
Abstract
Ischemic strokes are associated with a high rate of disability and death globally. Cerebral ischemia/reperfusion (I/R) injury is a type of brain damage associated with oxidative stress after an ischemic stroke. Beta-boswellic acid (β-BA) reportedly exerts antioxidant and neuroprotective effects, but its role in cerebral I/R injury is unclear. The aim of this research was to investigate the neuroprotective effects, as well as the mechanisms of β-BA in cerebral I/R injury. In vivo experiments were conducted using a rat middle cerebral artery occlusion and reperfusion (MCAO/R) model, and in vitro experiments were performed using a rat neuronal oxygen–glucose deprivation and reoxygenation (OGD/R) model. Triphenyltetrazolium chloride staining, neurological function scores, terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick end labeling, hematoxylin and eosin staining, and antioxidant levels in the brain were used to assess the effects of β-BA. Flow cytometry was used to detect reactive oxygen species and apoptotic cells. Western blotting and immunofluorescence staining were used to measure protein levels. The results showed that β-BA markedly improved neurological deficits and decreased infarct volume and necrotic neurons in rats. The in vitro results showed that β-BA protected neurons against OGD/R-induced injury. Additionally, β-BA significantly increased the phosphorylation of protein kinase C epsilon (PRKCE) at S729, the translocation of nuclear factor erythroid 2-like 2 (NFE2L2), and expression of heme oxygenase-1 (HMOX1). This study demonstrates that β-BA exerts neuroprotective effects against cerebral I/R via the activation of the PRKCE/NFE2L2/HMOX1 pathway and is a potential therapeutic candidate for ischemic stroke.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ischemic strokes caused by vascular obstruction are associated with a high rate of disability and death globally [1]. Thrombolysis is an effective stroke therapeutic approach, but owing to the narrow time window and applicability, it is recommended for only a small percentage of patients. In addition, recanalization of occluded cerebral vessels allows the rapid restoration of blood flow and increases tissue oxygenation levels, leads to reactive oxygen species (ROS) burst and reperfusion injury [2]. Excessive formation of ROS during cerebral ischemia/reperfusion (I/R) injury can cause changes in lipids and proteins, as well as DNA oxidative damage in the brain, leading to tissue damage and neurological disorders [3]. Therefore, antioxidant measures, such as antioxidant enzymes and free radical scavengers, are viable strategies for ischemic stroke prevention and therapy [4].
Beta‐boswellic acid (β-BA), the principal component of the pentacyclic triterpene boswellic acids (BAs) derived primarily from Boswellia serrata resin, has traditionally been used to enhance learning and memory. Recent studies have reported the antioxidant effects of BAs [5]. Studies have shown that β-BA significantly increases neurodevelopmental processes, including elongation of neurites, number of branches, and formation of tubulin polymers in embryonic hippocampal cells [6]. Thus, β-BA may have a protective effect against neurological diseases caused by neuronal damage. However, the precise mechanism underlying its actions remains unclear. Boswellic acid can mitigate fipronil-induced neurobehavioral toxicity by suppressing oxidation [7], and β-BA can significantly reduce serum malondialdehyde level and increase serum superoxide dismutase level in diabetic rats [8]. Based on these observations, we hypothesized that β-BA plays a neuroprotective role in cerebral I/R injury by influencing oxidative stress injury.
Protein kinase C epsilon (PRKCE) belongs to the classical protein kinase C family (plays a key role in neurological diseases such as stroke, Parkinson’s disease, and Alzheimer’s disease); activation of PRKCE can inhibit ROS production, reduce neuronal death, encourage cells to resist oxidative stress injury [9], and reduce the cerebral infarction area in rats with cerebral I/R injury [10]. PRKCE is considered an upstream signal and a key regulator for nuclear factor erythroid 2-like 2 (NFE2L2) activation [11]. NFE2L2 is an important redox-sensitive transcription factor that contributes to the improvement of the oxidative stress status of the body, promotion of cell survival, and maintenance of cellular redox homeostasis by regulating the constitutive and inducible expression of phase II detoxification and antioxidant enzymes in cells [12]. NFE2L2 further regulates antioxidant enzyme heme oxygenase-1 (HMOX1) expression, which is an important antioxidant enzyme that catalyzes the catabolism of heme to ferrous iron, carbon monoxide, and biliverdin (possesses effective ROS-scavenging activity). In our previous study, we evaluated the neuroprotective effects of natural antioxidants mediated through the antioxidant NFE2L2/HMOX1 pathway and confirmed the important role of NFE2L2/ HMOX1 in protection against oxidative stress in experimental stroke animal models [13]. Activation of PKCε activates NFE2L2/HMOX1 and reduces the excessive accumulation of ROS caused by ischemic stroke [3]. Therefore, we speculated that the antioxidant effect of β-BA on cerebral I/R injury is associated with the PRKCE/NFE2L2/HMOX1 signaling pathway.
This study was conducted using in vivo middle cerebral artery occlusion and reperfusion (MCAO/R) and in vitro oxygen–glucose deprivation and reoxygenation (OGD/R) models to investigate the neuroprotective effects and the action mechanisms of β-BA in cerebral I/R injury. We believe that our findings will provide new ideas and experimental clues for the treatment of brain I/R injury.
Material and Methods
Reagents and Chemicals
β-BA (purity > 95%) was obtained from Shanghai Pureone Biological Technology Co., Ltd. (Shanghai, China). Neurobasal medium, glutamine, and B27 were obtained from Gibco Life Technologies (Rockville, MD, USA). Dulbecco’s modified Eagle’s medium was purchased from Hyclone Inc. (Logan, UT, USA). Cytarabine was purchased from Tokyo Chemical Industry Co. (Tokyo, Japan). Trypsin was purchased from Bio-Technology Co., Ltd. (Shanghai, China). Fetal bovine serum (FBS) was purchased from Biological Industries (Kibbutz Beit Haeme, Israel). Triphenyltetrazolium chloride (TTC) and hematoxylin were purchased from Solarbio Life Sciences (Beijing, China). Eosin was purchased from Sangon Biotech (Shanghai, China). 4′,6-Diamidino-2-phenylindole (DAPI) was purchased from Aladdin Bio-Chem Technology Co., Ltd. (Shanghai, China). Polybrene was purchased from Sigma Chemical Co. (St. Louis, MO, USA). Commercial kits for the detection of ROS, glutathione peroxidase (GSH-Px), and lactate dehydrogenase (LDH); 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay kit; terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling staining (TUNEL) kit; annexin V-FITC/PI apoptosis detection kit; and antibodies for NFE2L2, HMOX1, β-actin, and histone H3 were obtained from Wanleibio Co., Ltd. (Shenyang, China). 4-Hydroxynonenal (4-HNE) and 8-hydroxy-2-deoxyguanosine (8-OHdG) enzyme-linked immunosorbent assay kits were obtained from Fine Biotech Co., Ltd. (Wuhan, China). Antibodies for p-PRKCE and PRKCE were purchased from Abcam PLC. (Cambridge, UK).
Animals and Focal Cerebral Ischemia Model
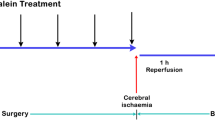
Six- to 8-week-old male Sprague–Dawley (SD) rats were obtained from the Animal Center of Fourth Military Medical University. The animal procedures for this study were conducted in accordance with internationally accepted principles for laboratory animal use and care (European community guideline) and were approved by the Animal Experimentation Ethics Committee of the Fourth Military Medical University. The rats were placed in a controlled environment at 22 °C ± 1 °C, 45–55% relative humidity, and a 12-h light–dark cycle, with free access to food and water. Before conducting the experiments, all rats were adapted to laboratory conditions for 1 week. The rats were randomly divided into the following five groups of six animals each: sham-operated (sham), I/R, and I/R + β-BA (10, 20, and 40 mg/kg). β-BA dose was determined based on the findings of a previous study [14]. The administration group rats received an intraperitoneal injection [13] of β-BA immediately after reperfusion, whereas the sham and I/R group rats received saline as a vehicle control.
The MCAO/R model was established as described previously [15]. The rats were intraperitoneally injected with 10% chloral hydrate (3 mL/kg) for anesthesia. A small incision was made near the bifurcation of the external carotid artery, and a 3–0 nylon suture (Beijing Cinontech Co., Ltd., China) was inserted into the right common carotid artery and advanced through the internal carotid artery to obstruct the middle cerebral artery. After 2 h of ischemia, we gently pulled the suture to restore blood flow and perfused for another 24 or 48 h. The same procedure was performed on the sham-operated rats, without blocking the middle cerebral artery.
Evaluation of Neurological Deficit
Neurological deficits were measured using Longa’s five-score system [16] as follows: 0 points for no deficit, 1 point for inability to fully straighten the forelimb on the opposite side from the lesion when lifting the tail, 2 points for rotational signs to the paralyzed side, 3 points for falling to the opposite side from the lesion, and 4 points for no spontaneous activity and impaired consciousness.
Evaluation of Infarct Size
After neurological evaluation, the rats were sacrificed, and the brain tissue was sampled and stored at − 70 °C for 20 min. The entire rat brain was cut into uniform 2-mm coronal sections, stained with 1% TTC for 30 min, and fixed with 4% formalin buffer solution for 24 h. A digital camera was used to image brain slices, and the infarct volume of each slice was assessed using ImageJ.
Histological Observation and TUNEL Staining
The brain tissues of the rats were fixed by transcardiac perfusion and paraffin blocks were prepared. First, the rats were anesthetized using an intraperitoneal injection of 10% chloral hydrate (3 mL/kg). The heart was then exposed by opening the thoracic cavity after anesthesia and the perfusion needle was inserted into the aorta through the left ventricle to fix it. Afterwards, a small incision was made in the right atrium and 150 mL of saline (4 °C) were perfused until the effluent from the right atrium became clear. Then, 300 mL of 4% paraformaldehyde (4 °C) were slowly dripped until the limbs, trunk, and tail became stiff. After severing the head, the brain was harvested, fixed in 4% paraformaldehyde, and soaked at 4 °C for 24 h. The fixed brain tissues were rinsed with tap water for 4 h, then dehydrated with different concentrations of alcohol. The dehydrated tissue blocks were placed in xylene until the specimens became transparent. The tissue blocks were transferred to xylene and paraffin wax and placed at 60 °C for 2 h to allow for the complete penetration of paraffin wax into the brain tissue. The dissolved paraffin wax was poured into a metal frame, and then the wax-impregnated tissues were buried in the center of the frame and cooled to become a hard wax block.
The wax blocks were cut into 5-μm coronal sections which were then dried in a 60 °C oven for 2 h. Subsequently, the brain sections were soaked in xylene and rehydrated through a series of graded ethanol solutions. The brain sections were soaked in hematoxylin for 5 min and stained with eosin solution for 3 min to observe morphological changes in the brain ischemia penumbra tissue. Following the manufacturer’s instructions, TUNEL staining was performed. Briefly, a drop of 0.1% Triton X-100 (50 μL) was added on the sections, which were then stored at 37 °C for 8 min. The sections were reacted with the TUNEL reaction mixture containing the label and enzyme solution in the dark at 37 °C for 60 min. The sections were washed three times with phosphate buffered saline (PBS) for 5 min each and stained with DAPI for 5 min at 37 °C. The total number of TUNEL-positive neurons in the ipsilateral hemisphere was determined in three different fields for each section by an investigator blinded to the experiments, using a fluorescence microscope (BX53; Olympus, Tokyo, Japan).
Measurement of GSH-Px, 4-HNE, and 8-OHdG
After 24 h of reperfusion, the rats were sacrificed and the peri-infarct brain tissues were collected. The brain tissues were weighed and mechanically homogenized with saline under ice bath conditions to prepare a 10% homogenate supernatant. The tissue homogenate supernatant was subjected to protein content determination. Thereafter, GSH-Px, 4-HNE, and 8-OHdG levels were determined using kits according to the manufacturer’s instructions, and the absorbance of the samples was measured at 570 nm using a microplate reader (Elx800 Bio-Tek, Winooski, VT, USA).
Immunofluorescence Staining
After 24 h of reperfusion, the brain tissue was fixed by transcardiac perfusion using 4% paraformaldehyde. Paraffin-embedded brain tissue was cut into 5-μm coronal sections, dewaxed with xylene, sequentially immersed in a graded series of ethanol solution, subsequently incubated with goat serum dropwise for 15 min, and then incubated at 4 °C overnight with the NFE2L2 antibody (1:100). Subsequently, the tissue was incubated with the corresponding secondary IgG/HRP polymer antibody (1:200) for 60 min. The sections were then stained with DAPI. Staining was observed under a BX53 fluorescence microscope.
Cell Culture
As previously described [4], 2-day-old SD rats were sterilized by immersion in 75% ethanol for 5 min, and hippocampal tissues were removed with the rats under aseptic conditions and then isolated in 0.125% trypsin solution. Primary hippocampal neurons were cultured with neurobasal medium containing 100 U/mL penicillin and streptomycin, 0.5 mM glutamine, and 2% B27. The medium was changed every 3 days. The cells were incubated for 7–10 days before experiments.
OGD/R Model
Neurons were treated with or without β-BA (10, 20, and 40 μM) for 24 h before in vitro exposure to OGD. To simulate ischemia-like conditions, the culture medium was replaced with glucose-free Dulbecco’s modified Eagle medium. Thereafter, the neurons were cultured in an incubator for 2 h with 95% N2 and 5% CO2. They were then incubated for 24 h at 37 °C with or without β-BA under 95% O2 and 5% CO2. Blank control neurons were not treated with OGD/R or β-BA.
Cell Viability Assay
Neurons were inoculated in 96-well plates (2 × 104 cells/well), and 20 μL of MTT solution (0.5 mg/mL) was added per well. The cells were then incubated in a carbon dioxide cell incubator for 4 h at 37 °C. Next, 150 μL of dimethyl sulfoxide was added to each well, and the plates were placed in dark for 10 min. An Elx800 microplate reader was used to measure the absorbance of the samples at 570 nm.
Cytotoxicity Assay
First, neurons were inoculated in 96-well plates (2 × 104 cells/well). After reperfusion, 50 µL of medium was taken and LDH release was determined using an assay kit following the manufacturer’s instruction. Using an Elx800 microplate reader, the absorbance of the samples was measured at 570 nm.
Flow Cytometric Analysis for Apoptosis
To measure neuronal death, neurons were first inoculated in six-well plates (7 × 105 cells/well). The neurons were harvested after 24 h of reperfusion, and annexin V-fluorescein isothiocyanate (FITC) (20 μg/mL, 5 μL) was added, and the solution was mixed well. Thereafter, propidium iodide (PI) (50 μg/mL, 10 μL) was added, and the solution was mixed well and incubated for 15 min while protected from light. Apoptosis was quantified using flow cytometry (ACEA Bioscience, San Diego, CA, USA).
Measurement of Intracellular ROS
Intracellular ROS levels were determined using the 2,7-dichlorofluorescein diacetate (DCFH-DA) assay. After reperfusion, neurons were harvested, centrifuged at 140 g for 5 min, the supernatant was aspirated, and the neurons were washed with PBS. We added 1 mL of DCFH-DA diluent separately, and the samples were mixed well and incubated at 37 °C for 30 min in an incubator. The neurons were then washed three times with PBS and resuspended in 500 μL of PBS, and fluorescence intensity was detected using flow cytometry.
For in vivo experiments, after reperfusion, the peri-infarct brain tissue was weighed, homogenized by adding homogenization medium (10 mM phosphate buffer), and then centrifuged at 1000 g for 10 min, and a portion of the supernatant was used for protein determination. The supernatant was incubated with 10 μL (1 mmol/L) DCFH-DA for 30 min at 37 °C. The fluorescence of the samples was read at 500 nm excitation wavelength and 525 nm emission wavelength with a fluorescence plate reader (SynergyH1, Biotek, USA).
Knockdown of Prkce Using Short Hairpin RNA
A Prkce-targeted lentiviral vector was constructed by Genepharma (Shanghai, China). Short hairpin RNA (shRNA) of rat Prkce was obtained against the target sequence: 5′-CCAAGAGAGATGTCAATAA-3′. The supernatant containing lentivirus was concentrated using an ultracentrifuge-based and sedimentation-based method and subsequently used for titration. The titer of the lentivirus was 1 × 109 TU/mL.
As previously described, rats and cells were transfected with the shRNA [17, 18]. Briefly, 60–80% confluent cells were transfected with either lentivirus (the multiplicity of infection was 10) or a negative control. Meanwhile, 5 μg/mL polybrene was added to the culture medium to improve the efficiency of virus infection. After 48 h of transfection, the efficiency of RNA interference was assessed using western blotting. The rats were anesthetized, and a fine hole was bored in their skulls. Thereafter, 1.5 μL of lentiviral suspension or a negative control was slowly injected (0.2 μL/min) into the lateral ventricle (3.6 mm depth from the pial surface, 0.8 mm behind the bregma, and 1.4 mm left and right). The experiment was performed 7 days later.
Western Blotting
After reperfusion, proteins were extracted from the brain tissue (ischemic penumbra) and primary hippocampal neurons. Proteins were transferred onto a nitrocellulose membrane. After blocking, the membranes were incubated with primary antibodies against NFE2L2 (1:500), HMOX1 (1:500), PRKCE (1:1000), p-PRKCE (1:1000), histone H3 (1:5000), and β-actin (1:5000) overnight at 4 °C. The membranes were washed, and then incubated with the corresponding secondary antibodies at 37 °C for 45 min. The membranes were visualized using electrochemiluminescence (Wanleibio) and analyzed using ImageJ (Version 1.44).
Quantitative Reverse Transcription-Polymerase Chain Reaction Assay
TRIpure reagent (Bio Teke, Beijing, China) was used to isolate the total RNA from neurons. Super M-MLV reverse transcriptase was used to obtain cDNA (BioTeke). Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) was performed on the Exicycler 96 real-time PCR system (Bioneer Corporation, Daejeon, Korea) using SYBR Green (Solarbio, Beijing, China), 2 × Power Taq PCR Master Mix (BioTeke), and specific Nfe2l2 and β-actin primers. The 2−ΔΔCT method was used to calculate relative mRNA levels. Gene expression analyses were performed using the following primers: Nfe2l2 Forward: 5′-CCATTTACGGAGACCCAC-3′, Nfe2l2 Reverse: 5′-AGCACTGTGCCCTTGAGC-3′, β-actin Forward: 5′-TTCCAGCCTTCCTTCCTG-3′, β-actin Reverse: 5′-GGCATAGAGGTCTTTACGG-3′.
Statistical Analysis
Results are expressed as mean ± standard deviation (n = 6 per group). Differences between groups were compared using a one-way ANOVA or an appropriate t-test. Data analysis was conducted using GraphPad Prism Version 8 (San Diego, CA, USA). Results with P < 0.05 were considered statistically significant.
Results
β-BA Reduced Infarct Volume and Ameliorated Neurological Deficits
Neurological score and infarct volume are markers for cerebral injury. Here, neurological scores were examined 24 h after reperfusion (Fig. 1a). The results revealed no neurological deficits in the sham group, whereas a significant increase in neurological scores was observed in the I/R group (P < 0.001 vs. sham). β-BA considerably decreased the neurological scores at doses of 20 and 40 mg/kg (P < 0.05, P < 0.001 vs. I/R group, respectively), whereas 10 mg/kg β-BA had no remarkable effect. The sham group showed no signs of injury, whereas the I/R group showed significant infarct volume (P < 0.001 vs. sham) (Fig. 1b and c). Administration of 20 and 40 mg/kg β-BA significantly reduced infarct volume (P < 0.05, P < 0.001 vs. I/R group, respectively), but 10 mg/kg β-BA did not show a significant improvement in infarct volume. In addition, to more accurately evaluate the effect of β-BA on neuroprotection in rats with cerebral I/R injury, we examined the infarct volume and neurological deficits at 48 h after reperfusion. The results showed that the brain infarct volume and neurological score were significantly reduced in the β-BA group compared with those in the I/R group (Fig. 1d–f) (P < 0.01).

β-Boswellic acid (β-BA) reduced infarct volume and neurological scores in cerebral I/R rats. Neurological deficit scores 24 h after reperfusion (a); TTC staining of rat brain sections (b); the infarct volume was quantitatively evaluated for each group (c), data shown as mean ± SD (n = 6). Neurological deficit scores 48 h after reperfusion (d); TTC staining of rat brain sections (e), the infarct volume was quantitatively evaluated for each group (f), data are shown as mean ± SD (n = 8). ###P < 0.001 vs. sham group; *P < 0.05, **P < 0.01, ***P < 0.001 vs. I/R group
β-BA Reduced Cerebral I/R-Induced Oxidative Stress Injury in Rats
To investigate the protective effect of β-BA on neuronal morphology, we performed hematoxylin and eosin staining and TUNEL staining. The observed histopathological changes are shown in Fig. 2a. No morphological changes were observed in the sham group, and the neuronal structures were clear and regularly arranged. In the I/R group, neurons were arranged irregularly, and karyopyknosis and neuronal loss were observed. The damage was substantially reduced in cerebral I/R rats at β-BA doses of 20 and 40 mg/kg compared with those in the I/R group. Additionally, TUNEL-positive cell numbers were clearly increased in I/R rats compared with those in the sham group (P < 0.001). β-BA doses of 20 and 40 mg/kg significantly decreased the number of TUNEL-positive cell (P < 0.01, P < 0.001 vs. I/R group, respectively) (Fig. 2b and c).

β-BA treatment attenuated oxidative stress injury caused by cerebral I/R in rats. a Hematoxylin and eosin staining was performed. b Representative images of each group subjected to TUNEL staining (scale bar = 100 μm). c TUNEL-positive cells were quantified. d Levels of reactive oxygen species (ROS), e glutathione peroxidase, f 8-hydroxy-2-deoxyguanosine, and g 4-hydroxynonenal for each group. Data are shown as mean ± SD (n = 6); ###P < 0.001 vs. sham group; *P < 0.05, **P < 0.01, ***P < 0.001 vs. I/R group
Oxidative damage was evaluated by measuring ROS, GSH-Px, 8-OHdG, and 4-HNE production. ROS production considerably increased in the I/R group (P < 0.001 vs. sham), whereas treatment with β-BA at doses of 20 and 40 mg/kg clearly reduced the production of ROS (P < 0.01, P < 0.001 vs. I/R group, respectively) (Fig. 2d). In the I/R group, the GSH-Px activity was significantly reduced (P < 0.001 vs. sham), and the administration of 20 and 40 mg/kg β-BA significantly increased the GSH-Px level (P < 0.01, P < 0.001 vs. I/R group, respectively) (Fig. 2e). The levels of 8-OHdG and 4-HNE were obviously increased in the I/R group (P < 0.001 vs. sham). β-BA at doses of 20 and 40 mg/kg markedly decreased the 4-HNE and 8-OHdG levels (P < 0.05, P < 0.001 vs. I/R group, respectively). Based on these results, we concluded that β-BA mediates neuroprotective effects via its antioxidant properties.
β-BA Induced NFE2L2 Nuclear Translocation and Promoted the Expression of p-PRKCE and HMOX1
As shown in Fig. 3a and b, we found that β-BA at doses of 20 and 40 mg/kg markedly increased the expression of p-PRKCE (S729) (P < 0.001 vs. I/R group). β-BA increased nuclear NFE2L2 expression, compared with that in the I/R group (P < 0.05 and P < 0.001, respectively) (Fig. 3a and c), whereas cytoplasmic NFE2L2 expression was downregulated with the administration of β-BA (P < 0.001 vs. I/R group) (Fig. 3a and d). Corroborating this, immunofluorescence staining indicated that NFE2L2 expression was upregulated in the β-BA group (P < 0.05 and P < 0.01 vs. I/R group) (Fig. 3f and g). As shown in Fig. 3a and e, HMOX1 expression was considerably enhanced in the β-BA group (P < 0.05 and P < 0.001 vs. I/R group). These data indicate that β-BA induced NFE2L2 nuclear translocation and upregulated p-PRKCE and HMOX1 expression.

Effect of β-BA on p-PRKCE, NFE2L2, and HMOX1 protein levels. a Western blotting detected the expression of p-PRKCE, cytoplasmic NFE2L2, nuclear NFE2L2, and HMOX1. b–e Relative expressions are shown. f Representative double immunofluorescent staining for NFE2L2/DAPI (400 ×). g Quantitative analysis of NFE2L2 immunofluorescence staining. Data are expressed as mean ± SD (n = 6); *P < 0.05, ***P < 0.001 vs. I/R group
β-BA Treatment Protected Neuronal Cells from Oxidative Stress Injury Caused by OGD/RIn Vitro
Detection of apoptosis was performed using annexin V/PI staining. In the OGD/R group, the percentage of apoptotic cells increased, whereas treatment with β-BA distinctly reduced the percentage of apoptotic cells (P < 0.05, P < 0.001 vs. OGD group) (Fig. 4a and b). Additionally, treatment with β-BA significantly improved cell viability (P < 0.01, P < 0.001 vs. OGD group) (Fig. 4c). The cellular release of LDH was tested to study the protective effect of β-BA on the cytotoxicity caused by OGD/R. As shown in Fig. 4d, the release of LDH significantly increased in the OGD/R group, whereas β-BA treatment reduced LDH release (P < 0.01, P < 0.001 vs. OGD group).

β-BA treatment protects neuronal cells from damage caused by oxygen–glucose deprivation and reoxygenation (OGD/R)-induced oxidative stress injury in vitro. a Apoptosis detection of neuronal cells using annexin V/PI staining. b The percentage of apoptosis cells in all groups was quantified. c MTT assay for cell viability. d LDH assay to reflect cell injury in different groups. e–f Analysis of neuronal ROS production and relative fluorescent intensity using flow cytometry. g Determination of p-PRKCE, NFE2L2, and HMOX1 expression using western blotting. h Relative expressions are shown. Data are presented as mean ± SD of triplicate independent experiments. #P < 0.05, ###P < 0.001 vs. control group; *P < 0.05, **P < 0.01, ***P < 0.001 vs. OGD/R group
The results of the flow cytometric analyses of intracellular ROS production in neuronal cells are presented in Fig. 4e and f. Treatment with β-BA significantly decreased ROS production, compared with that in the OGD/R group (P < 0.001).
To explore the effects of β-BA on p-RKCE, NFE2L2, and HMOX1 expression in neuronal cells, we performed western blotting (Fig. 4g and h). The findings demonstrated that p-RKCE, nuclear NFE2L2, and cytoplasmic HMOX1 expression were slightly increased in the OGD/R group. β-BA treatment considerably upregulated the expression of p-RKCE, nuclear NFE2L2, and cytoplasmic HMOX1, whereas the cytoplasmic NFE2L2 level was significantly downregulated (P < 0.05, P < 0.01 vs. OGD group).
Prkce Knockdown Attenuated the Protective Effect of β-BA
We knocked down Prkce to explore whether the protective action of β-BA against I/R was mediated via the regulation of Prkce expression. Subsequently, compared with the negative control (NC), the lentivirus interference group showed a considerable reduction in PRKCE expression, indicating successful lentiviral RNA interference (P < 0.01) (Fig. 5a and b). Compared with those in the NC + OGD/R + β-BA group, shPRKCE administration evidently decreased nuclear NFE2L2 and cytoplasmic HMOX1 expression under OGD/R and β-BA treatment, whereas the expression of cytoplasmic NFE2L2 clearly increased (P < 0.01) (Fig. 5c–f). These results demonstrated that NFE2L2 nuclear translocation and HMOX1 expression were significantly reduced with Prkce knockdown. To verify this result, we analyzed the mRNA expression of Nfe2l2 in neurons using qRT-PCR. As shown in Fig. 5g, treatment of neurons with β-BA had no effect on Nfe2l2 mRNA expression. Additionally, compared with that in the NC + OGD/R + β-BA group, the knockdown of Prkce increased LDH leakage and ROS levels (P < 0.001) (Fig. 5h and i). These results confirm that β-BA plays a role in cellular resistance to OGD/R-induced injury in vitro by regulating PRKCE protein level and upregulating NFE2L2 and downstream HMOX1 expression.

Prkce knockdown attenuated the protection of β-BA on cells subjected to OGD/R treatment. Neurons were transfected with negative control (NC) or Prkce shRNA, followed by β-BA 10 μM treatment for 24 h, and then OGD treatment for 2 h; this was followed by an additional 24-h treatment with β-BA. a and b Determination of PRKCE expression using western blotting after the knockdown of Prkce and the relative expression was calculated. c–f NFE2L2 and HMOX1 levels were measured using western blotting and the relative expression was calculated. g Nfe2l2 mRNA expression. h LDH assay reflecting cell injury in different groups. i Intracellular ROS levels in different groups. Data are presented as mean ± SD of triplicate independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 vs. NC + OGD/R group; ※※P < 0.01, ※※※P < 0.001 vs. NC + OGD/R + β-BA group
For in vivo experiments, we injected a lentiviral vector into the lateral ventricles of rats to knockdown Prkce. Subsequently, the expression of PRKCE was analyzed using western blotting. The results showed that the expression of PRKCE in the brain tissue of rats in the shPRKCE group was significantly lower than that in the NC group, indicating successful lentiviral RNA interference (P < 0.01 vs. NC) (Fig. 6a and b). The neurological scores were examined 24 h after reperfusion (Fig. 6c). The results revealed no neurological deficits in the NC group, whereas a significant increase in the neurological scores was observed in the NC + I/R group compared to the NC group (P < 0.001). In addition, there was a significant decrease in the neurological scores in the NC + I/R + β-BA group compared with the NC + I/R group (P < 0.001). Furthermore, the shPRKCE + I/R + β-BA group showed an increase in the neurological scores compared with the NC + I/R + β-BA group (P < 0.05). TTC staining revealed no injured area in the NC group, but the NC + I/R group had a large infarct volume (P < 0.001). The NC + I/R + β-BA group presented significantly reduced infarct volume compared with the NC + I/R group (P < 0.001), but the shPRKCE + I/R + β-BA group presented increased infarct volume compared with the NC + I/R + β-BA group (P < 0.05) (Fig. 6d and e). Collectively, these findings imply that an increase in PRKCE expression is required for the protective effect of β-BA.

Prkce knockdown attenuated the protection of β-BA on cerebral I/R injury. a Determination of PRKCE expression using western blotting after the knockdown of Prkce in rats. b The relative expressions are shown. c Neurological deficit scores 24 h after reperfusion. TTC staining results (d) and quantitative evaluation of infarct volume (e) in each group. Data are shown as mean ± SD (n = 6); ##P < 0.01, ###P < 0.001 vs. NC group; ***P < 0.001 vs. NC + I/R group; ※P < 0.05 vs. NC + I/R + β-BA group
Discussion
Stroke is the leading cause of disability and mortality worldwide, and ischemic stroke caused by vascular occlusion accounts for 61.4% of all causes [1]. As neuronal necrosis in the central region of the infarct occurs within minutes, restoration of blood flow by early thrombolytic therapy can reduce the lethality and disability of the disease. Paradoxically, reperfusion itself causes additional damage to the ischemic penumbra (the area bordering the infarct core), leading to I/R injury. Oxidative stress is closely associated with ischemic stroke. After acute ischemic stroke, ROS production increases rapidly, which in turn disrupts the antioxidant defense system and leads to neuronal damage. In addition, the rapid restoration of blood flow increases tissue oxygenation levels and leads to a second burst of ROS, resulting in reperfusion injury [19, 20]. Therefore, investigating antioxidant strategies that can reduce oxidative damage is essential to improve ischemic stroke. Several drugs with antioxidant effects have been shown to have neuroprotective effects [21].
Studies have shown that β-BA can penetrate the blood–brain barrier, promote the growth and branching of hippocampal neurites [6], improve memory performance [22, 23], and reduce peritumoral edema of the brain [24]. The neuroprotective effects of pentacyclic triterpene BAs may be associated with their antioxidant activity [25]. We speculated that β-BA may alleviate cerebral I/R injury and could be utilized to treat or prevent neurological diseases. Here, we demonstrated for the first time that β-BA exerts a neuroprotective effect in ischemic stroke, via the inhibition of oxidative stress.
In our study, β-BA significantly decreased infarct volume, reduced behavioral scores, ameliorated neurological deficits, and reduced the number of necrotic neurons in experimental rats. Furthermore, β-BA significantly improved cell viability and reduced neuronal apoptosis, thereby showing protective effects against OGD/R-induced neuronal injury. We also evaluated the possible signaling pathways involved; owing to the overproduction of ROS during cerebral I/R pathogenesis following stroke, oxidative stress is considered to be a cause of neuronal damage [26]. Accumulation of ROS can cause oxidative damage to cellular components including proteins, lipids, and nucleic acids, leading to excessive production of 4-HNE and 8-OHdG, and their concentrations represent the severity of oxidative damage in cells [27]. GSH-Px is a widespread peroxidase-decomposing enzyme, which can reduce toxic peroxides to non-toxic hydroxyl compounds and promote the decomposition of H2O2, thus protecting the structure and function of cell membranes from damage caused by peroxides [28]. Here, the ROS, 4-HNE, and 8-OHdG levels were apparently elevated in the I/R group, but they were notably reduced after β-BA treatment. In contrast, the GSH-Px level was markedly reduced in the I/R group but increased after β-BA treatment. Collectively, these findings suggest that β-BA inhibits the production of free radicals and thus reduces I/R-induced oxidative brain injury.
PRKCE/NFE2L2 and the downstream protein HMOX1 are key to regulating oxidative stress. PRKCE, a member of the PRKC subfamily, is an important upstream signal that activates NFE2L2, which protects SH-SY5Y cells against OGD/R-induced apoptosis and exhibits neuroprotective effects [29]. In our study, β-BA markedly enhanced p-PRKCE (S729) expression in I/R rats. NFE2L2 mediates its response to oxidative stress via PRKC activation, and it is a pivotal signaling molecule in antioxidant defense mechanisms. NFE2L2 is mostly found in the cytoplasm at low levels under normal physiological settings. However, when cells are subjected to severe oxidative stress (e.g., cerebral ischemia), NFE2L2 is transferred from the cytoplasm to the nucleus, then binds to Au-rich elements, thus upregulating the expression of target genes encoding antioxidant enzymes (including HMOX1) [30]. HMOX1 catalyzes the catabolism of heme to ferrous iron, carbon monoxide, and biliverdin. On one hand, the degradation of the heme moiety facilitates the blocking of its pro-oxidant effect. On the other hand, the byproduct biliverdin and its reduced form bilirubin have effective ROS-scavenging activity to counteract peroxides, peroxynitrite, and hydroxyl and superoxide radicals [31]. Therefore, the nuclear translocation and expression of NFE2L2 combats damaging stressors such as ROS and offers a viable cellular survival mechanism target [32]. Our results showed that, in the β-BA group, HMOX1 and nuclear NFE2L2 expression increased, and that the protective effect of β-BA was mediated via p-PRKCE stimulation, NFE2L2 nuclear translocation activation, and HMOX1 upregulation.
To further study the involvement of PRKCE in β-BA-mediated neuroprotection, RNA interference targeting Prkce was used. Lentiviral transfection of shRNA targeting Prkce markedly blocked the neurocytoprotective effect of β-BA against OGD/R injury. Moreover, the knockdown of Prkce attenuated the expression-promoting effect of β-BA on NFE2L2 and HMOX1, suggesting that Prkce expression affects the activation of NFE2L2 and HMOX1. In vivo, Prkce knockdown attenuated the protective effect of β-BA in rats with cerebral I/R and increased the neurological score and infarct volume. These results indicate that the protection of β-BA against cerebral I/R injury is dependent on Prkce expression.
However, several issues need to be further investigated. In the present study, we focused on the protective effect of β-BA on brain I/R injury through inhibition of oxidative stress, but we did not explore in detail whether other signaling pathways were involved in the action of β-BA. Therefore, whether the neuroprotective effect of β-BA is related to other signaling pathways still needs to be investigated. We confirmed the protective effect of β-BA in acute phase stroke in a short-term study; therefore, the possible long-term therapeutic effect of β-BA and the therapeutic time window of β-BA on brain I/R injury in rats need to be further determined. In addition, the current findings are limited to in vivo animal and in vitro models, and they need to be confirmed in clinical studies.
Conclusions
In summary, to the best of our knowledge, this is the first study to elucidate that β-BA can exert neuroprotective effects in cerebral I/R injury in rats via the PRKCE/NFE2L2/HMOX1 signaling pathway. Moreover, the highest neuroprotective effect was observed in the group receiving the optimal high β-BA dose of 40 mg/kg.
Data Availability
All data generated in this study are provided in this manuscript.
References
GBD 2019 Stroke Collaborators (2021) Global, regional, and national burden of stroke and its risk factors, 1990–2019: a systematic analysis for the global burden of disease study 2019. Lancet Neurol 20:795–820. https://doi.org/10.1016/S1474-4422(21)00252-0
Ya BL, Liu Q, Li HF, Cheng HJ, Yu T, Chen L, Wang Y, Yuan LL, Li WJ, Liu WY, Bai B (2018) Uric acid protects against focal cerebral ischemia/reperfusion-induced oxidative stress via activating Nrf2 and regulating neurotrophic factor expression. Oxid Med Cell Longev 2018:6069150. https://doi.org/10.1155/2018/6069150
Guo C, Wang S, Duan J, Jia N, Zhu Y, Ding Y, Guan Y, Wei G, Yin Y, Xi M, Wen A (2017) Protocatechualdehyde protects against cerebral ischemia-reperfusion-induced oxidative injury via protein kinase cepsilon/Nrf2/HO-1 pathway. Mol Neurobiol 54:833–845. https://doi.org/10.1007/s12035-016-9690-z
Cao Y, Zhang L, Sun S, Yi Z, Jiang X, Jia D (2016) Neuroprotective effects of syringic acid against OGD/R-induced injury in cultured hippocampal neuronal cells. Int J Mol Med 38:567–573. https://doi.org/10.3892/ijmm.2016.2623
Barakat BM, Ahmed HI, Bahr HI, Elbahaie AM (2018) Protective effect of boswellic acids against doxorubicin-induced hepatotoxicity: impact on Nrf2/HO-1 defense pathway. Oxid Med Cell Longev 2018:8296451. https://doi.org/10.1155/2018/8296451
Karima O, Riazi G, Yousefi R, Movahedi AA (2010) The enhancement effect of beta-boswellic acid on hippocampal neurites outgrowth and branching (an in vitro study). Neurol Sci 31:315–320. https://doi.org/10.1007/s10072-010-0220-x
Khafaga AF, El-Kazaz SE, Noreldin AE (2021) Boswellia serrata suppress fipronil-induced neuronal necrosis and neurobehavioral alterations via promoted inhibition of oxidative/inflammatory/apoptotic pathways. Sci Total Environ 785:147384. https://doi.org/10.1016/j.scitotenv.2021.147384
Khan A, Khan I, Halim SA, Ur Rehman N, Karim N, Ahmad W, Khan M, Csuk R, Al-Harrasi A (2022) Anti-diabetic potential of β-boswellic acid and 11-keto-β-boswellic acid: mechanistic insights from computational and biochemical approaches. Biomed Pharmacother 147:112669. https://doi.org/10.1016/j.biopha.2022.112669
Gundimeda U, McNeill TH, Elhiani AA, Schiffman JE, Hinton DR, Gopalakrishna R (2012) Green tea polyphenols precondition against cell death induced by oxygen-glucose deprivation via stimulation of laminin receptor, generation of reactive oxygen species, and activation of protein kinase cepsilon. J Biol Chem 287:34694–34708. https://doi.org/10.1074/jbc.M112.356899
Xue Z, Zhao K, Sun Z, Wu C, Yu B, Kong D, Xu B (2021) Isorhapontigenin ameliorates cerebral ischemia/reperfusion injury via modulating kinase cepsilon/Nrf2/HO-1 signaling pathway. Brain Behav 11:e02143. https://doi.org/10.1002/brb3.2143
Zhu Y, Di S, Hu W, Feng Y, Zhou Q, Gong B, Tang X, Liu J, Zhang W, Xi M, Jiang L, Guo C, Cao J, Fan C, Ma Z, Yang Y, Wen A (2017) A new flavonoid glycoside (APG) isolated from Clematis tangutica attenuates myocardial ischemia/reperfusion injury via activating PKCepsilon signaling. Biochim Biophys Acta Mol Basis Dis 1863:701–711. https://doi.org/10.1016/j.bbadis.2016.12.013
Ma Q (2013) Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53:401–426. https://doi.org/10.1146/annurev-pharmtox-011112-140320
Ding Y, Chen M, Wang M, Li Y, Wen A (2015) Posttreatment with 11-keto-beta-boswellic acid ameliorates cerebral ischemia-reperfusion injury: Nrf2/HO-1 pathway as a potential mechanism. Mol Neurobiol 52:1430–1439. https://doi.org/10.1007/s12035-014-8929-9
Siemoneit U, Koeberle A, Rossi A, Dehm F, Verhoff M, Reckel S, Maier TJ, Jauch J, Northoff H, Bernhard F, Doetsch V, Sautebin L, Werz O (2011) Inhibition of microsomal prostaglandin E2 synthase-1 as a molecular basis for the anti-inflammatory actions of boswellic acids from frankincense. Br J Pharmacol 162:147–162. https://doi.org/10.1111/j.1476-5381.2010.01020.x
Kuts R, Frank D, Gruenbaum BF, Grinshpun J, Melamed I, Knyazer B, Tarabrin O, Zvenigorodsky V, Shelef I, Zlotnik A, Boyko M (2019) A novel method for assessing cerebral edema, infarcted zone and blood–brain barrier breakdown in a single post-stroke rodent brain. Front Neurosci 13:1105. https://doi.org/10.3389/fnins.2019.01105
Liu P, Tang YY, Yang XS, Dai J, Yang M, Zhang H, Liu Y, Yan H, Song XY (2018) Validation of a preclinical animal model to assess brain recovery after acute stroke. Eur J Pharmacol 835:75–81. https://doi.org/10.1016/j.ejphar.2018.07.035
Zheng YQ, Liu JX, Li XZ, Xu L, Xu YG (2009) RNA interference-mediated downregulation of Beclin1 attenuates cerebral ischemic injury in rats. Acta Pharmacol Sin 30:919–927. https://doi.org/10.1038/aps.2009.79
Zhou Y, Zhou Y, Yu S, Wu J, Chen Y, Zhao Y (2015) Sulfiredoxin-1 exerts anti-apoptotic and neuroprotective effects against oxidative stress-induced injury in rat cortical astrocytes following exposure to oxygen-glucose deprivation and hydrogen peroxide. Int J Mol Med 36:43–52. https://doi.org/10.3892/ijmm.2015.2205
Su XT, Wang L, Ma SM, Cao Y, Yang NN, Lin LL, Fisher M, Yang JW, Liu CZ (2020) Mechanisms of acupuncture in the regulation of oxidative stress in treating ischemic stroke. Oxid Med Cell Longev 2020:7875396. https://doi.org/10.1155/2020/7875396
Rodrigo R, Fernández-Gajardo R, Gutiérrez R, Matamala JM, Carrasco R, Miranda-Merchak A, Feuerhake W (2013) Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS Neurol Disord Drug Targets 12:698–714. https://doi.org/10.2174/1871527311312050015
Liu Y, Zhu C, Guo J, Chen Y, Meng C (2020) The neuroprotective effect of irisin in ischemic stroke. Front Aging Neurosci 12:588958. https://doi.org/10.3389/fnagi.2020.588958
Jebelli A, Khalaj-Kondori M, Rahmati-Yamchi M (2020) The effect of beta-boswellic acid on the expression of Camk4 and Camk2alpha genes in the PC12 cell line. Adv Pharm Bull 10:437–443. https://doi.org/10.34172/apb.2020.053
Jebelli A, Khalaj-Kondori M, Bonyadi M, HosseinpourFeizi MA, Rahmati-Yamchi M (2019) Beta-boswellic acid and ethanolic extract of olibanum regulating the expression levels of CREB-1 and CREB-2 genes. Iran J Pharm Res 18:877–886. https://doi.org/10.22037/ijpr.2019.1100665
Gerbeth K, Hüsch J, Fricker G, Werz O, Schubert-Zsilavecz M, Abdel-Tawab M (2013) In vitro metabolism, permeation, and brain availability of six major boswellic acids from Boswellia serrata gum resins. Fitoterapia 84:99–106. https://doi.org/10.1016/j.fitote.2012.10.009
Ebrahimpour S, Fazeli M, Mehri S, Taherianfard M, Hosseinzadeh H (2017) Boswellic acid improves cognitive function in a rat model through its antioxidant activity: -neuroprotective effect of boswellic acid. J Pharmacopuncture 20:10–17. https://doi.org/10.3831/KPI.2017.20.001
Zeng J, Zhu L, Liu J, Zhu T, Xie Z, Sun X, Zhang H (2019) Metformin protects against oxidative stress injury induced by ischemia/reperfusion via regulation of the lncRNA-H19/miR-148a-3p/Rock2 axis. Oxid Med Cell Longev 2019:8768327. https://doi.org/10.1155/2019/8768327
Mu X, Zhang Y, Li J, Xia J, Chen X, Jing P, Song X, Wang L, Wang Y (2017) Angelica sinensis polysaccharide prevents hematopoietic stem cells senescence in d-galactose-induced aging mouse model. Stem Cells Int 2017:3508907. https://doi.org/10.1155/2017/3508907
Bazmandegan G, Boroushaki MT, Shamsizadeh A, Ayoobi F, Hakimizadeh E, Allahtavakoli M (2017) Brown propolis attenuates cerebral ischemia-induced oxidative damage via affecting antioxidant enzyme system in mice. Biomed Pharmacother 85:503–510. https://doi.org/10.1016/j.biopha.2016.11.057
Gao K, Liu M, Ding Y, Yao M, Zhu Y, Zhao J, Cheng L, Bai J, Wang F, Cao J, Li J, Tang H, Jia Y, Wen A (2019) A phenolic amide (LyA) isolated from the fruits of Lycium barbarum protects against cerebral ischemia-reperfusion injury via PKCepsilon/Nrf2/HO-1 pathway. Aging (Albany, NY) 11:12361–12374. https://doi.org/10.18632/aging.102578
Loboda A, Damulewicz M, Pyza E, Jozkowicz A, Dulak J (2016) Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci 73:3221–3247. https://doi.org/10.1007/s00018-016-2223-0
Ayer A, Zarjou A, Agarwal A, Stocker R (2016) Heme oxygenases in cardiovascular health and disease. Physiol Rev 96:1449–1508. https://doi.org/10.1152/physrev.00003.2016
Huang L, Wang J, Chen L, Zhu M, Wu S, Chu S, Zheng Y, Fan Z, Zhang J, Li W, Chen D, Yang X, Wang S, Qiu P, Wu J (2018) Design, synthesis, and evaluation of NDGA analogues as potential anti-ischemic stroke agents. Eur J Med Chem 143:1165–1173. https://doi.org/10.1016/j.ejmech.2017.09.028
Acknowledgements
We are grateful to our colleagues in the laboratory for technical guidance and reagent sharing for this study.
Funding
This work was supported by the National Natural Science Foundation of China (Grant numbers 81803749, 81870893, and 81503285), the Key Research and Development Plan of Shaanxi Province (Grant number 2021KW-56), and Shandong Traditional Chinese Medicine Science and Technology Project (Grant number 2021M079).
Author information
Authors and Affiliations
Contributions
MW: investigation, writing—original draft. JY, CG, and QY: investigation, visualization. WZ and WL: methodology, formal analysis. YW: data curation. YD and JW: conceptualization, methodology.
Corresponding authors
Ethics declarations
Ethics Approval
The animal procedures in this study were approved by the Animal Experimentation Ethics Committee of the Fourth Military Medical University.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wang, M., Yu, J., Yang, Q. et al. Beta-Boswellic Acid Protects Against Cerebral Ischemia/Reperfusion Injury via the Protein Kinase C Epsilon/Nuclear Factor Erythroid 2-like 2/Heme Oxygenase-1 Pathway. Mol Neurobiol 59, 4242–4256 (2022). https://doi.org/10.1007/s12035-022-02848-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-022-02848-w