we evaluate pro-apoptotic proteins Bax, caspase-3, cytochrome c expression, and neuronal apoptosis by TUNEL staining. At least, TSA-induced autophagy could reduce neuronal apoptosis partially by inhibiting pro-apoptotic proteins expression. However, whether TSA-induced autophagy could exert anti-apoptosis through other ways need to be addressed in our further experiment; specially, the possible involvement of nonmitochondrial apoptotic pathway in TSA’s neuroprotective mechanism needs further investigation.
Indeed, this study has several other limitations. First, the time course study of histone acetylation status after SAH in the brain has not been conducted. Besides, the mechanisms involved in autophagy activation by TSA in the context of SAH have not been totally elucidated, and the possibilities of other mechanisms about TSA-mediated neuroprotection have not been excluded. Furthermore, the molecular basis underlying anti-apoptotic mechanism of autophagy is still obscure. Considering the central roles of key moleculars, such as Beclin-1, the Bcl-2 family member, and p53, in the cross talk between autophagy and apoptosis [38–40], further SAH research should focus on these proteins, in order to elucidate the anti-apoptotic mechanisms of TSA-induced autophagy following SAH. Additionally, the therapeutic time window, the optimal dosage, the reliable drug administration route, and long-term effects of TSA treatment in SAH need to be addressed in our future studies.
Conclusion
In summary, we demonstrated, for the first time, the protective effects of TSA on EBI in the experimental SAH model. TSA treatment enhanced autophagy activities, which contributes to inhibition of caspase-dependent neuronal apoptosis, attenuation of brain edema, and improvement of neurological status following SAH. These results indicate that TSA could be a potential therapeutic candidate for preventing SAH-induced EBI.
Abbreviations
- SAH:
-
Subarachnoid hemorrhage
- TSA:
-
Trichostatin A
- EBI:
-
Early brain injury
- HDAC:
-
Histone deacetylase
- 3-MA:
-
3-Methyladenine
- TUNEL:
-
Terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end-labeling
- LC3:
-
Light chain 3
- Apaf-1:
-
Apoptotic protease-activating factor 1
- Ac-H3:
-
Acetylated histones H3
- CC3:
-
Cleaved caspase-3
References
Sehba FA, Hou J, Pluta RM et al (2012) The importance of early brain injury after subarachnoid hemorrhage. Prog Neurobiol 97:14–37
Chen S, Feng H, Sherchan P et al (2014) Controversies and evolving new mechanisms in subarachnoid hemorrhage. Prog Neurobiol 115:64–91
Cahill J, Calvert JW, Zhang JH (2006) Mechanisms of early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab 26:1341–53
Rubinsztein DC, Codogno P, Levine B (2012) Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 11:709–30
Lee JY, He Y, Sagher O et al (2009) Activated autophagy pathway in experimental subarachnoid hemorrhage. Brain Res 1287:126–35
Wang Z, Shi XY, Yin J et al (2012) Role of autophagy in early brain injury after experimental subarachnoid hemorrhage. J Mol Neurosci 46:192–202
Liu Y, Li J, Wang Z et al (2014) Attenuation of early brain injury and learning deficits following experimental subarachnoid hemorrhage secondary to cystatin C: possible involvement of the autophagy pathway. Mol Neurobiol 49:1043–54
Jing CH, Wang L, Liu PP et al (2012) Autophagy activation is associated with neuroprotection against apoptosis via a mitochondrial pathway in a rat model of subarachnoid hemorrhage. Neuroscience 213:144–53
Chen J, Wang L, Wu C et al (2014) Melatonin-enhanced autophagy protects against neural apoptosis via a mitochondrial pathway in early brain injury following a subarachnoid hemorrhage. J Pineal Res 56:12–9
Grunstein M (1997) Histone acetylation in chromatin structure and transcription. Nature 389:349–52
Shein NA, Shohami E (2011) Histone deacetylase inhibitors as therapeutic agents for acute central nervous system injuries. Mol Med 17:448–56
Hahnen E, Hauke J, Trankle C et al (2008) Histone deacetylase inhibitors: possible implications for neurodegenerative disorders. Expert Opin Investig Drugs 17:169–84
Leng Y, Chuang DM (2006) Endogenous alpha-synuclein is induced by valproic acid through histone deacetylase inhibition and participates in neuroprotection against glutamate-induced excitotoxicity. J Neurosci 26:7502–12
Chen PS, Wang CC, Bortner CD et al (2007) Valproic acid and other histone deacetylase inhibitors induce microglial apoptosis and attenuate lipopolysaccharide-induced dopaminergic neurotoxicity. Neuroscience 149:203–12
Meisel A, Harms C, Yildirim F et al (2006) Inhibition of histone deacetylation protects wild-type but not gelsolin-deficient neurons from oxygen/glucose deprivation. J Neurochem 98:1019–31
Maruoka H, Sasaya H, Sugihara K et al (2011) Low-molecular-weight compounds having neurotrophic activity in cultured PC12 cells and neurons. J Biochem 150:473–5
Marinova Z, Ren M, Wendland JR et al (2009) Valproic acid induces functional heat-shock protein 70 via Class I histone deacetylase inhibition in cortical neurons: a potential role of Sp1 acetylation. J Neurochem 111:976–87
Francisco R, Perez-Perarnau A, Cortes C et al (2012) Histone deacetylase inhibition induces apoptosis and autophagy in human neuroblastoma cells. Cancer Lett 318:42–52
Xie M, Kong Y, Tan W et al (2014) HDAC inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation 129:1139–51
Gammoh N, Lam D, Puente C et al (2012) Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc Natl Acad Sci U S A 109:6561–5
Chen S, Ma Q, Krafft PR et al (2013) P2X7 receptor antagonism inhibits p38 mitogen-activated protein kinase activation and ameliorates neuronal apoptosis after subarachnoid hemorrhage in rats. Crit Care Med 41:e466–74
Garcia JH, Wagner S, Liu KF et al (1995) Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke 26:627–34, discussion 35
Sugawara T, Ayer R, Jadhav V et al (2008) A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model. J Neurosci Methods 167:327–34
Zhou F, Chen G, Zhang J (2009) Edaravone reduces brain oedema and attenuates cell death after intracerebral haemorrhage in mice. Brain Inj 23:353–7
Chen S, Ma Q, Krafft PR et al (2013) P2X7R/cryopyrin inflammasome axis inhibition reduces neuroinflammation after SAH. Neurobiol Dis 58:296–307
Hong Y, Yan W, Chen S et al (2010) The role of Nrf2 signaling in the regulation of antioxidants and detoxifying enzymes after traumatic brain injury in rats and mice. Acta Pharmacol Sin 31:1421–30
He Z, Ostrowski RP, Sun X et al (2012) Targeting C/EBP homologous protein with siRNA attenuates cerebral vasospasm after experimental subarachnoid hemorrhage. Exp Neurol 238:218–24
Kazantsev AG, Thompson LM (2008) Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov 7:854–68
Ren M, Leng Y, Jeong M et al (2004) Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: potential roles of histone deacetylase inhibition and heat shock protein induction. J Neurochem 89:1358–67
Sinn DI, Kim SJ, Chu K et al (2007) Valproic acid-mediated neuroprotection in intracerebral hemorrhage via histone deacetylase inhibition and transcriptional activation. Neurobiol Dis 26:464–72
Shein NA, Grigoriadis N, Alexandrovich AG et al (2009) Histone deacetylase inhibitor ITF2357 is neuroprotective, improves functional recovery, and induces glial apoptosis following experimental traumatic brain injury. FASEB J 23:4266–75
Kim HJ, Rowe M, Ren M et al (2007) Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther 321:892–901
Altay O, Hasegawa Y, Sherchan P et al (2012) Isoflurane delays the development of early brain injury after subarachnoid hemorrhage through sphingosine-related pathway activation in mice. Crit Care Med 40:1908–13
Reed JC (2002) Apoptosis-based therapies. Nat Rev Drug Discov 1:111–21
Li F, Vierstra RD (2012) Autophagy: a multifaceted intracellular system for bulk and selective recycling. Trends Plant Sci 17:526–37
Smith CM, Chen Y, Sullivan ML et al (2011) Autophagy in acute brain injury: feast, famine, or folly? Neurobiol Dis 43:52–9
Zhao H, Ji Z, Tang D et al (2013) Role of autophagy in early brain injury after subarachnoid hemorrhage in rats. Mol Biol Rep 40:819–27
Zhou F, Yang Y, Xing D (2011) Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J 278:403–13
Kang R, Zeh HJ, Lotze MT et al (2011) The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 18:571–80
Maiuri MC, Zalckvar E, Kimchi A et al (2007) Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 8:741–52
Acknowledgments
This work was supported by Grant 81371369 and 81371433 from the National Natural Science Foundation of China; Grant 20120101120030 from the Doctoral Program of the Ministry of Education; Grant 2013KYA088 from the Zhejiang Provincial Medical Science and Technology Planning Project; and Grant Y13H090007 and LY13H090002 from the Zhejiang Provincial Natural Science Foundation of China.
Conflict of Interest
The authors declare that there is no conflicts of interest regarding the publication of this paper.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Anwen Shao and Zhen Wang contributed equally to this work.
Rights and permissions
About this article

Cite this article
Shao, A., Wang, Z., Wu, H. et al. Enhancement of Autophagy by Histone Deacetylase Inhibitor Trichostatin A Ameliorates Neuronal Apoptosis After Subarachnoid Hemorrhage in Rats. Mol Neurobiol 53, 18–27 (2016). https://doi.org/10.1007/s12035-014-8986-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-014-8986-0