
Abstract
Elevated iron levels are considered to play a role in the neurodegenerative mechanisms that underlie Alzheimer's and Parkinson's disease. The linkage between hepcidin (Hepc) and ferroportin-1 (FPN1), the divalent metal transporter 1 (DMT1), and ceruloplasmin (CP) in the brain is unknown. To discern the role of Hepc in regulating the expression of these proteins, we investigated FPN1, DMT1, and CP protein and mRNA expression in the brain after the intracerebroventricular injection of Hepc. Our results show that after Hepc injection, expression of FPN1 mRNA and FPN1 protein was inhibited in the cerebral cortex and hippocampus. Furthermore, we showed a clear change of DMT1 and CP protein and mRNA levels in the brain. The immunohistochemical analysis revealed an increase of DMT1 and a decrease of CP levels. Semi-quantitative analysis using PCR methods showed an increase of DMT1(+IRE) mRNA, and a decrease of DMT1(−IRE) mRNA and CP mRNA levels. Since alterations in iron levels in the brain are causally linked to degenerative conditions such as Alzheimer's disease, an improved understanding of the regulation of iron transport protein expression such as FPN1, DMT1, and CP could lead to novel strategies for treatments.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Recent studies have shown that hepcidin (Hepc) plays an essential role in maintaining normal iron homeostasis [1]. Hepc levels increase with iron loading and inflammation and decreases with anemia and hypoxia. Regulation of Hepc plays a central role in some diseases. For example, Hepc regulation is in part responsible for the degree of iron overload in aceruloplasminemia, which is a rare autosomal recessive neurodegenerative disease associated with brain and liver iron accumulation, and which typically presents with movement disorders, retinal degeneration, and diabetes mellitus [2].
Ferroportin-1 (FPN1) is the sole exporter of iron and is responsible for iron absorption from the intestine, recycling of erythrocyte iron by macrophages, and maternal delivery of iron to the fetus [3, 4]. Although Hepc is predominantly expressed in the liver, it is also detected in the brain [5]. Hepc is widely expressed in different brain areas, including the olfactory bulb, cortex, hippocampus, amygdala, thalamus, hypothalamus, mesencephalon, cerebellum, pons, spinal cord, as well as in dorsal root ganglia of the peripheral nervous system [6]. FPN1 is the first transport protein identified in vertebrates that specifically removes iron from cells. FPN1 is also found in the brain, mainly in the choroid plexus, in ependymal cells lining the ventricles, and in neuromelanin cells in the substantia nigra [7]. Iron levels in the brain are of central importance for neuronal function and survival. Increased levels of iron is considered to be responsible for the neurotoxic properties of l-dopa [8]. Iron-generated hydroxyl radicals are neurotoxic [9], and elevated iron levels are considered to play a role in the neurodegenerative mechanisms that underlie Alzheimer's and Parkinson's disease [10]. The exact mechanisms of Hepc activity and the linkage between Hepc, FPN1 expression, and other iron transport proteins in the brain are unknown.
The divalent metal transporter 1 (DMT1) is a transmembrane protein [11]. There are four DMT1 isoforms that differ in their N- and C-termini. Based on the presence or absence of the iron response element (IRE), DMT1 mRNA encodes two different proteins altered at the C-terminal: DMT1 with IRE (+IRE) and DMT1 without IRE (−IRE). DMT1 is a key protein for transmembrane iron transport as well as iron translocation from endosomes. High levels of DMT1 are extensively expressed in the brain: the nuclei of the basal ganglia, particularly the caudate nucleus, putamen, and substantia nigra pars reticulata. It may explain why high levels of iron are found in these regions [12]. Berg [13] demonstrated that both + IRE and −IRE forms of DMT1 are distributed in neurons and astrocytes in the rat SN, the same region where iron deposits are found in Parkinson's disease (PD) patients and animal models of PD.
Ceruloplasmin (CP) is a serum ferroxidase and contains over 95% of the copper present in plasma of PD patients [14]. The human CP gene has 20 exons and spans about 65 kb. It encodes a 132-kDa glycoprotein containing 1,046 amino acids. CP is synthesized predominantly by hepatocytes and undergoes N-linked glycosylation prior to secretion into circulation. CP expression has also been detected in the spleen, lung, testis, and brain. CP acts in conjunction with FPN1 to mediate iron release from hepatic cells. CP is synthesized and secreted by hepatocytes, astrocytes, choroid plexus, and Sertoli cells in addition is expressed as a glycosylphosphatidylinositol (GPI)-linked CP on both astrocytes and Sertoli cells. CP does not bind to transferrin directly. The main roles and functions of CP are (1) to regulate iron efflux, (2) to stabilize FPN1 membrane expression, (3) to express in form of GPI-linked CP, (4) to function as a ferroxidase and to regulate the oxidation of Fe2+ to Fe3+.
Mena et al. [15] showed that the effect of Hepc is cell dependent: in macrophages, it inhibits iron export by inducing FPN1 degradation, whereas in enterocytes, it inhibits apical iron uptake by inhibiting DMT1 transcription. In cultural intestinal epithelial cells which were exposed to synthetic Hepc for 24 h, apical iron uptake was decreased, accompanied by a decrease in DMT1 protein and mRNA expression [16]. Ge et al. [17] showed a decrease of FPN-1 levels induced by Hepc in the H9C2 cardiomyocyte cell line, but no significant change in CP levels was found. Chung et al. [18] showed that Hepc decreases iron transporter expression in vivo in mouse duodenum and spleen, and in vitro in THP-1 macrophages and intestinal caco-2 cells. Evidence from the in vivo uptake studies suggests that the primary effect of Hepc is to downregulate iron efflux across the basolateral membrane of enterocytes [19].
The mechanisms by which Hepc regulates FPN1, DMT1, and CP in the brain are unclear. To discern the role of Hepc in regulating FPN1, DMT1, and CP expression in the brain, we investigated FPN1, DMT1, and CP protein and mRNA expression in the brain after intracerebroventricular (icv) injection of Hepc.
Materials and Methods
Drugs
Purified Hepc for injection: HepcC81-P Mouse HepcC 25-aa was purchased from AlphaDiagnostic. HepcC was synthesized, purified to >95% by HPLC. Mouse HepcC-25 was also synthesized with N-terminal Biotin. Cysteines were air-oxidized assuming that they will assume natural cys–cys bridging.
Animals
Twenty-four male Wistar rats (170–220 g) purchased from the animal Center in Shanxi Medical University were grouped: saline-treated control and Hepc-treated rats. Twelve rats were treated by saline, and 12 rats were treated by Hepc icv. And then rats were subgroups: six rats in saline-treated control and six Hepc-treated rats were used for immunohistochemistric analysis; six rats in saline-treated control and six Hepc-treated rats were used for reverse transcription PCR (RT-PCR) analysis. mRNA or protein of DMT1, FPN1, and CP in every animal was measured.
The rats were anesthetized with 8% chloral hydrate by intra-abdominal injection, and their heads were placed in a stereotaxic apparatus. A guide cannula was inserted into the left lateral ventricle (posterior, 1.3 mm and lateral, 1.5 mm to the bregma as a reference and 4.0 mm ventral to the brain surface). Hepc solution (5 μl) (6 μg/μl) in Hepc-treated rats and 5 μl saline in control rats was administered.
After injection of Hepc (or saline), rats were subgrouped. (1) Twelve rats were killed and brains were fixed by transcardial perfusion using 4% paraformaldehyde after 12 h of Hepc injection icv. The brain was further fixed by immersion in a solution of 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.2 or (2) The brains of the second group of 12 rats were taken immediately after 12 injections of Hepc icv, and cerebral cortex and hippocampus was frozen for RT-PCR analysis.
All experiments received the appropriate ethical permission. The NIH Guide regulations for the care and use of laboratory animals (National Institutes of Health Publications, No. 80–23, revised 1978) were adhered to.
Immunohistochemistry
For immunohistochemical staining, the sections were immersed in 0.3% H2O2 in methanol to inhibit endogenous peroxidase and then precoated with 1% nonfat milk in PBS to block nonspecific binding. For immunohistochemical staining of FPN1, a rabbit polyclonal antibody (Ab) to the anti-rat FPN1 (1:500, San Antonio company, USA) was used.
For immunohistochemical staining of DMT1 and CP, a rabbit polyclonal Ab to the anti-rat DMT1 (1:500, San Antonio company, USA) and the anti-rat CP (1:200, San Antonio company, USA) was applied to the sections as the primary Ab. The sections were incubated with a biotin-conjugated anti-rabbit IgG as the secondary Ab. The Ab reactions were visualized by using a standard ABC secondary antibody staining Kit which was used according to the manufacturers' instructions. For negative control, the primary antibody was omitted. Positive cells were stained by brown deposition.
All morphometric parameters were counted using the image analyzing computer program Image-Pro Plus Version 6.0. The cross-sectional profile of each pulp using a magnification of ×5 and representative fields under a ×100 oil-immersion objective were photographed with a digital camera. Total cross-sectional area was measured using a magnification of ×5, and the density was denoted as mean density of object, mean density of field of view field; and sum area of objects; and sum area of field of view. A positive unit (PU) was calculated according to the formula as follows:
RT-PCR
Total RNA was purified using the Trizol reagent (Shanghai Sangon Biological Engineering Technology and Services Co., Ltd.), and RT-PCR was performed using the Takara two-step RT-PCR kits (Takara Biotechnology (Dalian) Co., Ltd.), following the protocol of manufacturer. The primers for FPN1 mRNA analysis were as follows: FP(AF394785), forward 5′-TAA TGG GAA CTG TGG CCT TC-3′, reverse 5′-GGA GAT TAT GGG GAC GGA TT-3′; GAPDH (NM_017008), forward 5′-TCC CTC AAG ATT GTC AGC AA-3′, reverse 5′-AGA TCC ACA ACG GAT ACA TT-3′ (Takara Biotechnology Co. Ltd., Dalian, China). PCR was performed under the following conditions: step 1, 2 min at 94°C for one cycle; and step 2, 30 s at 94°C, 30 s at 57°C (50°C for GAPDH), 1 min at 72°C, for 30 cycles.
For DMT1 analysis, the methods according to Roth et al. [20] were employed. The primers FOR DMT1(+IRE) were as follows: forward 5′-CGGTAAGCATCTCTAAAG-3′, reverse 5′-TAGCAGCATGCTATTTGAC-3′. The primers FOR DMT1(−IRE) were as follows:), forward 5′-TCTAGATGACCAACAGCC-3′, reverse 5′-GCAGACACAAGCCTGCGT-3′; GAPDH primer see for FPN1. PCR was performed using the methods published by Chen et al. [21].
The primers for CP were as follows: forward 5′-GTATGTGATGGCTATGGGCAA TGA-3′, reverse 5′-CCTGGATGGAACTGGTGATGGA-3′; GAPDH primer see for FPN1.
mRNA bands were analyzed using imaging densitometry. The density of mRNA was expressed as absorbance (optical density, OD × square millimeter).
Statistical Analysis
Data were expressed as mean ± SD. To analyze the data, one-way ANOVA was used. Calculations were performed with the SPSS program, version 11.5.
Results
Expression of FPN1 Mediated by Hepc icv
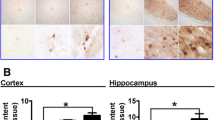
FPN1 expression was significantly decreased by 26.83% and 30.87% in the cerebral cortex (ANOVA p < 0.01) and hippocampus (ANOVA p < 0.01), respectively, after Hepc icv injection (Table 1; Fig. 1). An extensive expression of FPN1 was shown in the cerebral cortex and hippocampus. Positive staining was present predominantly on the cell membrane and some in the cytoplasm. A strong expression of FPN1 was shown in the cerebral cortex and hippocampus of control rats, and a significant decrease of FPN1 expression in hippocampus and cerebral cortex was induced by Hepc icv injection (Fig. 1).

FPN1 neurons was shown by immunohistochemistry in different brain regions after Hepc injection from lateral cerebral ventricle. a FPN1 positive staining of cerebral cortex. b FPN1 positive staining of hippocampus in the control groups, respectively. Positive staining is evident in cell bodies. c FPN1 positive staining of cerebral cortex. d FPN1 positive staining of hippocampus in the Hepc injected groups. There is a weaker reactivity in the Hepc injected groups than the control. Bar a,b = 25 μm, bar c,d = 50 μm. Quantitatively, the mean density of positive straining measured using the image analyzing computer program Image-Pro Plus Version 6.0. Right-hand histogram shows that decrease of FPN1 expression was from 0.25 down to 0.17 in the cerebral cortex and from 0.22 down to 0.16 in hippocampus after treatment by Hepc (*p < 0.01). See “Materials and Methods” for technical details
Expression of FPN1 mRNA in the Cerebral Cortex and Hippocampus
There was a significant decrease of expression of FPN1 mRNA in the cortex and hippocampus of rats treated by Hepc. FPN1 mRNA density expressed as in OD × square millimeter was a decreased by 8.32% and 9.47% in cerebral cortex and hippocampus (ANOVA p < 0.01), respectively, after Hepc icv injection (Table 2; Fig. 2).

Expression of FPN1 mRNA levels in the cerebral cortex, hippocampus after Hepc injection into the lateral cerebral ventricle. Inhibition of FP1 mRNA expression induced by Hep icv injection. Less FP1 mRNA expression in cerebral cortex and hippocampus was shown in rats treated by Hepc (H) as compared with control rats (C). Quantificationally, right-hand histogram shows that degree of decrease of FPN1 expression was from 1.08 down to 0.99 in cerebral cortex and from 1.01 down to 0.91 in hippocampus after treatment with Hepc (*p < 0.01). See “Materials and Methods” for technical details
Expression of DMT1 Induced by Hepc icv
An extensive expression of DMT1 was shown in the cerebral cortex and hippocampus. In the cerebral cortex, immunoreactive staining was found in neurons and glia cells. Immunoreactive granules were predominantly seen in cytoplasm in neurons. In the hippocampus, immunoreactive granules were seen in the cytoplasm and axons of pyramidal cells. The expression of DMT1 increased after treatment with Hepc. A 28.31% increase was seen in cerebral cortex and a 25.61% increase was seen in the hippocampus (Table 1; Fig. 3).

DMT1 neurons was shown by immunohistochemistry in different brain regions after Hepc injection from lateral cerebral ventricle. a DMT1 positive staining of cerebral cortex. b DMT1 positive staining of hippocampus in the control groups, respectively. Positive staining is evident in cell bodies. c DMT1 positive staining of cerebral cortex, d DMT1 positive staining of hippocampus in the Hepc-injected groups. There is a stronger reactivity in the Hepc-injected groups than the control. Bar a,b = 25 μm, bar c,d = 50 μm. Quantitatively, the mean density of positive straining measured using the image analyzing computer program Image-Pro Plus Version 6.0. Right-hand histogram shows that increase of DMT1 expression was from 0.21 down to 0.26 in the cerebral cortex and from 0.23 down to 0.26 in hippocampus after treatment by Hepc (*p < 0.01). See “Materials and Methods” for technical details
The Expression of DMT1 mRNA
Expression of DMT1 mRNA was evaluated by semi-quantitative analysis in the cerebral cortex and hippocampus. Results showed that DMT1 (+IRE) density expressed as in OD × square millimeter was increased by 53.34% in the cerebral cortex and by 16.97% in the hippocampus (p < 0.01) after treatment with Hepc icv. Furthermore, DMT1 (−IRE) density was decreased by 10.95% in the cerebral cortex and by 19.04% in the hippocampus (p < 0.01) after treatment with Hepc icv (Table 2; Figs. 4 and 5).

Expression of DMT1(+) mRNA levels in the cerebral cortex, hippocampus after Hepc injection into the lateral cerebral ventricle. Increase of DMT1(+) mRNA expression induced by Hepc. More DMT1(+) mRNA expression in cerebral cortex and hippocampus was shown in rats treated with Hep (H) as compared to control rats (C). Quantificationally, right-hand histogram shows that the degree of increase of DMT1(+) mRNA expression was from 0.56 up to 0.86 in cerebral cortex and from 0.75 up to 0.88 in hippocampus after treated by Hep (*p < 0.01). See “Materials and Methods” for technical details

Expression of DMT1 (−) mRNA levels in the cerebral cortex, hippocampus after Hepc injection into the lateral cerebral ventricle. Decrease of DMT1 (−) mRNA expression induced by Hepc. Less DMT1 (−) mRNA expression in cerebral cortex and hippocampus was shown in rats treated with Hepc (H) as compared to control rats (C). Quantificationally, right-hand histogram shows that the degree of decrease of DMT1 (−) mRNA expression was from 0.43 down to 0.38 in cerebral cortex and from 0.65 down to 0.52 in hippocampus after treated by Hep (*p < 0.01). See “Materials and Methods” for technical details
Expression of CP Induced by Hepc icv
An increased expression of CP was shown in the cerebral cortex and hippocampus. In the cerebral cortex, immunoreactive staining was found in astrocytes. The expression of CP decreased after treatment with Hepc icv. A 20% decrease was seen in the cerebral cortex and a 21.71% decrease was seen in the hippocampus (Table 1; Fig. 6).

CP neurons was shown by immunohistochemistry in different brain regions after Hepc injection from lateral cerebral ventricle. a CP positive staining of cerebral cortex. b CP positive staining of hippocampus in the control groups, respectively. Positive staining is evident in cell bodies. c CP positive staining of cerebral cortex. d CP positive staining of hippocampus in the Hepc-injected groups. There is a weaker reactivity in the CP-injected groups than the control. Bar a,b = 25 μm, bar c,d = 50 μm. Quantitatively, the mean density of positive straining measured using the image analyzing computer program Image-Pro Plus Version 6.0. Right-hand histogram shows that decrease of CP expression was from 0.27 down to 0.22 in the cerebral cortex and from 0.27 down to 0.21 in hippocampus after treatment by Hepc (*p < 0.01). See “Materials and Methods” for technical details
The Expression of CP mRNA
An extensive expression of CP in the cerebral cortex and hippocampus was found. Quantitatively, CP density expressed as in OD × square millimeter was decreased by 33.14% in the cerebral cortex (p < 0.01) and by 13.17% in the hippocampus (p < 0.01) after treatment by Hepc icv (Table 2; Fig. 7).

Expression of CP mRNA levels in the cerebral cortex, hippocampus after Hepc injection into the lateral cerebral ventricle. Inhibition of CP mRNA expression induced by Hepc. Less CP mRNA expression in cerebral cortex and hippocampus was shown in rats treated by Hepc (H) as compared to control rats (C). Quantificationally, right-hand histogram shows that the degree of decrease of CP mRNA expression was from 0.78 down to 0.52 in cerebral cortex and from 0.76 down to 0.66 in hippocampus after treatment with Hepc (*p < 0.01). See “Materials and Methods” for technical details
Discussion
Our results show that after Hepc injection, expression of FPN1 mRNA was inhibited and FPN1 expression in neurons was reduced in the cerebral cortex and hippocampus. FPN1 is the sole exporter of iron which is responsible for iron absorption from the intestine, recycling of erythrocyte iron by macrophages, and other processes [3, 4]. In most physiological conditions, the transport of iron by FPN1 is regulated by Hepc by inducing the internalization of FPN1 from cell surfaces [1]. Similar changes were seen in present study in the brain: FPN1 was downregulated after Hepc icv. It is to be assumed that in the brain, Hepc also binds to the iron exporter FPN1, induces its internalization and degradation, and thereby blocks cellular iron efflux from neurons. In the situation of an unphysiological elevation of iron levels in the body, Hepc combined with FPN1 is taken up by cells. Once internalized, FPN1 is degraded in the lysosome, resulting in decreased cellular iron export [22]. The FPN1 internalization in both macrophages and duodenal epithelial cells leads to a decrease of available iron in the bloodstream. This negative feedback system is thought to be part of a basic mechanism to maintain iron homeostasis [23, 24]. The disturbance of Hepc and FPN1 activity results in the dysregulation of iron absorption relating to both iron deficiency and excessive iron accumulation [23]. Most hereditary iron overload diseases can be explained by inadequate production of Hepc in response to iron stores or by mutations in FPN1 [25].
Zechel et al. [6] showed a wide-spread distribution of Hepc in different brain areas, including the neocortex and hippocampus. FPN1 is also found in the brain, e.g., in the substantia nigra [7]. Iron levels are tightly controlled in the brain and chronically elevated or reduced iron levels have toxic effects [19]. Elevated neuronal iron levels play a role in the neurodegenerative mechanisms that underlie Alzheimer's and Parkinson's disease [8, 21] very likely by producing toxic amounts of free radicals [20, 26, 27]. It is feasible that a lack of control of iron levels in the brain is linked to disturbed FPN1 or Hepc expression. Similarly, Wang et al. [28] showed an inhibition of FPN1 expression in the murine brain treated by Hepc. Similarly, in the present study, we showed an inhibition of FPN1 protein and mRNA expression. Further, we showed a clear change of DMT1 and CP protein and mRNA levels in the brains of rats which were treated by Hepc icv. An increase of DMT1 protein and a decrease of CP protein expression were seen. Semi-quantitative analysis of the DMT1 and CP mRNA expression using PCR methods revealed that an increase of DMT1 (+IRE) mRNA and a decrease of DMT1 (−IRE) mRNA, and a decrease of CP mRNA in rats took place after Hepc treatment. However, we found an increase of DMT1 protein and a decrease of DMT1 (−IRE) mRNA in rats treated by Hepc icv in the present study. A possible explanation for this apparent discrepancy between DMT1 protein upregulation and DMT1 −IRE mRNA downregulation is that the DMT1 +IRE mRNA isoform contributes the most to total DMT1 protein expression as opposed to the −IRE mRNA counterpart. A decreased expression of DMT1 protein levels after Hepc treatment was reported in a cell culture study [16]. That study showed a decrease of DMT1 protein and mRNA expression in Caco-2 cells, an intestinal epithelial cell line, when exposed to synthetic Hepc for 24 h. Contrary to this report, our study showed an increase of DMT1 protein and (DMT +IRE) mRNA expression and a decrease of DMT1 (−IRE) levels. One reason for this difference might be linked to the conditions that the cells were in. Iron transport is tightly regulated and will depend on the iron load status of the cells in the study. Ke et al. [29] showed that low concentrations of CP can promote iron influx into iron-deficient neurons, but not into neurons with normal iron status. The same concentrations of CP had no effect on iron efflux from neurons with normal iron levels. Ge et al. [17] showed that Hepc has the ability to reduce FPN1 levels as well as iron release in heart cells. The study also found that Hepc has no significant effects on CP levels in heart cells. This suggests that DMT1 expression is regulated by iron levels, and that the iron concentrations in vivo can differ greatly from those in vitro. The change of DMT1 protein observed in our study correlated with the changes of DMT1 (+IRE) and (−IRE) mRNA expression. An increased expression was shown in DMT1 (+IRE) mRNA and a decreased expression was shown in DMT1 (−IRE) mRNA in rat brains treated by Hepc. This highlights the complexity of DTM1 expression regulation and underscores the different roles that DMT1 (+IRE) and (−IRE) play in iron transport. CP and DMT1 are essential for brain iron homeostasis. In conclusion, the changes induced by Hepc icv in the present study: DMT1 protein upregulation combined with FPN1 and CP downregulation may increase cellular iron levels in rat brains treated by Hepc. This will have to be investigated in further detail in future experiments.
To our knowledge, this is the first report of changes of FPN1, CP, or DMT1 levels in the brain induced by Hepc. However, the precise functions of Hepc in neuronal iron transport has not been determined. Additionally, we lack understanding of how exactly Hepc regulates FPN1, CP, and DMT1 expression, and further research is required to cast light on these mechanisms that may be tissue specific. In our study, FPN1 and CP mRNA and protein expression in cerebral cortex and hippocampus was inhibited by Hepc icv. Hence, we speculate that Hepc can decrease iron influx by inhibiting FPN1 or CP in the cerebral cortex and hippocampus.
Since alterations in iron levels in the brain are causally linked to a number of diseases and degenerative conditions, an improved understanding of the regulation of iron transport protein expression such as FPN1, DMT1, and CP could lead to novel strategies of treatment.
References
Nemeth E, Ganz T (2006) Regulation of iron metabolism by hepcidin. Annu Rev Nutr 26:323–342
Finkenstedt A, Wolf E, Höfner E, Gasser BI, Bösch S, Bakry R, Creus M, Kremser C, Schocke M, Theurl M, Moser P, Schranz M, Bonn G, Poewe W, Vogel W, Janecke AR, Zoller H (2010) Hepatic but not brain iron is rapidly chelated by deferasirox in aceruloplasminemia due to a novel gene mutation. J Hepatol 53:1101–1107
De Domenico I, Nemeth E, Nelson JM, Phillips JD, Ajioka RS, Kay MS, Kushner JP, Ganz T, Ward DM, Kaplan J (2008) The hepcidin-binding site on ferroportin is evolutionarily conserved. Cell Metab 8:146–156
Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, Andrews NC (2005) The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab 1:191–200
Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P, Loréal O (2001) A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem 276:7811–7819
Zechel S, Huber-Wittmer K, von Bohlen und Halbach O (2006) Distribution of the iron-regulating protein hepcidin in the murine central nervous system. J Neurosci Res 84:790–800
Clardy SL, Wang X, Boyer PJ, Earley CJ, Allen RP, Connor JR (2006) Is ferroportin-hepcidin signaling altered in restless legs syndrome? J Neurol Sci 247:173–179
Chang YZ, Ke Y, Du JR, Halpern GM, Ho KP, Zhu L, Gu XS, Xu YJ, Wang Q, Li LZ, Wang CY, Qian ZM (2006) Increased divalent metal transporter 1 expression might be associated with the neurotoxicity of L-DOPA. Mol Pharmacol 69:968–974
Chao HM, Chen YH, Liu JH, Lee SM, Lee FL, Chang Y, Yeh PH, Pan WH, Chi CW, Liu TY, Lui WY, Ho LT, Kuo CD, Lin DE, Chan CC, Yang DM, Lin AM, Chao FP (2008) Iron-generated hydroxyl radicals kill retinal cells in vivo: effect of ferulic acid. Hum Exp Toxicol 27:327–339
Barnham KJ, Bush AI (2008) Metals in Alzheimer's and Parkinson's diseases. Curr Opin Chem Biol 12:222–228
Song N, Jiang H, Wang J, Xie JX (2007) Divalent metal transporter 1 up-regulation is involved in the 6-hydroxydopamine-induced ferrous iron influx. J Neurosci Res 85:3118–3126
Knutson M, Menzies S, Connor J, Wessling-Resnick M (2004) Developmental, regional, and cellular expression of SFT/UbcH5A and DMT1 mRNA in brain. J Neurosci Res 76:633–641
Berg D, Hochstrasser H (2006) Iron metabolism in parkinsonian syndromes. Mov Disord 9:1299–1310
Hellman NE, Gitlin JD (2002) Ceruloplasmin metabolism and function. Annu Rev Nutr 22:439–458
Mena NP, Esparza A, Tapia V, Valdés P, Núñez MT (2008) Hepcidin inhibits apical iron uptake in intestinal cells. Am J Physiol Gastrointest Liver Physiol 294:192–198
Yamaji S, Sharp P, Ramesh B, Srai SK (2004) Inhibition of iron transport across human intestinal epithelial cells by hepcidin. Blood 104:2178–2180
Ge XH, Wang Q, Qian ZM, Zhu L, Du F, Yung WH, Yang L, Ke Y (2009) The iron regulatory hormone hepcidin reduces ferroportin 1 content and iron release in H9C2 cardiomyocytes. J Nutr Biochem 20:860–865
Chung B, Chaston T, Marks J, Srai SK, Sharp PA (2009) Hepcidin decreases iron transporter expression in vivo in mouse duodenum and spleen and in vitro in THP-1 macrophages and intestinal Caco-2 cells. J Nutr 139:1457–1462
Anderson GJ, Frazer DM, McKie AT, Vulpe CD, Smith A (2005) Mechanisms of haem and non-haem iron absorption: lessons from inherited disorders of iron metabolism. Biometals 18:339–348
Roth JA, Horbinski C, Feng L, Dolan KG, Higgins D, Garrick MD (2000) Differential localization of divalent metal transporter 1 with and without iron response element in rat PC12 and sympathetic neuronal cells. J Neurosci 20:7595–7601
Chen L, Dentchev T, Wong R (2003) Increase expression of ceruloplasmin in the retina following photic injury. Mol Vis 9:151–158
Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J (2004) Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306:2090–2093
Ganz T, Nemeth E (2006) Regulation of iron acquisition and iron distribution in mammals. Biochim Biophys Acta 1763:690–699
Mok H, Mlodnicka AE, Hentze MW, Muckenthaler M, Schumacher A (2006) The molecular circuitry regulating the switch between iron deficiency and overload in mice. J Biol Chem 281:7946–7951
Pietrangelo A (2006) Hereditary hemochromatosis. Biochim Biophys Acta 1763:700–710
Zecca L, Casella L, Albertini A, Bellei C, Zucca FA, Engelen M, Zadlo A, Szewczyk G, Zareba M, Sarna T (2008) Neuromelanin can protect against iron-mediated oxidative damage in system modeling iron overload of brain aging and Parkinson's disease. J Neurochem 106:1866–1875
Hölscher C (2005) Development of beta-amyloid-induced neurodegeneration in Alzheimer's disease and novel neuroprotective strategies. Rev Neurosci 16:181–212
Wang SM, Fu LJ, Duan XL, Crooks DR, Yu P, Qian ZM, Di XJ, Li J, Rouault TA, Chang YZ (2010) Role of hepcidin in murine brain iron metabolism. Cell Mol Life Sci 67:123–133
Ke Y, Ho K, Du J, Zhu L, Xu Y, Wang Q, Wang CY, Li L, Ge X, Chang Y, Qian ZM (2006) Role of soluble ceruloplasmin in iron uptake by midbrain and hippocampus neurons. J Cell Biochem 98:912–919
Acknowledgements
The authors wish to thank Dr. Zhong-Ming Qian in the Hong Kong Polytechnic University for his support. This work was supported by a Grant-in-Aid for Scientific Research (051015) from the ShanXi Science and Technology Department.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, L., Holscher, C., Chen, BB. et al. Hepcidin Treatment Modulates the Expression of Divalent Metal Transporter-1, Ceruloplasmin, and Ferroportin-1 in the Rat Cerebral Cortex and Hippocampus. Biol Trace Elem Res 143, 1581–1593 (2011). https://doi.org/10.1007/s12011-011-8967-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12011-011-8967-3