
Abstract
Oxidative stress is well known to play a pivotal role in cerebral ischemia–reperfusion injury. The nuclear factor erythroid-2-related factor 2 (Nrf2)/heme oxygenase-1 (HO-1) pathway has been considered a potential target for neuroprotection in stroke. 11-Keto-β-boswellic acid (KBA) is a triterpenoid compound from extracts of Boswellia serrata. The aim of the present study was to determine whether KBA, a novel Nrf2 activator, can protect against cerebral ischemic injury. Middle cerebral artery occlusion (MCAO) was operated on male Sprague–Dawley rats. KBA (25 mg/kg) applied 1 h after reperfusion significantly reduced infarct volumes and apoptotic cells as well as increased neurologic scores at 48 h after reperfusion. Meanwhile, posttreatment with KBA significantly decreased malondialdehyde (MDA) levels, restored the superoxide dismutase (SOD) activity, and increased the protein Nrf2 and HO-1 expression in brain tissues. In primary cultured astrocytes, KBA increased the Nrf2 and HO-1 expression, which provided protection against oxygen and glucose deprivation (OGD)-induced oxidative insult. But knockdown of Nrf2 or HO-1 attenuated the protective effect of KBA. In conclusion, these findings provide evidence that the neuroprotection of KBA against oxidative stress-induced ischemic injury involves the Nrf2/HO-1 pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ischemic stroke is a debilitating clinical disorder that affects millions of people, but effective neuroprotective treatments is lacking. Brain tissue is particularly susceptible to oxidative damage. Evidence has accumulated that excessive reactive oxygen species (ROS) are closely related to cerebral ischemia/reperfusion (I/R) injury in stroke [1]. Therefore, antioxidants have been considered in prevention and treatment of stroke. The expression of phase II enzymes is regulated by nuclear factor E2-related factor 2 (Nrf2), a transcription factor [2]. In particular, Nrf2 binds to antioxidant response element (ARE) localized in the promoter regions of a battery of antioxidant and detoxifying genes including heme oxygenase 1 (HO-1) [2]. Recently, studies have provided evidence for the therapeutic potential of targeting the Nrf2/HO-1 pathway in brain injury after ischemic stroke [3, 4]. In our previous studies, we also have demonstrated an important role of Nrf2 activation in prevention of cardiovascular and cerebrovascular diseases [5, 6].
11-Keto-β-boswellic acid (KBA), a pentacyclic triterpenoid compound, is one of the most important active principles within the multicomponent mixture of Boswellia serrata resin [7]. The plant B. serrata shows antioxidant activity in the cerebrovascular and digestive system [8, 9]. KBA protects against myocardial I/R injury in rats through mechanisms related to enhancement of antioxidant capacity and prevention of inflammatory cascade redox and inflammatory cascades [10]. The neuroprotective property of pentacyclic triterpenoid has drawn increasing interest during the past couple of years. By way of example, oleanolic acid shows protective effects on cerebral ischemic damage and H2O2-induced injury in vitro [11]. Similarly, ursolic acid, a naturally occurring pentacyclic triterpenoid, was reported to promote the neuroprotection after cerebral ischemia in mice by activating Nrf2 pathway [12].
Considering the beneficial properties of triterpenoids and the possible role of the Nrf2/HO-1 pathway, we hypothesized that KBA would provide neuroprotection against I/R injury induced by transient middle cerebral artery occlusion (MCAO) in rats and oxygen and glucose deprivation (OGD) in primary cultured astrocytes.
Material and Methods
Middle Cerebral Artery Occlusion Model
All procedures involving animals were approved by the Institutional Animal Care and Use Committee of the Fourth Military Medical University and were performed in accordance with published National Institutes of Health guidelines.
Adult male Sprague–Dawley rats from the Experimental Animal Center of Fourth Military Medical University (Xi’an, China) (280–300 g) were divided randomly into the following three groups using a random number table generated by SPSS 16.0 (SPSS Inc., Chicago, IL, USA): sham-operated group (sham), vehicle-treated I/R group (vehicle + I/R), and KBA-treated I/R group (KBA + I/R). Under chloral hydrate anesthesia, focal cerebral ischemia was performed using the method of right MCAO with an intraluminal filament as described previously [13]. Cerebral blood flow (CBF) was monitored using laser Doppler flowmetry (Perimed AB, PeriFlux System 5000, Stockholm, Sweden) in the ipsilateral cortex (2 mm posterior and 5 mm lateral to the bregma). Sham-operated rats were manipulated in the same way, but the MCA was not occluded. Animals that did not show a CBF reduction of at least 70 % and animals that died after ischemia induction were excluded from the groups (4 out of 50 in KBA + I/R group). At 2 h after the induction of ischemia, the filament was slowly withdrawn. The neck incision was closed and rats were allowed to recover. After revival from anesthesia, the animals were put back into cages with the room temperature maintained at 25 ± 2 °C. The animals were allowed to survive for 2 days with free access to food and water.
KBA (reagent grade, purity >95 %, Sigma-Aldrich) diluted with physiological saline (25 mg/kg) was administered by intraperitoneal injection. According to the published studies, 25 mg/kg KBA administered to rats (corresponding to about 350 mg B. serrata extract/kg) could reach to physiological concentrations in the plasma and brain [14, 15]. Vehicle of 2 ml/kg physiological saline (vehicle + I/R group) and 25 mg/kg KBA (KBA + I/R group) were given 1 h after reperfusion.
Measurement of Neurological Score and Infarct Size
At 48 h after reperfusion, an 18-point scoring system was used to evaluate the neurologic deficits in rats [16]. The scale was based on the following six tests: (1) spontaneous activity (0 to 3 points); (2) symmetry in the movement of four limbs (0 to 3 points); (3) forepaw outstretching (0 to 3 points); (4) climbing (1 to 3 points); (5) body proprioception (1 to 3 points); and (6) response to vibrissae touch (1 to 3 points). The six individual test scores were summed up at the end of the evaluation (minimum score, 3; maximum score, 18).
To calculate infarct volume, brains were removed at 48 h after MCAO and were cut into 2-mm-thick coronal sections and subjected to 2,3,5-triphenyltetrazolium chloride (TTC) staining. Unstained areas were defined as infarcts and were measured using image analysis software. The percentage of the infarct volume was calculated by the following formula: ([total contralateral hemispheric volume] − [total ipsilateral hemispheric stained volume]) / (total contralateral hemispheric volume) × 100 %.
HE and TUNEL Staining
Hematoxylin and eosin (HE) staining was performed to show the morphological features of injured neurons in the cerebral cortex. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed on paraffin-embedded sections. Commercially available reagents (Promega, DeadEnd Fluorometric TUNEL System) were used to perform TUNEL analysis. The total number of TUNEL-positive cells in the ipsilateral hemisphere was counted in five different fields for each section by light microscopy by an investigator who was blinded to the studies.
Assessment of Oxidative Stress
The right cerebral cortex tissue was homogenized in 2 ml of 10 mM phosphate buffer (pH 7.4). After centrifugation at 12,000g for 20 min, the superoxide dismutase (SOD), glutathione peroxidase (GPx), and malondialdehyde (MDA) content in the supernatant was assessed spectrophotometrically with the corresponding kits (Nanjing Jiancheng Biochemistry Co., Nanjing, China). The protein concentrations were determined by the Bradford method. The levels of SOD, GPx, and MDA were expressed as units per milligram protein, units per milligram protein, and nanomole per gram protein, respectively.
Immunofluorescence
The paraffin sections, 5 mm thick, which were drawn 48 h after reperfusion, were first deparaffinized in xylene, rehydrated with various grades of ethanol, and pretreated with 10 μg/ml proteinase K for 30 min at 37 °C. By incubating the sections in 10 % bovine serum albumin, nonspecific binding of immunoglobulins was blocked for 20 min. Then the sections were incubated overnight at room temperature in with anti-Nrf2 or anti-HO-1 and with anti-glial fibrillary acidic protein (GFAP) antibody to mark astroglial cell. Finally, the sections were coverslipped. The stained sections were examined under a fluorescence microscope (Olympus, Tokyo, Japan).
Western Blot
Tissues in cortex or cells were homogenized in RAPI lysis buffer (Beyotime, China), centrifuged at 14,000g at 4 °C for 30 min, and then the supernatants were collected as total proteins. To prepare the cytoplasmic and nuclear proteins, cells were lysed using a nuclear and cytoplasmic protein extraction kit (Beyotime, China) according to the manufacturer’s instructions. Equal amounts of protein extracts were separated by SDS-PAGE and transferred onto polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA) by electrophoresis, and membranes were blocked with 5 % nonfat milk in 0.1 % Tween 20 in TBS (TBST) for 1 h at room temperature. Blots were then incubated with the antibody for HO-1 (Stressgen), Nrf2 (Abcam), histone H3 (Cell Signaling Technology), and β-actin (Cell Signaling Technology).
Primary Culture and In Vitro Model of Ischemia
Primary astrocyte cultures were obtained from neonatal rats as described previously [17]. Cerebral cortices were harvested from neonatal Sprague–Dawley rats. To model ischemia-like conditions in vitro, primary cultured astrocytes were exposed to transient OGD for 60 min. Then, the astrocytes were incubated again in the incubator with 95 % air and 5 % CO2 with or without KBA for an additional 24 h. OGD-induced cell death was quantified using the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma).
Intracellular ROS Measurement
The level of oxidative stress was determined by measuring intracellular ROS generation. The production of cellular ROS was detected using the 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) fluorescence assay. Cells were washed with phosphate-buffered saline (PBS) and fresh medium containing DCFH-DA (10 μM) was added. After incubating at 37 °C for 30 min, the cells were washed with PBS, trypsinized, and resuspended in PBS. The level of ROS generated was measured using flow cytometry based upon the fluorescence intensity of DCF at 525 nm after excitation at 485 nm. The ROS production was expressed as a percentage of the control sample.
Nrf2 and HO-1 siRNA Transfection
Neuronal cells were transiently transfected with small interfering RNA (siRNA) targeting to Nrf2 or HO-1 by Lipofectamine 2000 TM (Invitrogen) according to the manufacturer’s protocol. Knockdown efficiency of Nrf2 and HO-1was determined by Western blot analysis using anti-Nrf2 and HO-1 antibodies. Cells were transfected with siRNA or with a nontargeting scramble control siRNA for 48 h, followed by treatment with KBA for the indicated times. Rat Nrf2-specific siRNA (5′-ACGCAGGAGAGGGAAGAAUAAAGUU-3′ and 5′-AACUUUAUUCUUCCCUCUCCUGCGU-3′), rat HO-1-specific siRNA (5′-AUGGCAUAAAUUCCCACUGCCACGG-3′ and 5′-CCGUGGCAGUGGGAAUUUAUGCCAU-3′), and a nonspecific siRNA (5′-AUGCACGAUAUAACCUCACCGUCGG-3′ and 5′-CCGACGGUGAGGUUAUAUCGUGCAU-3′) were provided by Invitrogen (Carlsbad, CA, USA). Cell samples were then collected for Western blot analysis, MTT assay, and measurement of intracellular ROS levels.
Statistical Analysis
The statistical analyses were performed using SPSS 16.0 (SPSS Inc., Chicago, IL, USA). All of the values were presented as mean ± standard deviation (SD), except for the neurological deficit score, and differences between groups were compared with one-way ANOVA followed by followed by Fisher’s post hoc test. Neurological deficit scores were expressed as the median with range. This data was analyzed by the Kruskal–Wallis test followed by the Mann–Whitney U test and Bonferroni post hoc correction. P < 0.05 was regarded statistically significant.
Results
KBA Protects Against Cerebral Ischemic Injury
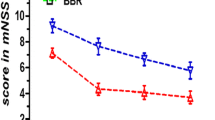
KBA (25 mg/kg) was injected intraperitoneally to rats 1 h after reperfusion to assess the in vivo neuroprotective effect. The infarct volume of the ipsilateral brain was measured 48 h later with TTC staining. KBA significantly decreased infarct volumes 48 h after reperfusion (Fig. 1a). As shown in Fig. 1b, a remarkably decreased pale-colored region was observed in the KBA + IR-treated rats (37.1 ± 5.0 %) compared with rats in the vehicle + IR group (23.2 ± 2.9 %) (P < 0.05; n = 6 animals per group). Neurological dysfunction after MCAO was characterized by impaired motility, grasping reflex, and placing reaction. In agreement with infarct volume measurement, KBA treatment significantly reduced the neurological deficit score compared with the vehicle treatment (Fig. 1b) (n = 6 animals per group; P < 0.05).

Posttreatment with 25 mg/kg 11-keto-β-boswellic acid (KBA) protects against cerebral ischemia reperfusion injury in middle cerebral artery occlusion (MCAO) rats. a Neurological deficit scores at 48 h after reperfusion (n = 6 animals per group). Data are presented as the median (range). b The percentage of infarct volume and representative 2,3,5-triphenyltetrazolium chloride (TTC) staining of the cerebral infarct in the rat brain. *P < 0.05 vs sham and #P < 0.05 vs vehicle + I/R
The protective effect of KBA against cerebral ischemic damage was supported by TUNEL staining and HE staining on sections from the ischemic cortex at 48 h after ischemia/reperfusion in rats (Fig. 2). Representative photomicrographs of HE staining are shown. The cells of cortex in sham rats showed an orderly arrangement. In addition, the cell outline was clear as well as the structure was compact. In vehicle + I/R group, the cells were arranged irregularly in ischemic peri-infarct of the cerebral cortex. Most of them were shrunken with a triangulated pycnotic nucleus. In contrast, damage was substantially reduced in the KBA + I/R group.

Representative images of HE staining and TUNEL staining performed on sections from the ischemic cortex at 48 h after ischemia/reperfusion in rats (TUNEL-positive cells in green, DAPI in blue; Scale bar = 20 μm). Quantitative analysis of TUNEL-positive cells is also exhibited. n = 6 animals for each group. *P < 0.05 vs sham and #P < 0.05 vs vehicle + I/R
To detect DNA fragmentation in situ, we performed TUNEL staining in brain sections. KBA reduced ischemia-induced DNA damage after 48 h of reperfusion. In the control group, TUNEL-positive cells were densely distributed in the ischemic cortex. The percentage of TUNEL-positive cells in the ischemic cortex was decreased from 55.0 ± 7.2 to 36.5 ± 4.4 % by KBA treatment (n = 6 animals per group; P < 0.05). The KBA-treated group exhibited a smaller amount of TUNEL-positive cells.
KBA Attenuated Oxidative Stress
SOD activity in the cortex was decreased in the vehicle group compared with the sham group, which was restored by KBA (n = 8 animals per group; P < 0.05) (Table 1). GPx was decreased after I/R (P < 0.05), while KBA could upregulate the activity of GPx significantly (P < 0.05 versus vehicle + I/R) (Table 1). The MDA level in the cortex, which is an index of lipid peroxidation, was significantly increased in the vehicle group compared with the sham-operated group. An evident reduction of the MDA level was observed in the KBA + I/R group compared with the vehicle + I/R group (P < 0.05) (Table 1).
KBA Promoted the Expression of Nrf2 and HO-1 in Cortex
To identify whether Nrf2/HO-1 signaling is involved in the neuroprotective effect of KBA, we analyzed ischemic brain tissue by Western blot and immunofluorescence staining. Western blot analysis in cortical tissues at 48 h after MCAO showed that Nrf2 and HO-1 protein expressions were increased at 48 h after ischemia while they were significantly further increased in KBA-treated group (Fig. 3a) (n = 6 animals per group; P < 0.05).

Effect of 25 mg/kg KBA on expression of Nrf2 and HO-1 in the brain. a Brain cortex tissues were collected at 48 h after cerebral ischemia/reperfusion injury and brain homogenates were evaluated by Western blot for Nrf2, HO-1, and actin. (n = 6 animals per group). *P < 0.05 vs sham and #P < 0.05 vs vehicle + I/R. Representative double immunofluorescent stainings for Nrf2/GFAP (b) and HO-1/GFAP (c) are shown in the ischemic cortex at 48 h after reperfusion and in the corresponding regions of sham controls. Scale bar = 20 μm
Consistently, immunofluorescence staining also showed that the expression of Nrf2 and HO-1 in astrocytes of the cortex was upregulated by KBA at 48 h after ischemia (Fig. 3b, c). In the sham group, few cells were stained by Nrf2 and HO-1. In the vehicle + I/R group, the number of cells stained by Nrf2 and HO-1 increased in the ischemic cortex. The number of cells labeled with Nrf2 and HO-1 in the KBA + I/R group was significantly increased compared with the vehicle + I/R group, which indicated that the Nrf2/HO-1 pathway may have a critical role in the KBA-mediated neuroprotection against I/R injuries in rats.
KBA Promoted Nrf2 Nuclear Translocation and Increased HO-1 Expression in a Concentration-Dependent Manner in Cells
In vitro, Fig. 4a showed the nuclear translocation of Nrf2 when cells were treated with the indicated concentrations of KBA for 24 h. Nrf2 was accumulated in the nucleus in a concentration-dependent manner while cytoplasmic Nrf2 levels were decreased gradually. We also found that the treatment of astrocyte cells with 10, 30, and 50 μM of KBA for 24 h increased HO-1 protein expression in a concentration-dependent manner (Fig. 4b). The highest induction level of HO-1 (up to sixfold protein expression) was observed after 50 μM of KBA treatment for 24 h (P < 0.05). Therefore, a 50 mM concentration of KBA was used in the following experiments. However, no significant effect was found in HO-1 expression after treatment with 50 μM of KBA in cells transfected with Nrf2 siRNA (P > 0.05). Knockdown of Nrf2 abolished HO-1 induction by KBA treatment (Fig. 4c).

KBA induces expression of Nrf2 and HO-1 in primary cultured astrocytes. All data represent the mean ± SD of triplicate independent experiments. a KBA induced increase nuclear localization and Nrf2 expression in a concentration-dependent manner. *P < 0.05 vs control and #P < 0.05 vs control. b KBA induced HO-1 expression in a concentration-dependent manner. *P < 0.05 vs control. c Cells were transiently transfected with control or Nrf2 siRNA for 48 h, followed by treatment with 50 μM of KBA for an additional 8 h. *P < 0.05 vs si-control group without KBA and #P < 0.05 vs si-control group with KBA
KBA Protects Astrocytes Against Injury Induced by OGD
OGD treatment resulted in 58 % cell death (P < 0.05). KBA significantly blocked OGD-induced cell death dose dependently (P < 0.05; Fig. 5a). Compared to the OGD group, the viability of the cells treated with 50 μM KBA was increased by approximately 30 % (P < 0.05). As shown in Fig. 5, ROS production of OGD model group markedly increased (up to 2.5-fold) when compared with the control group. Compared to the OGD model group, no difference was found in the ROS level of the groups treated with 10 mM KBA, while in the groups treated with 30 and 50 mM KBA, ROS level significantly decreased by approximately 32 and 44 %, respectively (P < 0.05). KBA effectively reduced OGD-induced increase of intracellular ROS level in a dose-dependent manner (Fig 5b).

KBA protects primary cultured astrocytes against OGD-induced cell death. KBA affords cell protection through the Nrf2/HO-1 pathway. All data represent the mean ± SD of triplicate independent experiments. KBA protects primary cultured astrocytes against OGD-induced cell death. a Cell viability assay using MTT; b intracellular ROS level. *P < 0.05 vs. control. c Cells were treated for 48 h with control or siRNA, then subjected to 60 min OGD followed by the MTT assay; d intracellular ROS level. (a, P < 0.05 vs. control; b, P < 0.05 vs. OGD; and c, P < 0.05 vs. KBA-treated OGD group)
The Protection of KBA Involves the Nrf2/HO-1 Pathway
To confirm the role of Nrf2/HO-1 pathway in KBA-mediated neuroprotection, we transfected control (si-control), HO-1-specific (si-HO-1), or Nrf2-specific (si-Nrf2) siRNA in primary cultured astrocytes for 48 h. Knockdown of Nrf2 or HO-1 inhibited cell viability that was increased by KBA under OGD (Fig. 5c). In consistent, knockdown of Nrf2 or HO-1 increased ROS level by 25 and 19 % compared with the si-control group, respectively (Fig. 5d). Taken together, KBA effectively prevented astrocytes from oxidative damage involved activation of Nrf2/HO-1 pathway.
Discussion
There is a considerable current interest in the neuroprotective effects of natural antioxidants against oxidative stress and the different defense mechanisms involved, such as sulforaphane [18] and resveratrol [19], can activate Nrf2 and increase expression of antioxidative genes. Due to its antioxidant properties, KBA has potential implications in treating oxidative injuries [9]; however, the mechanism of KBA protection was poorly understood. Our study shows for the first time that KBA has protective effects against cerebral I/R injury in an MCAO model demonstrated by improved neurologic scores, reduced infarct volume, and ameliorated neuronal apoptosis. The mechanism is possibly attributed to activating Nrf2/HO-1 pathway. More recently, triterpenoids structurally similar to KBA, such as maslinic acid and oleanolic acid, have been reported to significantly increase Nrf2, leading to neuroprotection [20, 11]. In another approach, stimulating Nrf2 by triterpenoid could effectively reduce 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced oxidative stress in the mouse model while Nrf2 knockout mice failed to block against MPTP neurotoxicity, implying a direct protective role of Nrf2/ARE against MPTP neurotoxicity [21]. These data are noteworthy and suggest that specific triterpenoid compounds could be beneficial for the treatment of ischemic stroke related to oxidative stress.
Nrf2 is a master regulator of the antioxidative defense responses. Our in vivo and in vitro studies found that KBA regulates Nrf2, thereby enhancing the protective defense mechanisms through the Nrf2/HO-1 pathway. KBA markedly increased the expression of Nrf2 and HO-1 in the ischemic cortex at 48 h after MCAO as well as in primary culture astrocytes. Consistent with this, the double immunofluorescent studies revealed that in the cortex, the HO-1 was clearly upregulated. On the other hand, consistent to our findings in this study, Nrf2 and HO-1 induction had been reported previously in the rat brain following transient focal ischemia [22, 23]. In primary astrocyte cells, we found that KBA caused Nrf2 nuclear translocation in a concentration-dependent manner. Activation and nuclear accumulation of Nrf2 upregulates endogenous antioxidant defenses to restore cellular redox homeostasis via the induction of phase II defense enzymes and antioxidant stress proteins [24, 25]. Existing data has demonstrated that Nrf2-deficient mice are more susceptible to oxidative stress [26]. Another study on wild-type and Nrf2 knockout mice also showed that Nrf2 reduces ischemic brain injury by protecting against oxidative stress [27]. The HO-1 promoter is known to have a large number of ARE sequences to which Nrf2 can bind to induce its expression in a preferential manner [28]. As shown in our in vitro study, the si-Nrf2 treatment significantly decreased the level of Nrf2 in nuclear extracts from cells treated with KBA and reduced the upregulation of its target gene HO-1.
Noteworthy, the expression of HO-1 is mediated by several signaling pathways and transcription factors, including Nrf2, AP-1, and NF-kB as the most prominent [29]. Among them, redox-dependent Keap1/Nrf2 system plays a central role for HO-1 induction in response to oxidative stress [30]. HO-1 has been reported to protect tissues by restoring redox homeostasis and reducing inflammation due to its antioxidant, antiapoptotic, and anti-inflammatory effects. HO-1 is more likely to exert a central role in neuroprotection because it is inducible to degrade free heme, and its metabolites, CO or biliverdin/bilirubin, can directly provide cytoprotection [31, 32]. HO-1 has been implicated to be particularly important in neuroprotection against cerebral ischemia, as evidenced by HO-1 knockout mice exhibiting greater ischemic damage as compared to wild-type mice [33].
With KBA treatment in MCAO, enhanced activities of superoxide dismutase (SOD) and glutathione peroxidase (GPx) and a decreased level of MDA were observed. SOD dismutates superoxide to hydrogen peroxide and oxygen, and GPx eliminates hydrogen peroxide, which is potentially converted to other radicals [34]. MDA is not only produced by oxidative stress-induced peroxidation but also by enzymatically produced lipid peroxidation during the arachidonic acid cascade which is an important element of postischemic secondary injury [34]. The reduced activities of SOD and GPx and enhanced MDA level in the vehicle group imply that severe oxidative stress occurred during permanent MCAO that increased free radical activity and reciprocally reduced endogenous antioxidants occurring during cerebral ischemia. Our results are consistent with previous work [9, 35]. We propose that an in vivo therapeutic effect of KBA is related to an antioxidant effect by enhancing Nrf2 regulation and, therefore, alleviates oxidative stress during ischemic stroke.
Activation of Nrf2 in astrocytes protects neurons from a wide array of potentially toxic insults. Nrf2 activation in astrocytes has thus been proposed as a novel therapeutic target for neuroprotection [36]. Astrocytes, the major glial nonneuronal cells, play an important role in the cellular antioxidant defense in the brain. They are the main source of glutathione (GSH) and supply the neurons with substrate for GSH synthesis to improve the neuronal antioxidative reserves [37]. ARE-regulated genes are preferentially activated in astrocytes, which consequently have more efficient detoxification and antioxidant defense than neurons.
According to the MTT assay and ROS measurement in vitro, pretreatment with KBA before OGD damage can significantly reduce cell death. We speculate that the protective effect was due to the antioxidant properties of KBA. In cell viability assays, KBA reduced astrocytes death triggered by OGD. The fact that KBA could inhibit the increase in ROS also shows that KBA may have the ability to rescue cells from OGD. These in vitro data support our in vivo results and show that KBA promotes an antioxidative effect.
The present study also demonstrated that knockdown of Nrf2 or HO-1 in primary cultured astrocytes that were subjected to OGD partly diminished KBA’s neuroprotective effect. These observations strongly suggest that Nrf2/HO-1 is required for KBA-dependent cytoprotection against oxidative stress, although other enzymes can also assist with KBA neuroprotection. Although the mechanisms leading to nuclear translocation of Nrf2 are poorly defined, we believe that Nrf2 and HO-1 induced by KBA decreased ROS in astrocytes and that it is responsible, at least in part, for the protective effects against OGD.
This study focused on the protective effect of KBA against oxidative stress-induced ischemic injury and did not explore in detail whether other signaling pathways contribute to the effects of KBA. Moreover, because we only performed mechanism research in astrocytes involved in the Nrf2/HO-1 pathway, we should consider the neurons or other cell types may also involve in this process as a previous study concluded [38]. Another limitation was that, in this study, just merely a single dose of KBA was used in animal experiments. In myocardial I/R injury rats, KBA exerted a dose-dependent cardioprotective effect through mechanisms related to enhancement of antioxidant capacity [39]. Therapeutic time window and the dose–response relationship of KBA injection against cerebral I/R injury in rat should be determined.
To the best of our knowledge, this is the first demonstration of the therapeutic potential of KBA on permanent MCAO in rats. We revealed that posttreatment with KBA 1 h after reperfusion could attenuate ischemic injury in an MCAO model. Furthermore, in vivo studies showed that AKBA protects astrocytes against OGD-induced cell death activating the Nrf2/HO-1 pathway. However, knockdown of HO-1 or Nrf2 partly blocked the protective effect of AKBA. Therefore, our study provides evidence about the therapeutic potential of targeting the Nrf2/HO-1 pathway with KBA to prevent brain injury after ischemic stroke.
Abbreviations
- KBA:
-
11-Keto-β-boswellic acid
- ARE:
-
Antioxidant response elements
- CBF:
-
Cerebral blood flow
- EMSA:
-
Electrophoresis mobility shift assay
- GPx:
-
Glutathione peroxidase
- I/R:
-
Ischemia–reperfusion
- HE:
-
Hematoxylin and eosin
- HO-1:
-
Heme oxygenase-1
- Keap1:
-
Kelch-like ECH-associated protein 1
- MCAO:
-
Middle cerebral artery occlusion
- MDA:
-
Malondialdehyde
- MTT:
-
3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide
- Nrf2:
-
Nuclear factor erythroid-2-related factor 2
- OGD:
-
Oxygen and glucose deprivation
- PBS:
-
Phosphate-buffered saline
- ROS:
-
Reactive oxygen species
- SD:
-
Standard deviation
- siRNA:
-
Small interfering RNA
- SOD:
-
Superoxide dismutase
- TTC:
-
2,3,5-Triphenyltetrazolium chloride
- TUNEL:
-
Terminal deoxynucleotidyl transferase dUTP nick end labeling
References
Chan PH (1994) Oxygen radicals in focal cerebral ischemia. Brain Pathol 4(1):59–65
Jaiswal AK (2004) Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med 36(10):1199–1207
Shah ZA, R-c L, Ahmad AS, Kensler TW, Yamamoto M, Biswal S, Doré S (2010) The flavanol (−)-epicatechin prevents stroke damage through the Nrf2/HO1 pathway. J Cereb Blood Flow Metab 30(12):1951–1961
Alfieri A, Srivastava S, Siow R, Cash D, Modo M, Duchen MR, Fraser PA, Williams SC, Mann GE (2013) Sulforaphane preconditioning of the Nrf2/HO-1 defense pathway protects the cerebral vasculature against blood–brain barrier disruption and neurological deficits in stroke. Free Radic Biol Med 65:1012–1022
Guo C, Zhu Y, Weng Y, Wang S, Guan Y, Wei G, Yin Y, Xi M, Wen A (2014) Therapeutic time window and underlying therapeutic mechanism of breviscapine injection against cerebral ischemia/reperfusion injury in rats. J Ethnopharmacol 151(1):660–666. doi:10.1016/j.jep.2013.11.026
Ding Y, Zhang B, Zhou K, Chen M, Wang M, Jia Y, Song Y, Li Y, Wen A (2014) Dietary ellagic acid improves oxidant-induced endothelial dysfunction and atherosclerosis: role of Nrf2 activation. Int J Cardiol. doi:10.1016/j.ijcard.2014.06.045
Safayhi H, Mack T, Sabieraj J, Anazodo MI, Subramanian LR, Ammon HP (1992) Boswellic acids: novel, specific, nonredox inhibitors of 5-lipoxygenase. J Pharmacol Exp Ther 261(3):1143–1146
Assimopoulou A, Zlatanos S, Papageorgiou V (2005) Antioxidant activity of natural resins and bioactive triterpenes in oil substrates. Food Chem 92(4):721–727
Hartmann RM, Morgan Martins MI, Tieppo J, Fillmann HS, Marroni NP (2012) Effect of Boswellia serrata on antioxidant status in an experimental model of colitis rats induced by acetic acid. Dig Dis Sci 57(8):2038–2044. doi:10.1007/s10620-012-2134-3
Elshazly SM, El Motteleb DMA, Nassar NN (2013) The selective 5-LOX inhibitor 11-keto-β-boswellic acid protects against myocardial ischemia reperfusion injury in rats: involvement of redox and inflammatory cascades. Naunyn Schmiedeberg’s Arch Pharmacol 386(9):823–833
Rong ZT, Gong XJ, Sun HB, Li YM, Ji H (2011) Protective effects of oleanolic acid on cerebral ischemic damage in vivo and H(2)O(2)-induced injury in vitro. Pharm Biol 49(1):78–85. doi:10.3109/13880209.2010.499130
Li L, Zhang X, Cui L, Wang L, Liu H, Ji H, Du Y (2013) Ursolic acid promotes the neuroprotection by activating Nrf2 pathway after cerebral ischemia in mice. Brain Res 1497:32–39. doi:10.1016/j.brainres.2012.12.032
Yan W, Fang Z, Yang Q, Dong H, Lu Y, Lei C, Xiong L (2013) SirT1 mediates hyperbaric oxygen preconditioning-induced ischemic tolerance in rat brain. J Cereb Blood Flow Metab: Off J Int Soc Cereb Blood Flow Metab 33(3):396–406. doi:10.1038/jcbfm.2012.179
Kruger P, Daneshfar R, Eckert GP, Klein J, Volmer DA, Bahr U, Muller WE, Karas M, Schubert-Zsilavecz M, Abdel-Tawab M (2008) Metabolism of boswellic acids in vitro and in vivo. Drug Metab Dispos: Biol Fate Chem 36(6):1135–1142. doi:10.1124/dmd.107.018424
Reising K, Meins J, Bastian B, Eckert G, Mueller WE, Schubert-Zsilavecz M, Abdel-Tawab M (2005) Determination of boswellic acids in brain and plasma by high-performance liquid chromatography/tandem mass spectrometry. Anal Chem 77(20):6640–6645. doi:10.1021/ac0506478
Garcia JH, Wagner S, Liu KF, Hu XJ (1995) Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke; J Cereb Circ 26(4):627–634, discussion 635
Halim ND, McFate T, Mohyeldin A, Okagaki P, Korotchkina LG, Patel MS, Jeoung NH, Harris RA, Schell MJ, Verma A (2010) Phosphorylation status of pyruvate dehydrogenase distinguishes metabolic phenotypes of cultured rat brain astrocytes and neurons. Glia 58(10):1168–1176. doi:10.1002/glia.20996
Zhao J, Kobori N, Aronowski J, Dash PK (2006) Sulforaphane reduces infarct volume following focal cerebral ischemia in rodents. Neurosci Lett 393(2–3):108–112. doi:10.1016/j.neulet.2005.09.065
Ungvari Z, Bagi Z, Feher A, Recchia FA, Sonntag WE, Pearson K, de Cabo R, Csiszar A (2010) Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am J Physiol Heart Circ Physiol 299(1):H18–H24. doi:10.1152/ajpheart.00260.2010
Qian Y, Guan T, Tang X, Huang L, Huang M, Li Y, Sun H (2011) Maslinic acid, a natural triterpenoid compound from Olea europaea, protects cortical neurons against oxygen-glucose deprivation-induced injury. Eur J Pharmacol 670(1):148–153. doi:10.1016/j.ejphar.2011.07.037
Kaidery NA, Banerjee R, Yang L, Smirnova NA, Hushpulian DM, Liby KT, Williams CR, Yamamoto M, Kensler TW, Ratan RR, Sporn MB, Beal MF, Gazaryan IG, Thomas B (2013) Targeting Nrf2-mediated gene transcription by extremely potent synthetic triterpenoids attenuate dopaminergic neurotoxicity in the MPTP mouse model of Parkinson’s disease. Antioxid Redox Signal 18(2):139–157. doi:10.1089/ars.2011.4491
Tanaka N, Ikeda Y, Ohta Y, Deguchi K, Tian F, Shang J, Matsuura T, Abe K (2011) Expression of Keap1-Nrf2 system and antioxidative proteins in mouse brain after transient middle cerebral artery occlusion. Brain Res 1370:246–253. doi:10.1016/j.brainres.2010.11.010
Nimura T, Weinstein PR, Massa SM, Panter S, Sharp FR (1996) Heme oxygenase-1 (HO-1) protein induction in rat brain following focal ischemia. Brain Res Mol Brain Res 37(1–2):201–208
Ishii T, Itoh K, Ruiz E, Leake DS, Unoki H, Yamamoto M, Mann GE (2004) Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: activation by oxidatively modified LDL and 4-hydroxynonenal. Circ Res 94(5):609–616. doi:10.1161/01.RES.0000119171.44657.45
Taguchi K, Motohashi H, Yamamoto M (2011) Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells: Devoted Mol Cell Mech 16(2):123–140. doi:10.1111/j.1365-2443.2010.01473.x
Zhao Z, Chen Y, Wang J, Sternberg P, Freeman ML, Grossniklaus HE, Cai J (2011) Age-related retinopathy in NRF2-deficient mice. PLoS One 6(4):e19456. doi:10.1371/journal.pone.0019456
Shah ZA, Li RC, Thimmulappa RK, Kensler TW, Yamamoto M, Biswal S, Dore S (2007) Role of reactive oxygen species in modulation of Nrf2 following ischemic reperfusion injury. Neuroscience 147(1):53–59. doi:10.1016/j.neuroscience.2007.02.066
Kensler TW, Wakabayashi N, Biswal S (2007) Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol 47:89–116. doi:10.1146/annurev.pharmtox.46.120604.141046
Durante W (2010) Targeting heme oxygenase-1 in vascular disease. Curr Drug Targets 11(12):1504–1516
Paine A, Eiz-Vesper B, Blasczyk R, Immenschuh S (2010) Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem Pharmacol 80(12):1895–1903. doi:10.1016/j.bcp.2010.07.014
Zeynalov E, Dore S (2009) Low doses of carbon monoxide protect against experimental focal brain ischemia. Neurotox Res 15(2):133–137. doi:10.1007/s12640-009-9014-4
Dore S, Takahashi M, Ferris CD, Zakhary R, Hester LD, Guastella D, Snyder SH (1999) Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc Natl Acad Sci U S A 96(5):2445–2450
Kim YM, Pae HO, Park JE, Lee YC, Woo JM, Kim NH, Choi YK, Lee BS, Kim SR, Chung HT (2011) Heme oxygenase in the regulation of vascular biology: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal 14(1):137–167. doi:10.1089/ars.2010.3153
Warner DS, Sheng H, Batinic-Haberle I (2004) Oxidants, antioxidants and the ischemic brain. J Exp Biol 207(Pt 18):3221–3231. doi:10.1242/jeb.01022
Yin MC, Lin MC, Mong MC, Lin CY (2012) Bioavailability, distribution, and antioxidative effects of selected triterpenes in mice. J Agric Food Chem 60(31):7697–7701. doi:10.1021/jf302529x
Bell KF, Al-Mubarak B, Fowler JH, Baxter PS, Gupta K, Tsujita T, Chowdhry S, Patani R, Chandran S, Horsburgh K (2011) Mild oxidative stress activates Nrf2 in astrocytes, which contributes to neuroprotective ischemic preconditioning. Proc Natl Acad Sci 108(1):E1–E2
Dringen R, Pfeiffer B, Hamprecht B (1999) Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione. J Neurosci Off J Soc Neurosci 19(2):562–569
Zhang M, Wang S, Mao L, Leak RK, Shi Y, Zhang W, Hu X, Sun B, Cao G, Gao Y (2014) Omega-3 fatty acids protect the brain against ischemic injury by activating Nrf2 and upregulating heme oxygenase 1. J Neurosci 34(5):1903–1915
Elshazly SM, Abd El Motteleb DM, Nassar NN (2013) The selective 5-LOX inhibitor 11-keto-beta-boswellic acid protects against myocardial ischemia reperfusion injury in rats: involvement of redox and inflammatory cascades. Naunyn Schmiedeberg’s Arch Pharmacol 386(9):823–833. doi:10.1007/s00210-013-0885-9
Acknowledgments
This work received the support of the grant from the Key Technologies for New Drug Innovation and Development of China (No. 2011ZXJ09202-13; No. 2012BAK25B00) and National Natural Science Foundation of China (No.81373947; No.81201985).
Conflict of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Ding, Y., Chen, M., Wang, M. et al. Posttreatment with 11-Keto-β-Boswellic Acid Ameliorates Cerebral Ischemia–Reperfusion Injury: Nrf2/HO-1 Pathway as a Potential Mechanism. Mol Neurobiol 52, 1430–1439 (2015). https://doi.org/10.1007/s12035-014-8929-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-014-8929-9