
Abstract
Neuroinflammation is closely related to brain iron homeostasis. Our previous study demonstrated that lipopolysaccharides (LPS) can regulate expression of iron-regulatory peptide hepcidin; however, the mechanism is undefined. Here, we demonstrated that intracerebroventricular injection of LPS in rat brain upregulated hepcidin and downregulated ferroportin 1 in the cortex and substantia nigra. LPS increased hepcidin expression in neurons only when they were co-cultured with BV-2 microglia, and the upregulation was suppressed by IL-6 neutralizing antibody in vitro. In addition, IL-6 but not IL-1α, IL-1β, or tumor necrosis factor-alpha increased hepcidin expression and signal transducer and activator of transcription 3 (STAT3) phosphorylation in cortical neurons and MES23.5 dopaminergic neurons. These effects were blocked by the STAT3 inhibitor, stattic. Our results show that neurons are the major source of increased hepcidin expression in response to LPS challenge but microglia play a key mediator role by releasing IL-6 and recruiting the STAT3 pathway. We conclude that LPS upregulates hepcidin expression in neurons via microglia and the IL-6/STAT3 signaling pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Iron serves as an essential trace element in various physiological activities in multiple types of cells, including those in the brain [1]. As both iron deficiency and iron overload can lead to cellular dysfunctions, maintaining normal iron homeostasis is crucial [2, 3]. Accumulated data have shown that the peptide hepcidin plays an essential role in maintaining normal iron homeostasis in peripheral organs [4, 5]. This hormone regulates plasma iron concentration and tissue iron distribution by controlling intestinal iron absorption, iron recycling by macrophages, iron mobilization from hepatic stores, and iron utilization by the bone marrow cells [4–6]. Hepcidin is mainly synthesized by the liver, distributed in extracellular fluid, and excreted in urine [7–10]. Mice that fail to express hepcidin have elevated body iron stores, presumably due to hyperabsorption associated with decreased iron in tissue macrophages [10–13]. On the other hand, overexpression of hepcidin causes hypoferremia and anemia [11, 14, 15]. The synthesis of hepcidin in the periphery is homeostatically regulated by multiple signals, including iron store, erythropoietic activity, hypoxia, and inflammation [16, 17]. Recent studies have confirmed that hepcidin is also widely expressed in the brain and that this peptide can regulate the expression of brain iron transport proteins [18, 19]. These results imply that hepcidin has a central role in brain iron homeostasis.
Both misregulation in brain iron metabolism and inflammation have been considered as primary causes of neuronal death in some neurodegenerative disorders such as AD, PD, and Hallervorden–Spatz syndrome [20–22]. Studies have shown that defective brain iron metabolism found in neurodegenerative disorders has a multifactorial pathogenesis, and inflammation has been implicated as one of the factors [21]. Hepcidin is not only an iron regulatory hormone but also an antimicrobial peptide that exerts functions in host defense outside the brain [6, 8, 9, 11]. However, at present, little is known on what the effects of inflammation on brain iron metabolism are and a role in the development of neurological disorders. The studies on these issues are critically important for understanding the pathogenesis of neurological disorders. In a recent study [23], we demonstrated that lipopolysaccharide (LPS) administration regulated the expression of hepcidin mRNA and protein not only in peripheral organs such as the liver but also in the brain. Specifically, LPS induced a significant increase in the expression of hepcidin mRNA and protein in the cortex and substantia nigra. However, the mechanism involved in this observation is still unknown. It has been demonstrated that during inflammation and infection, the inflammatory cytokine interleukin-6 (IL-6) directly regulate hepcidin expression through induction and subsequent promoter binding of signal transducer and activator of transcription 3 (STAT3) [24–26]. Based on our understanding of the regulation of hepcidin in the periphery [24–26] and its actions on brain iron metabolism [18, 19, 23], we speculated that IL-6 and STAT3 signaling are involved in the expression of hepcidin in the central nervous system. Experiments were designed to test this hypothesis and further elucidate the nature of the response of the brain to LPS. Given that neuroinflammation is associated with various common neurodegenerative diseases, we focused our study on the cortex and substantia nigra, two well-known susceptible brain regions in these diseases.
Materials and Methods
Animals and Materials
Male Sprague-Dawley (SD) rats weighing 230–250 g were supplied by the Laboratory Animal Services Center of The Chinese University of Hong Kong and were housed under a standard 12-h light–dark cycle with water and food supplied ad libitum. All animal handling, surgical procedures were carried out in accordance with university guidelines on the animal ethics. Except the followings, all chemicals were obtained from Sigma Chemical Co (St. Louis, MO, USA). These include IL-1α, IL-1β, and tumor necrosis factor-alpha (TNF-α) from Calbiochem (San Diego, CA, USA) and IL-6 from Peprotech (Rocky Hill, NJ, USA). Antibodies were obtained from the following sources: Abcam (Cambridge, MA, USA): rabbit polyclonal anti-hepcidin-25 and anti-IL-6, Cell Signaling Technology, Inc. (Danvers, MA, USA): rabbit polyclonal anti-phospho-STAT3 (Tyr705) and mouse monoclonal anti-STAT3, Alpha Diagnostic International Company (San Antonio, TX, USA): rabbit polyclonal anti-mouse ferroportin 1 (Fpn1), Chemicon International (Temecula, CA, USA): mouse monoclonal anti-GFAP, Invitrogen (Carlsbad, CA, USA): Alexa Fluor 546-conjugated goat anti-rabbit IgG and FITC-conjugated goat anti-mouse IgG, Millipore (Billerica, MA, USA): mouse anti-rat neuronal nuclear antigen (NeuN), and Wako (Osaka, Japan): anti-Iba1. Rat hepcidin ELISA kit and rat IL-6 ELISA kit were from Novatein Biosciences (Cambridge, MA, USA) and R&D systems (Minneapolis, MN, USA), respectively.
Intracerebroventricular Injection of LPS
SD rats were anesthetized with 4 % chloral hydrate (1 ml/100 g body weight) by i.p. injection and secured in a stereotaxic instrument. LPS (3 μg/3 μl of sterile saline) or normal saline (3 μl, the control) were injected bilaterally into the lateral ventricles according to a standard stereotaxic atlas (0.8 mm posterior to the bregma, 1.5 mm lateral to the midline, 3.8 mm ventral to the dura bregma) at a rate 1 μl/min. The syringe was left in place for an additional 5 min before removal.
Animal Sacrifice and Sample Collection
After different treatments, rats were deeply anesthetized with 4 % chloral hydrate (2 ml/100 g body weight, i.p.) and then transcardially perfused with ice-cold normal saline through the left ventricle. The cortex and substantia nigra in the left hemisphere were dissected out as previously described [27] and stored at −80 °C for western blot analysis. The right hemisphere was immersion-fixed with 4 % paraformaldehyde (pH 7.4) for 1 week and cryoprotected in 30 % sucrose (pH 7.4) at room temperature overnight. The hemi-brains were used for immunofluorescence double staining.
Cell Cultures
Primary Cortical Neuron Culture
Cortical neurons were prepared from E16–17 rats as previously described [28]. Dissociated cortical cells were suspended in Dulbecco’s modified Eagle’s medium (DMEM) containing 10 % fetal bovine serum (FBS; Invitrogen-Life Technologies) with antibiotics (penicillin 100 U/ml, streptomycin 100 μg/ml) and then seeded on six-, 12-, or 96-well plates pre-coated with poly-l-lysine at a density of 1.5 × 106 cells/ml. The cultures were maintained at 37 °C in a humidified environment with 5 % CO2 in a CO2 incubator (NAPCO 5400). After 24 h, the cultural medium was replaced with neurobasal medium supplemented with 2 % B-27 and antibiotics (penicillin 100 U/ml, streptomycin 100 μg/ml). The cultured neurons were maintained in the neurobasal–B27 medium for 6 days. The purity of the cultures was assessed by staining with the neuron-specific antibodies against microtubule associated protein 2 (MAP2; Temecula, CA, USA) and the astrocyte marker glial fibrillary acidic protein (GFAP). It approached ≧98.5 % positive cells.
BV-2 Microglia Cells
BV-2 microglia cells (a murine microglia cell line) were grown in a 5 % CO2 incubator at 37 °C in DMEM supplemented with 10 % FBS and antibiotics (penicillin 100 U/ml, streptomycin 100 μg/ml). The cultural medium was changed every 2 days. For the co-culture experiments, BV-2 cells were seeded on 12-well cell culture inserts (BD FalconTM, USA) at a density of 1 × 105 cells/ml overnight, and then the inserts were transferred onto the primary cortical neurons that were seeded on 12-well plates. BV-2 cells were treated with LPS (10 μg/ml) dissolved in fresh DMEM without serum.
MES23 5 Dopaminergic Cells
MES23.5 cells (a dopaminergic cell line) were grown in a 5 % CO2 incubator at 37 °C in DMEM supplemented with 10 % FBS, 1× N-2 supplement (Invitrogen-Life Technologies) and antibiotics (penicillin 100 U/ml, streptomycin 100 μg/ml). The cultural medium was changed every 2 days. MES23.5 cells were seeded on six- or 96-well cell culture plates pre-coated with poly-l-lysine at a density of 5 × 105 cells/ml. MES23.5 cells were treated with IL-6 (20 ng/ml) and/or stattic (5 μM) dissolved in basal DMEM without serum or N-2 supplement.
Double Immunofluorescence Staining
The fixed brain tissues were immersed in cryoembedding media (OCT) and frozen on liquid nitrogen prior to be sectioned into 20-μm-thick slices using the cryotome. The slices were mounted onto super frost slides coated with poly-l-lysin, incubated with the mixture of two primary antibodies, rabbit polyclonal anti-hepcidin-25 (1:100) and mouse against rat NeuN (1:1,000) or mouse monoclonal anti-GFAP (1:2,000), in a humidified chamber at room temperature for 24 h and then with the mixture of two secondary antibodies with two different fluorochromes, Alexa Fluor 546-conjugated goat anti-rabbit IgG (1:1,000) and FITC-conjugated goat anti-mouse IgG (1:50), at room temperature for 60 min. For the double staining with hepcidin and Iba1, the slices were first incubated with primary antibody against Iba1 (1:1,000) and then with rabbit polyclonal anti-hepcidin-25. After three washes, the sections were cover-slipped in mounting medium (M1289, Sigma) and then seal coverslip with nail polish to prevent drying and movement under microscope. The colocalization of hepcidin with the markers of neuron, astrocytes, and microglia was performed by laser scanning confocal microscopy (C-1, Nikon Corporation, Tokyo, Japan).
Western Blot Analysis
The neurons or brain tissues were washed and homogenized and then sonicated as described previously [29]. Aliquots of the extract containing about 20 μg of protein are loaded and run on a single track of 10 % SDS–PAGE under reducing conditions and subsequently transferred to a pure nitrocellulose membrane (Bio-Rad). The blots were blocked and incubated with primary antibodies against rabbit polyclonal anti-hepcidin-25 (1:400), rabbit polyclonal anti-mouse Fpn1 (1:5,000), rabbit polyclonal anti-phospho-STAT3 (Tyr705) (1:500), and mouse monoclonal anti-STAT3 (1:1,000) overnight at 4 °C. After the incubation, the blots were washed three times and incubated with goat anti-rabbit or anti-mouse IRDye 800 CW secondary antibody (1:5,000) for 1 h at room temperature. The intensity of the specific bands was detected and analyzed by Odyssey infrared image system (Li-Cor). To ensure even loading of the samples, the same membrane was probed with rabbit anti-actin polyclonal antibody at a 1:2,000 dilution.
ELISA
The concentrations of hepcidin and IL-6 were determined using ELISA kits according to suppliers’ instructions. Briefly, brain tissues were homogenized in phosphate-buffered saline (PBS) (1:5, w/v), or cell samples were scraped off with 50 μl PBS/well (12-well plate), followed by sonication using Soniprep 150. The samples were centrifuged at 3,000 × g for 10 min at 4 °C, and the supernatant was collected. A 2-μl aliquot was subjected to detect the protein concentration. One hundred microliters of assay buffer and 50 μl of each standard, control, or samples were added into appropriate wells, and 100 μl biotin conjugate was dispensed into each well. Following incubation for 2 h at room temperature, the wells were washed with diluted wash solution, and 100 μl of enzyme complex was added to each well. After incubation for 1 h at room temperature, 100 μl substrate solution was added to each well. Finally, after reaction for 30 min at room temperature, the enzymatic reaction was stopped by adding 100 μl of stop solution, and the OD at 450 nm was read by using an ELX-800 microplate assay reader (Bio-tek, USA). The average absorbance values for each set of standards, controls, and samples were calculated, and a standard curve was constructed. The concentrations of the samples were then calculated from the standard curve.
Statistical Analysis
Statistical analyses were performed using SPSS software for Windows (version 10.0). Data were presented as mean ± SEM. The difference between the means was determined by one-way ANOVA followed by a Student–Newman–Keuls test for multiple comparisons. Two groups of data were considered to be statistically different when the P value was less than 0.05.
Results
Effects of LPS on Hepcidin Expression in the Rat Brain Neurons
We first investigated the effects of LPS on the expression of hepcidin in rat brain by ELISA. The concentration of hepcidin in the cortex, substantia nigra, and hippocampus was measured 24 h after the injection of LPS into the lateral ventricle. Consistent with our previous findings [23], the results demonstrated that LPS treatment induced a significant increase in hepcidin expression in both cortex and substantia nigra (Fig. 1a) but not the hippocampus (data not shown). It has been reported that, in the brain, the expression of hepcidin is not confined to neurons but could also be detected in other cell types such as GFAP-positive astrocytes [30]. Therefore, we examined the effects of LPS on the expression of hepcidin in neurons, astrocytes, and microglia by double immunofluorescence labeling. In the control group, there was almost no expression of hepcidin in the astrocytes in the substantia nigra, but there was a low expression of hepcidin in the astrocytes in the cortex. On the other hand, microglia in both regions showed a low expression of hepcidin. Nevertheless, LPS treatment did not affect the expression patterns and levels of hepcidin in both the astrocytes and microglia in these regions (Supplementary Fig. 1A, B). In contrast, by double staining with NeuN, we found that there was a significant increase in hepcidin expression in neurons within the cortex and substantia nigra (Fig. 1b). Also, hepcidin was mainly expressed in the cytosol of neurons. In addition, double staining with tyrosine hydroxylase (TH) antibody showed that increased hepcidin occurred in, although not confined to, TH-positive neurons (Supplementary Fig. 2). These results suggest that neurons, but not astrocytes or microglia, are mainly responsible for the regulation of hepcidin expression in the brain. These results were confirmed by western blot analysis showing that at 24 h after LPS injection, hepcidin expression was upregulated in the cortex and substantia nigra. At the same time, the iron exporter Fpn1 was downregulated by LPS, a finding consistent with the known inhibitory effect of hepcidin on Fpn1 (Fig. 2a). Furthermore, IL-6 was also upregulated by LPS treatment in these brain regions (Fig. 2b), suggesting that this cytokine may mediate the action of LPS in the brain.

Effects of LPS on the expression of hepcidin in the neurons of rat cortex and substantia nigra in rat brain. Twenty-four hours after LPS was injected into the lateral ventricle of SD rat brain, the concentration of hepcidin in the cortex and substantia nigra was measured by using ELISA. A significant increase in hepcidin content was found in these two regions (a). The expression of hepcidin in the neurons in these two regions was determined using double immunofluorescence with NeuN (b). Data are presented as mean ± SEM (n = 4 in control group; n = 5 in LPS-treated group). *P < 0.05

Effects of LPS on hepcidin and ferroportin 1 in the cortex and substantia nigra. a The effects of LPS on hepcidin and Fpn1 in the cortex and substantia nigra at 24 h after injection into the lateral ventricle were examined by western blot. LPS induced upregulation of hepcidin and simultaneous downregulation of Fpn1 in both areas. b LPS treatment also increased the expression of the cytokine IL-6 in these two brain regions. Data are normalized by β-actin and expressed as mean ± SEM (n = 4). *P < 0.05, **P < 0.01 versus the control group
LPS-Induced Upregulation of Hepcidin Is Dependent on Microglia
It is believed that Toll-like receptor 4 (TLR4) is the major receptor that recognizes LPS. In the brain, although it has been reported that astrocytes [31], endothelial cells [32], and neurons [33, 34] do express TLR4, the major cell type that expresses this receptor and mediates the host response is the microglia [35–37]. Given that our data showed that neurons were the major brain cells responsible for expressing the hepcidin induced by LPS, we speculated that the microglia play a mediator role of this LPS response in neurons. To test this hypothesis, we co-cultured primary cortical neurons with BV-2 microglia and examined their interaction during LPS-induced hepcidin regulation. We measured hepcidin expression of primary cortical neurons with or without co-culturing with BV-2 microglia after treatment with LPS (10 μg/ml) for 18 h. The results by ELISA demonstrated that LPS treatment had no effect on hepcidin expression on the cultured cortical neurons alone. However, in the co-culture of neurons and microglia, LPS induced a 1.8-fold increase in the expression of hepcidin (P < 0.01; Fig. 3a). This result strongly suggests that LPS-induced increase of hepcidin requires the presence of microglia.

Microglia and IL-6-dependent upregulation of hepcidin in primary cortical neurons. a Primary cortical neurons were treated with LPS (10 μg/ml) in the absence or presence of BV-2 microglia cells for 18 h, and hepcidin levels were then measured using ELISA. LPS significantly increased the cellular level of hepcidin in neurons only in the presence of BV-2 cells. b When the cultured neurons were incubated with IL-1α, IL-1β, IL-6, or TNF-α (all at 10 ng/ml) for 2 h, only IL-6 significantly increased the cellular expression of hepcidin. c In the presence of anti-IL-6 neutralizing antibody (anti-IL6), LPS failed to increase the level of hepcidin at the end of 2 h. Anti-IL6 itself had no effect on hepcidin. **P < 0.05
The Role of IL-6 Released by Microglia in LPS-Induced Upregulation of Hepcidin
How do microglia help regulate hepcidin expression in neurons? It is known that, in response to LPS challenge, microglia can release a battery of pro-inflammatory cytokines including IL-1α, IL-1β, IL-6, and TNF-α [38]. It has also been demonstrated that the cytokine IL-6 plays a key role in the regulation of hepcidin expression during inflammation in the periphery [24–26]. To determine the involvement of IL-6 in LPS-induced central hepcidin upregulation, we first examined its expression in the brain with or without LPS treatment. ELISA analysis demonstrated that LPS injection induced a significant increase in the production of IL-6 in the cortex and substantia nigra (Fig. 3b) (all P < 0.01). The specific role of IL-6 in neuronal hepcidin upregulation was also demonstrated by examining the effects of IL-1α, IL-1β, IL-6, and TNF-α on hepcidin expression in the primary cortical neurons. Among these cytokines, only IL-6 induced a significant increase in hepcidin level (P < 0.01; Fig. 3c). Furthermore, in the neuron and BV-2 microglia co-culture, the stimulatory effect of LPS was completely suppressed by anti-IL-6 antibodies while the antibody itself had no effect hepcidin level (P < 0.01; Fig. 3d). Together, these results revealed a key role of microglia released IL-6 in mediating the hepcidin response of the nervous system to LPS.
Intriguingly, when we examined the time course of the effects of IL-6 on hepcidin expression (Fig. 4a) in primary cortical neurons, a peak response was found at 2 h, while at later time points (8 and 18 h) the hepcidin levels returned to baseline. The same pattern was also found in the expression of Fpn1 (Fig. 4b). These results are consistent with the ELISA measurement of the cellular content and hepcidin (Fig. 4c) and its level in the medium (Fig. 4d).

Acute effect of IL-6 on hepcidin and ferroportin 1 in cortical neurons. Primary cortical neurons were treated with IL-6 (10 ng/ml) for 2, 8, and 18 h, and the expression of hepcidin (a) and Fpn1 (b) was determined by western blot. Representative blots are shown in the upper panels. A significant increase in hepcidin and a significant decrease in Fpn1 were found at 2 h only. The histograms show the pooled data (n = 6). c ELISA measurement of the cellular level of hepcidin (n = 5) and d hepcidin released into the medium (n = 6) exhibited similar patterns of time course. **P < 0.01
Inhibition of STAT3 Suppresses IL-6-Induced Hepcidin Upregulation in Neurons
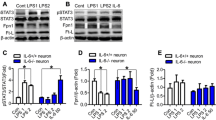
The transcription factor STAT3 is a major effector following IL-6 receptor activation and may therefore be responsible for upregulating the gene expression of neuronal hepcidin in response to LPS challenge and IL-6 release. We tested the role of STAT3 phosphorylation by first examining its expression following injection of LPS in the brain. As expected, pSTAT3 was robustly upregulated in the cortex and substantia nigra (Fig. 5a) in vivo 24 h after LPS administration. These effects were observed in vitro to be specific to IL-6 as other cytokines tested, including IL-1α, IL-1β, and TNF-α produced no effect (Fig. 5B). To confirm and better elucidate the role of STAT3 in mediating IL-6-induced hepcidin expression, we compared the effect of IL-6 in primary cortical neurons with or without 1 h pre-treatment of the STAT3 inhibitor, stattic. Western blot results demonstrated that stattic blocked the IL-6-induced STAT3 phosphorylation (Fig. 6a) and at the same time suppressed hepcidin upregulation (Fig. 6B). Stattic alone had no effect on either the pSTAT3 or hepcidin levels. Consistently, ELISA analysis showed that stattic significantly inhibited the elevation of intracellular hepcidin level and the amount of hepcidin released into the medium (Fig. 6c, d).

Only IL-6 induces LPS-mediated phosphorylation of STAT3. a Western blot analysis of the effects of LPS on STAT3 in the cortex and substantia nigra at 24 h post-injection revealed a strong degree of STAT3 phosphorylation in both regions. The histograms show pSTAT3/STAT3 ratio normalized with the control group (n = 4). b Among the different cytokines tested, only IL-6 upregulated the phosphorylation of STAT3 in primary cortical neurons measured after 30 min of incubation (n = 4). **P < 0.01 versus the control or IL-6 group

IL-6 induces hepcidin expression via STAT3 pathway. a–d Primary cortical neurons were pretreated with 20 μM of stattic (ST; a STAT3 inhibitor) for 1 h followed by incubation with 10 ng/ml of IL-6 for 2 h. Western blot analysis showed that the elevated expression of hepcidin was suppressed by stattic that alone had no effect (n = 5). This blocking action of static was also revealed by ELISA measurement of cellular hepcidin (c) and hepcidin in the medium (d). n = 4. *P < 0.05, **P < 0.01 versus the control or IL-6 group
Since the substantia nigra consists mainly of dopamine neurons which are different from the cortical neurons, we wanted to confirm our observations in MES23.5 dopaminergic cells. These cells share many properties with those of primary neurons originated from substantia nigra [39]. The results were highly similar to those observed in cortical neurons. Thus, in these cells, western blot results also showed that stattic blocked IL-6-induced STAT3 phosphorylation (Fig. 7a) and suppressed IL-6-induced upregulation of hepcidin (Fig. 7b). The ELISA results also showed that stattic could significantly inhibit the elevated intracellular hepcidin level and suppressed hepcidin release after IL-6 treatment (Fig. 7c, d).

Similar mechanisms of hepcidin upregulation in MES23.5 dopaminergic neurons. MES23.5 dopaminergic cells were pretreated with 5 μM of stattic for 1 h followed by incubation with 20 ng/ml of IL-6 for 30 min. Western blot analysis shows that IL-6 increased STAT3 phosphorylation (a) and hepcidin expression (b), an action that was suppressed by static (n = 3). Similarly, ELISA measurement of the cellular content of hepcidin (c) in the cultured neurons or in the medium (d) showed that the stimulatory effect of IL-6 was blocked by static (n = 3). *P < 0.05, **P < 0.01
Discussion
Antimicrobial peptides such as hepcidin are an evolutionarily conserved component of the innate immune system but could serve as multifunctional molecules in the brain [40]. To gain insight into the nature of inflammation induced alteration of hepcidin and iron metabolism in the brain, in this study, we induced neuroinflammation with LPS by i.c.v. injection, which enabled the experimental inflammation to occur directly and locally within the brain when compared with peripheral administration [23, 41]. The dose and time point of LPS injection chosen were based on the neuroinflammation models reported previously [41–43]. In this model, we found that LPS injection upregulated the expression of hepcidin in the neurons of the cortex and substantia nigra, which was accompanied by a reduction in Fpn1. In vitro experiments on primary cortical neurons and MES 23.5 dopaminergic cells revealed the dependence of the hepcidin response on IL-6 released by microglia and also the STAT3 pathway. Figure 8 is a proposed scheme for the neuronal hepcidin regulation during inflammation. LPS activates microglia probably by its direct binding to TRL4 on microglia membrane. The activated microglia produce and release a series of inflammatory cytokines including IL-6. The IL-6 released from the activated microglia then acts on neurons to induce STAT3 phosphorylation. In turn, the phosphorylated STAT3 is translocated into the nucleus, where it regulates hepcidin gene expression. Increased hepcidin expression and release downregulates Fpn1 and alters other iron transport proteins like transferrin receptor 1 [18] in neurons, thus affecting iron metabolism in the brain. Our results therefore suggest that IL-6 and the STAT3 pathway play similar key roles in the regulation of hepcidin expression to inflammatory stimuli in both the periphery and central nervous system.

A proposed scheme for the mechanism of neuronal hepcidin regulation during inflammation. Microglia are activated by the binding of LPS with TLR4 on the membrane. The activated microglia produce and release a series of inflammatory factors including IL-6. IL-6 is the main factor that induces STAT3 phosphorylation in neurons. The phosphorylated STAT3 is translocated into the nucleus, where it regulates hepcidin gene (HAMP) expression. Increased hepcidin expression and its release could function as a microbial peptide and also downregulate Fpn1 expression in brain cells including neurons thus altering iron metabolism in the brain
The cellular origin of hepcidin in the brain is a debatable issue, at least in the case of neuroinflammation. It has been suggested that astrocytes and microglia, but not neurons, are the major source of hepcidin production when challenged by inflammatory stimuli [44]. In the present study, however, we demonstrated that while cultured neurons did not respond to LPS, a robust hepcidin upregulation was found when the neurons were co-cultured with microglia. We also showed by immunohistochemistry that in vivo, LPS administration could induce hepcidin expression in neurons in the cortex and substantia nigra, casting doubt on the non-neuronal origin of hepcidin.
A notable feature of the effect of IL-6 on hepcidin expression and release in cell cultures is the rapid response detected within the first 2 h, which progressively returned to baseline level at later hours. In fact, similar observation has been found in periphery [45] and also in the choroid plexus and cultured brain cells in which IL-6 or LPS caused a peak increase in hepcidin gene expression at around 2–3 h [44, 46]. Changes in the expression of iron-transport proteins apart from Fpn1, such as DMT-1, exhibited a slower time course (data not shown), which is consistent with the notion that regulation of hepcidin gene expression represents a primary response of inflammatory stimuli. However, in the in vivo brain, instead of an early peak response, we found that there was prolonged upregulation of hepcidin level by LPS. These findings together suggest that in vivo, chronic activation by LPS causes a prolonged release of IL-6 and therefore elevated hepcidin level.
Although IL-6 production and STAT3 pathway are implied in the upregulation of hepcidin in the CNS, regional difference of the response in the brain seems to be present. For example, we have shown that LPS-induced hepcidin increase is found in the cortex and substantia nigra but not the hippocampus [23]. Since we have shown that microglia plays an essential role in the hepcidin response acting as the source of IL-6, the abundance of microglia in different brain regions may underlie the regional specificity observed. Interestingly, in line with this idea, Kim et al. [47] demonstrated that there is regional difference in susceptibility to LPS-induced neurotoxicity in the rat brain, which is most likely dependent on the abundance of microglia in the specific region. For example, the substantia nigra, a susceptible region to LPS-induced neurotoxicity, contains the highest concentration of microglia [48]. In addition, the differential vulnerability of different neuronal populations [49, 50] and the distribution of iron-related proteins [1] in different brain regions may also be contributing factors.
There is substantial evidence that inflammation is associated with many neurological disorders, including AD [51–53], PD [54–56], cerebral ischemia and brain trauma [57], and infectious diseases such as bacterial meningitis [58]. However, whether inflammation in neurological disorders is beneficial or deleterious is so far uncertain. Inflammation plays a key role in host defense and tissue repair [59, 60], but on the other hand, it can cause neuronal damage and underlies the pathology of neurological disorders. Inflammation and iron metabolism is closely related, and misregulation in brain iron metabolism is regarded as one of the basic causes of neuronal death in some neurodegenerative disorders [20–22]. Since the iron exporter Fpn1 is downregulated in neurons by the increased hepcidin after LPS administration, intracellular iron may thus be raised and causes neuronal damage via increased oxidative stress, which is known to be associated with the endotoxicity of LPS [61]. On the other hand, it has been demonstrated that hepcidin can inhibit the expression of transferrin receptor 1 in astrocytes via a cyclic AMP-protein kinase A pathway [18]. Our unpublished data also suggested that hepcidin upregulation could reduce iron in the brain by inhibiting iron transport across the blood–brain barrier and uptake by neurons and other brain cells. This finding is in line with the reported action of LPS-induced hepcidin choroid plexus epithelial cells in suppressing iron entry into the cerebrospinal fluid [46]. In this regard, hepcidin may have a beneficial role in suppressing iron-related neurodegenerative process. Therefore, the significance of LPS or inflammation upregulation of hepcidin in the CNS is still unclear and awaits further investigation. Nevertheless, upregulation of hepcidin is likely to play a key role in the host defense mechanism of the CNS. Elucidating the mechanism and impact of alteration in hepcidin expression in neuroinflammation has strong implication in exploring novel therapeutic strategies in neurodegenerative diseases.
Abbreviations
- AD:
-
Alzheimer’s disease
- ELISA:
-
Enzyme-linked immunosorbent assay
- FBS:
-
Fetal bovine serum
- Fpn1:
-
Ferroportin 1
- IL-1α:
-
Interleukin-1 alpha
- IL-1β:
-
Interleukin-1 beta
- IL-6:
-
Interleukin-6
- LPS:
-
Lipopolysaccharides
- MAP2:
-
Microtubule-associated protein 2
- PD:
-
Parkinson’s disease
- STAT3:
-
Signal transducer and activator of transcription 3
- TLRs:
-
Toll-like receptors
- TNF-α:
-
Tumor necrosis factor-alpha
References
Qian ZM, Wang Q (1998) Expression of iron transport proteins and excessive iron accumulation in the brain in neurodegenerative disorders. Brain Res Rev 27:257–267
Andrews NC (1999) Disorders of iron metabolism. N Engl J Med 341:1986–1995
Fleming RE, Sly WS (2002) Mechanisms of iron accumulation in hereditary hemochromatosis. Annu Rev Physiol 64:663–680
Hentze MW, Muckenthale MU, Andrews NC (2004) Balancing acts: molecular control of mammalian iron metabolism. Cell 117:285–297
Nemeth E, Ganz T (2006) Regulation of iron metabolism by hepcidin. Annu Rev Nutr 26:323–342
Ganz T (2006) Hepcidin—a peptide hormone at the interface of innate immunity and iron metabolism. Curr Top Microbiol Immunol 306:183–198
Krause A, Neitz S, Magrert HJ, Schulz A, Forssmann WG, Schulz-Knappe P, Adermann K (2000) LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett 480:147–150
Park CH, Valore EV, Waring AJ, Ganz T (2001) Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem 276:7806–7810
Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P, Loreal O (2001) A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem 276:7811–7819
Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A, Vaulont S (2001) Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci U S A 98:8780–8785
Nicolas G, Bennoun M, Porteu A, Mativet S, Beaumont C, Grandchamp B, Sirito M, Sawadogo M, Kahn A, Vaulont S (2002) Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc Natl Acad Sci U S A 99:4596–4601
Nicolas G, Chauvet C, Viatte L, Danan JL, Bigard X, Devaux I, Beaumont C, Kahn A, Vaulont S (2002) The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest 110:1037–1044
Lesbordes-Brion JC, Viatte L, Bennoun M, Lou DQ, Ramey G, Houbron C, Hamard G, Kahn A, Vaulont S (2006) Targeted disruption of the hepcidin 1 gene results in severe hemochromatosis. Blood 108:1402–1405
Nicolas G, Viatte L, Lou D-Q, Bennoun M, Beaumont C, Kahn A, Andrews NC, Vaulont S (2003) Constitutive hepcidin expression prevents iron overload in a mouse model of hemochromatosis. Nat Genet 34:97–101
Rivera S, Liu L, Nemeth E, Gabayan V, Sorensen OE, Ganz T (2005) Hepcidin excess induces the sequestration of iron and exacerbates tumor-associated anemia. Blood 105:1797–1802
Hentze MW, Muckenthaler MU, Galy B, Camaschella C (2010) Two to tango: regulation of mammalian iron metabolism. Cell 142:24–38
Ganz T, Nemeth E (2011) Hepcidin and disorders of iron metabolism. Annu Rev Med 62:347–360
Du F, Qian C, Ming Qian Z, Wu X-M, Xie H, Yung W-H, Ke Y (2011) Hepcidin directly inhibits transferrin receptor 1 expression in astrocytes via a cyclic AMP-protein kinase a pathway. Glia 59:936–945
Li L, Holscher C, Chen B-B, Zhang Z-F, Liu Y-Z (2011) Hepcidin treatment modulates the expression of divalent metal transporter-1, ceruloplasmin, and ferroportin-1 in the rat cerebral cortex and hippocampus. Biol Trace Elem Res 143:1581–1593
Qian ZM, Shen X (2001) Brain iron transport and neurodegeneration. Trend Mol Med 7(3):103–108
Ke Y, Qian ZM (2003) Iron misregulation in the brain: a primary cause of neurodegenerative disorders. Lancet Neurol 2:246–253
Ke Y, Qian ZM (2007) Brain iron metabolism: neurobiology and neurochemistry. Prog Neurobiol 83(3):149–173
Wang Q, Du F, Qian Z-M, Ge XH, Zhu L, Yung WH, Yang L, Ke Y (2008) Lipopolysaccharide induces a significant increase in expression of iron regulatory hormone hepcidin in the cortex and substantia nigra in rat brain. Endocrinology 149:3920–3925
Wrighting DM, Andrews NC (2006) Interleukin-6 induces hepcidin expression through STAT3. Blood 108:3204–3209
Verga Falzacappa MV, Vujic Spasic M, Kessler R, Stolte J, Hentze MW, Muckenthaler MU (2007) STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood 109:353–358
Pietrangelo A, Dierssen U, Valli L, Garuti C, Rump A, Corradini E, Ernst M, Klein C, Trautwein C (2007) STAT3 Is required for IL-6-gp130–dependent activation of hepcidin in vivo. Gastroenterology 132:294–300
Ke Y, Chang YZ, Duan XL, Du JR, Zhu L, Wang K, Yang XD, Ho KP, Qian ZM (2005) Age-dependent and iron-independent expression of two mRNA isoforms of divalent metal transporter 1 in rat brain. Neurobiol Aging 26(5):739–748
Du F, Z-m Q, Zhu L, Wu XM, W-h Y, T-y T, Ke Y (2009) L-DOPA neurotoxicity is mediated by up-regulation of DMT1 − IRE expression. PLoS ONE 4:e4593
Chang YZ, Qian ZM, Wang K, Zhu L, Yang XD, Du JR, Jiang L, Ho KP, Wang Q, Ke Y (2005) Effects of development and iron status on ceruloplasmin expression in rat brain. J Cell Physiol 204(2):623–633
Zechel S, Huber-Wittmer K, von Bohlen und Halbach O (2006) Distribution of the iron-regulating protein hepcidin in the murine central nervous system. J Neurosci Res 84:790–800
Bowman CC, Rasley A, Tranguch SL, Marriott I (2003) Cultured astrocytes express toll-like receptors for bacterial products. Glia 43(3):281–291
Zhou H, Lapointe BM, Clark SR, Zbytnuik L, Kubes P (2006) A requirement for microglial TLR4 in leukocyte recruitment into brain in response to lipopolysaccharide. J Immunol 177(11):8103–8110
Rolls A, Shechter R, London A, Ziv Y, Ronen A, Levy R, Schwartz M (2007) Toll-like receptors modulate adult hippocampal neurogenesis. Nat Cell Biol 9(9):1081–1088
Shechter R, Ronen A, Rolls A, London A, Bakalash S, Young MJ, Schwartz M (2008) Toll-like receptor 4 restricts retinal progenitor cell proliferation. J Cell Biol 183(3):393–400
Lehnardt S, Lachance C, Patrizi S, Lefebvre S, Follett PL, Jensen FE, Rosenberg PA, Volpe JJ, Vartanian T (2002) The Toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J Neurosci 22:2478–2486
Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, Volpe JJ, Vartanian T (2003) Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci U S A 100:8514–8519
Glezer I, Lapointe A, Rivest S (2006) Innate immunity triggers oligodendrocyte progenitor reactivity and confines damages to brain injuries. FASEB J 20:750–752
Benicky J, Sánchez-Lemus E, Pavel J, Saavedra JM (2009) Anti-inflammatory effects of angiotensin receptor blockers in the brain and the periphery. Cell Mol Neurobiol 29(6–7):781–792
Crawford G, Le W, Smith R, Xie W, Stefani E, Appel S (1992) A novel N18TG2 x mesencephalon cell hybrid expresses properties that suggest a dopaminergic cell line of substantia nigra origin. J Neurosci 12:3392–3398
Su Y, Zhang K, Schluesener HJ (2010) Antimicrobial peptides in the brain. Arch Immunol Ther Exp 58:365–377
Milatovic D, Zaja-Milatovic S, Montine KS, Shie FS, Montine TJ (2004) Neuronal oxidative damage and dendritic degeneration following activation of CD14-dependent innate immune response in vivo. J Neuroinflammation 1(1):20. doi:10.1186/1742-2094-1-20
Montine TJ, Milatovic D, Gupta RC, Valyi-Nagy T, Morrow JD, Breyer RM (2002) Neuronal oxidative damage from activated innate immunity is EP2 receptor-dependent. J Neurochem 83:463–470
Delgado M, Ganea D (2003) Vasoactive intestinal peptide prevents activated microglia-induced neurodegeneration under inflammatory conditions: potential therapeutic role in brain trauma. FASEB J 17:1922–1924
Urrutia P, Aguirre P, Esparza A, Tapia V, Mena NP, Arredondo M, González-Billault C, Núñez MT (2013) Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J Neurochem. doi:10.1111/jnc.12244
Oliveira-Filho JP, Badial PR, Cunha PH, Peiró JR, Araújo JP Jr, Divers TJ, Winand NJ, Borges AS (2012) Lipopolysaccharide infusion up-regulates hepcidin mRNA expression in equine liver. Innate Immunity 18:438–446
Marques F, Falcao AM, Sousa JC, Coppola G, Geschwind D, Sousa N, Correia-Neves M, Palha JA (2009) Altered iron metabolism is part of the choroid plexus response to peripheral inflammation. Endocrinology 150:2822–2828
Kim W-G, Mohney RP, Wilson B, Jeohn G-H, Liu B, Hong J-S (2000) Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci 20:6309–6316
Lawson LJ, Perry VH, Dri P, Gordon S (1990) Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39:151–170
Willard LB, Hauss-Wegrzyniak G, Wenk GL (1999) Pathological and biochemical consequences of acute and chronic neuroinflammation within the basal forebrain cholinergic system of rats. Neuroscience 88:193–200
Kalman J, Engelhardt JI, Le WD, Xie W, Kovacs I, Kasa P, Appel SH (1997) Experimental immune-mediated damage of septal cholinergic neurons. J Neuroimmunol 77:63–74
Koistinaho M, Koistinaho J (2005) Interactions between Alzheimer's disease and cerebral ischemia—focus on inflammation. Brain Res Brain Res Rev 48(2):240–250
Salminen A, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T (2009) Inflammation in Alzheimer's disease: amyloid-beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog Neurobiol 87(3):181–194
von Bernhardi R (2010) Immunotherapy in Alzheimer's disease: where do we stand? Where should we go? J Alzheimers Dis 19(2):405–421
Centonze D, Finazzi-Agrò A, Bernardi G, Maccarrone M (2007) The endocannabinoid system in targeting inflammatory neurodegenerative diseases. Trends Pharmacol Sci 28(4):180–187
Marek K, Jennings D (2009) Can we image premotor Parkinson disease? Neurology 72(7 Suppl):S21–S26
Long-Smith CM, Sullivan AM, Nolan YM (2009) The influence of microglia on the pathogenesis of Parkinson's disease. Prog Neurobiol 89(3):277–287
Stoll G, Jander S, Schroeter M (1998) Inflammation and glial responses in ischemic brain lesions. Prog Neurobiol 56:149–171
Braun JS, Novak R, Herzog KH, Bodner SM, Cleveland JL, Tuomanen EI (1999) Neuroprotection by a caspase inhibitor in acute bacterial meningitis. Nat Med 5:298–302
Streit WJ (2002) Microglia as neuroprotective, immunocompetent cells of the CNS. Glia 40:133–139
Town T, Nikolic V, Tan J (2005) The microglial “activation” continuum: from innate to adaptive responses. J Neuroinflammation 2:24. doi:10.1186/1742-2094-2-24
Sebai H, Gadacha W, Sani M, Aouani E, Ghanem-Boughanmi N, Ben-Attia M (2009) Protective effect of resveratrol against lipopolysaccharide-induced oxidative stress in rat brain. Brain Inj 23:1089–1094
Acknowledgments
This work was supported by the Key Project Grant of the National Natural Science Foundation of China (NSFC) (31330035), the Competitive Earmarked Grants of The Hong Kong Research Grants Council (GRF 466713), The Hong Kong Health and Medical Research Fund (grant No. 0112014), National Basic Research Program of China (973 Program, 2011CB510004), the General Grant of NSFC (31271132, 31371092), and Direct Grant of the Chinese University of Hong Kong (4054042).
Conflict of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Figure 1
Effects of LPS on hepcidin expression in microglia and astrocytes in the cortex and substantia nigra in rat brain. Typical micrographs showing that the expression of hepcipin in microglia in these two regions was not changed after LPS treatment, revealed by double immunofluorescence labeling for Iba-1 (A). Similarly, the expression of hepcidin in astrocytes in these two regions was also not elevated, revealed by double immunofluorescence labeling for GFAP (B). Scale bar = 50 μm in main picture and 20 μm in inset (n = 4 in control group; n = 5 in LPS treated group). (TIFF 422 kb)
High resolution image
(JPEG 180 kb)
Supplementrary Figure 2
Effect of LPS on hepcidin expression in substantia nigra dopamine neurons. The expression of hepcidin in the substantia nigra dopamine neurons was elevated after LPS treatment, revealed by double immunofluorescent labeling for tyrosine hydroxylase (TH) Scale bar = 50 μm in main picture and 20 μm in inset (n = 4 in control group; n = 5 in LPS-treated group). (TIFF 175 kb)
High resolution image
(JPEG 98 kb)
Rights and permissions
About this article
Cite this article
Qian, ZM., He, X., Liang, T. et al. Lipopolysaccharides Upregulate Hepcidin in Neuron via Microglia and the IL-6/STAT3 Signaling Pathway. Mol Neurobiol 50, 811–820 (2014). https://doi.org/10.1007/s12035-014-8671-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-014-8671-3